Resumos
OBJETIVO: Avaliar características clínicas, bioquímicas, hormonais e genéticas de familiares de duas pacientes portadoras de lipodistrofia parcial familiar (FPLD) tipo 2. MATERIAIS E MÉTODOS: Foram avaliados 50 indivíduos de duas famílias brasileiras não relacionadas a partir de dois propósitos com fenótipo de FPLD. Foi confirmada a mutação no éxon 8 do gene LMNA em 18 destes e identificada a substituição em heterozigose no códon 482, resultando na mutação p.R482W. Com base na presença ou não da mutação, os indivíduos foram separados em afetados e não afetados, e comparados quanto a parâmetros clínicos, bioquímicos e hormonais. RESULTADOS: Indivíduos afetados tiveram 2,8 vezes mais chance de manifestar diabetes e síndrome dos ovários policísticos (SOP), maiores índices HOMA-IR, níveis de insulina e de triglicérides e menores níveis de leptina. Essas alterações precedem o início do diabetes, pois foram evidenciadas nos afetados diabéticos e não diabéticos. Foi constatada heterogeneidade fenotípica entre os portadores da mutação. CONCLUSÃO: A mutação no gene da LMNA é determinante de alterações clínicas, bioquímicas e hormonais que implicam deterioração metabólica nos portadores da mutação.
Lâminas A/C; gene LMNA; lipodistrofia; resistência insulínica
OBJECTIVE: To evaluate clinical, biochemical, hormonal and genetic characteristics of relatives of two patients with familial partial lipodystrophy (FPLD) type 2. MATERIALS AND METHODS: Fifty subjects, members of two non-related Brazilian families from two different probands with FPLD phenotype, were evaluated. A mutation in exon 8 of LMNA gene was confirmed in 18 of them, and a heterozygous substitution at codon 482 was identified, predicting a p.R482W mutation. Based on the presence or absence of the mutation, subjects were classified in affected and unaffected, and compared in terms of clinical, biochemical and hormonal parameters. RESULTS: Affected subjects were 2.8 times more likely to manifest diabetes and PCOS, higher HOMA-IR, insulin and triglyceride levels, and lower levels of leptin. These changes preceded the onset of diabetes, because they were observed in diabetic and non-diabetic affected patients. A phenotypic heterogeneity was found among mutation carriers. CONCLUSION: A mutation in the LMNA gene is a determinant of clinical, biochemical and hormonal changes that imply in metabolic deterioration in mutation carriers.
Lamin A/C; LMNA gene; lipodystropy; insulin resistance
ARTIGO PARCIALMENTE RETRATADO
[ARTIGO PARCIALMENTE RETRATADO] Avaliação bioquímica, hormonal e genética das famílias de duas pacientes brasileiras portadoras de lipodistrofia parcial familiar tipo 2
[ARTICLE PARTIAL RETRACTION] Biochemical, hormonal and genetic evaluation of the families of two Brazilian patients with type 2 familial partial lipodystrophy
Dayse CaldasI; Wellington Santana da Silva JúniorII; José Pascoal SimonettiIII; Eliane Veiga da CostaII; Maria Lucia Fleiuss de FariasIV
O Conselho Editorial dos Arquivos Brasileiros de Endocrinologia e Metabologia, ISSN 0004-2730, decidiu, após análise, retratar as figuras 1 e 2 do seguinte artigo original: Caldas D, Silva Júnior WS, Simonetti JP, Costa EV, de Farias MLF. Avaliação bioquímica, hormonal e genética das famílias de duas pacientes brasileiras portadoras de lipodistrofia parcial familiar tipo 2. ABEM. 2013;57/8:583-93.
Trata-se de solicitação das pacientes retratadas.
Lamentamos qualquer mal-entendido causado ao nosso leitor.
ARTIGOS ORIGINAIS
Avaliação bioquímica, hormonal e genética das famílias de duas pacientes brasileiras portadoras de lipodistrofia parcial familiar tipo 2
Biochemical, hormonal and genetic evaluation of the families of two Brazilian patients with type 2 familial partial lipodystrophy
Dayse CaldasI; Wellington Santana da Silva JúniorII; José Pascoal SimonettiIII; Eliane Veiga da CostaII; Maria Lucia Fleiuss de FariasIV
IInstituto Estadual de Diabetes e Endocrinologia Luiz Capriglione (IEDE), Rio de Janeiro, RJ, Brasil
IIIEDE; Programa de Pós-Graduação em Fisiopatologia Clínica e Experimental (FISCLINEX), Universidade do Estado do Rio de Janeiro (UERJ), Rio de Janeiro, RJ, Brasil
IIIInstituto Oswaldo Cruz (IOC/Fiocruz), Rio de Janeiro, RJ, Brasil
IVUniversidade Federal do Rio de Janeiro (UFRJ), Rio de Janeiro, RJ, Brasil
Correspondência para Correspondência para: Dayse Caldas Rua Conde de Bonfim, 370, sala 502 20520-054 Rio de Janeiro, RJ, Brasil caldasdayse@hotmail.com
RESUMO
OBJETIVO: Avaliar características clínicas, bioquímicas, hormonais e genéticas de familiares de duas pacientes portadoras de lipodistrofia parcial familiar (FPLD) tipo 2.
MATERIAIS E MÉTODOS: Foram avaliados 50 indivíduos de duas famílias brasileiras não relacionadas a partir de dois propósitos com fenótipo de FPLD. Foi confirmada a mutação no éxon 8 do gene LMNA em 18 destes e identificada a substituição em heterozigose no códon 482, resultando na mutação p.R482W. Com base na presença ou não da mutação, os indivíduos foram separados em afetados e não afetados, e comparados quanto a parâmetros clínicos, bioquímicos e hormonais.
RESULTADOS: Indivíduos afetados tiveram 2,8 vezes mais chance de manifestar diabetes e síndrome dos ovários policísticos (SOP), maiores índices HOMA-IR, níveis de insulina e de triglicérides e menores níveis de leptina. Essas alterações precedem o início do diabetes, pois foram evidenciadas nos afetados diabéticos e não diabéticos. Foi constatada heterogeneidade fenotípica entre os portadores da mutação.
CONCLUSÃO: A mutação no gene da LMNA é determinante de alterações clínicas, bioquímicas e hormonais que implicam deterioração metabólica nos portadores da mutação.
Descritores: Lâminas A/C; gene LMNA; lipodistrofia; resistência insulínica
ABSTRACT
OBJECTIVE: To evaluate clinical, biochemical, hormonal and genetic characteristics of relatives of two patients with familial partial lipodystrophy (FPLD) type 2.
MATERIALS AND METHODS: Fifty subjects, members of two non-related Brazilian families from two different probands with FPLD phenotype, were evaluated. A mutation in exon 8 of LMNA gene was confirmed in 18 of them, and a heterozygous substitution at codon 482 was identified, predicting a p.R482W mutation. Based on the presence or absence of the mutation, subjects were classified in affected and unaffected, and compared in terms of clinical, biochemical and hormonal parameters.
RESULTS: Affected subjects were 2.8 times more likely to manifest diabetes and PCOS, higher HOMA-IR, insulin and triglyceride levels, and lower levels of leptin. These changes preceded the onset of diabetes, because they were observed in diabetic and non-diabetic affected patients. A phenotypic heterogeneity was found among mutation carriers.
CONCLUSION: A mutation in the LMNA gene is a determinant of clinical, biochemical and hormonal changes that imply in metabolic deterioration in mutation carriers.
Keywords: Lamin A/C; LMNA gene; lipodystropy; insulin resistance
INTRODUÇÃO
As lipodistrofias representam um grupo heterogêneo de doenças raras caracterizadas por alterações na distribuição do tecido adiposo, apresentando seletiva e variável perda tecidual (1). Os indivíduos afetados constituem-se em modelos humanos de resistência insulínica monogênica (2). A severidade das alterações clínicas e metabólicas, as quais incluem hiperinsulinemia, diabetes mellitus, hipertrigliceridemia, síndrome dos ovários policísticos (SOP) e esteatose hepática, entre outras, é diretamente relacionada à extensão da perda tecidual adiposa (1,2).
As lipodistrofias são classificadas, quanto à sua origem, nas formas adquiridas ou familiares, as quais podem ser subdivididas, de acordo com o padrão de perda do tecido adiposo subcutâneo, em formas generalizadas ou parciais (2).
As lipodistrofias adquiridas, formas generalizada e parcial, apresentam origem principalmente autoimune e exibem anormalidades do complemento. Atualmente, o subtipo de lipodistrofia adquirida mais prevalente está relacionado à terapia antirretroviral de longa duração com inibidores da protease em pacientes infectados pelo HIV (1).
Os principais subtipos de lipodistrofias familiares são a forma generalizada autossômica recessiva, ligada a mutações nos genes seipin ou AGPAT2, e a forma parcial autossômica dominante devido a mutações no gene codificador da lâmina A/C (LMNA) ou no gene PPARG. A lipodistrofia parcial familiar (FPLD), cuja forma mais prevalente é a tipo 2 ou variante de Dunnigan (FPLD2), é caracterizada por perda de tecido adiposo subcutâneo no glúteo e nas extremidades (com perdas variadas no tronco) e acúmulo de tecido adiposo na face e no pescoço, iniciando-se na puberdade (3).
A gênese da FPLD2 está intimamente relacionada a alterações na estrutura e na função do envelope nuclear das células eucarióticas. São três os seus componentes distintos: a membrana nuclear, os complexos de poros nucleares (NPCs) e a lâmina nuclear (4). A membrana nuclear é pontuada pelos NPCs, os quais regulam a passagem de macromoléculas entre o núcleo e o citoplasma (5). A lâmina nuclear, organizada como uma malha entrelaçada de fibras na face interna da membrana nuclear, interage com os NPCs e com proteínas transmembrana do envelope nuclear (NETs), promovendo a organização e a interconexão dos NPCs e o ancoramento dos NETs à membrana nuclear. Dessa forma, a lâmina nuclear desempenha numerosas funções biológicas, incluindo a manutenção da integridade estrutural nuclear, organização da cromatina, regência da divisão celular durante a mitose e a meiose e regulação do acesso dos fatores de transcrição ao núcleo (6).
O gene LMNA está situado no cromossomo 1q21-22, apresenta 12 éxons e codifica as lâminas do tipo A e do tipo C. Mutações em sua estrutura foram evidenciadas em alguns estudos em portadores de FPLD2 (7-11).
As doenças relacionadas a mutações no gene LMNA são altamente heterogêneas, incluindo distrofias neuromusculares e cardíacas, lipodistrofias e síndromes de envelhecimento precoce (3). A primeira constatação de mutação no gene LMNA associada à FPLD2 foi relatada em 2000, confirmando a mutação p.R482Q. As mutações no gene LMNA relatadas na literatura estão mais frequentemente localizadas no éxon 8, podendo ou não se associar a mutações no éxon 11 (7-10,12-14).
Neste artigo, serão relatados aspectos clínicos, laboratoriais e a respectiva mutação no gene LMNA em duas famílias brasileiras distintas de indivíduos com fenótipo de FPLD.
PACIENTES E MÉTODOS
Foram avaliados cinquenta indivíduos de duas famílias não relacionadas (FI e FII) a partir de duas pacientes do sexo feminino que apresentam o fenótipo de FPLD, designadas como propósito. Todos deram seu consentimento livre e esclarecido por escrito. O estudo foi aprovado pelo Comitê de Ética em Pesquisa do Instituto Estadual de Diabetes e Endocrinologia Luiz Capriglione (CEP-IEDE) e ratificado pela Comissão Nacional de Ética em Pesquisa (CONEP) do Conselho Nacional de Saúde do Ministério da Saúde.
Avaliação clínica
A avaliação clínica dos propósitos deu-se conforme questionário e roteiro clínico predeterminados, visando à elaboração de anamnese e à obtenção de parâmetros clínicos e antropométricos.
Análise molecular do gene LMNA
O DNA genômico foi extraído de células do sangue periférico (coletado em tubos contendo EDTA) e submetido ao método de extração em coluna (QIAmp DNA Midi-QIAGEN Inc). O método da reação em cadeia da polimerase (PCR) foi utilizado para a identificação e amplificação das regiões genômicas do éxon 8 (primer sense 5'-CCA AGA GCC TGG GTG AGC CTC-3' e primer antisense 5'-GAC ACT TAC CCC AGC GCT CC-3') e do éxon 11 (primer sense 5'-GGA GCC TGC AGG AGC CTG GAG C-3' e primer antisense 5'-CGC CTG CAG GAT TTG GAG A-3') do gene LMNA. O método PCR-RFLP (polimorfismo dos comprimentos dos fragmentos de restrição) foi utilizado para a avaliação da presença de mutação nos 50 indivíduos avaliados. Para o éxon 8, utilizou-se a enzima MspI e, para o éxon 11, a enzima DraIII. Na preparação da reação para sequenciamento, utilizou-se o Kit Big Dye Terminator versões 2 e 3 e a análise do material foi realizada por sequenciamento direto utilizando-se o sequenciador ABI 310 Prism. As sequências nucleotídicas obtidas foram analisadas por meio do programa Sequence Analysis 3.3 (Biosystems) e comparadas pelo programa BLAST (National Center Blast Informatics).
Após o estudo genético, os indivíduos foram separados em afetados (n = 18) e não afetados (n = 32) de acordo com a presença ou não de mutação no gene LMNA.
Avaliação bioquímica e hormonal
Após jejum de 12 horas, todos os 50 indivíduos foram submetidos à coleta de amostras de sangue para as dosagens de glicemia plasmática (método enzimático da glicose-oxidase) e HbA1c (cromatografia líquida de alta performance HPLC). Os triglicérides, colesterol total e HDL-c foram realizados por ensaio enzimático automatizado, sendo LDL-c e VLDL-c calculados pela fórmula de Friedwald. Foram ainda medidos os valores de CPK, TGO e TGP (cinética UV-IFCC); fosfatase alcalina (ensaio colorimétrico); ureia (método cinético em ultravioleta); creatinina (método de Jaffé). As dosagens de TSH, T3 total, T4 livre e peptídeo C foram realizadas por ensaio imunoenzimático; a insulina, por fluoroimunoensaio (reação cruzada menor que 0,5% tanto com a pró-insulina intacta quanto com as frações "32-33 split proinsulin"); e a leptina, por radioimunoensaio.
O teste de tolerância oral à glicose (TTOG) com 75 g de glicose anidra foi realizado nos voluntários com glicemia de jejum ≥ 110 e < 126 mg/dL em dieta normal. Definiu-se como intolerância à glicose a presença de glicemia ≥ 140 e < 200 mg/dL após TOTG. Foram considerados diabéticos os indivíduos com glicemia de jejum ≥ 126 mg/dL ou glicemia após TTOG ≥ 200 mg/dL.
A estimativa da sensibilidade à insulina e da função da célula β foi calculada por meio do índice HOMA "homeostasis model assessment". O índice para avaliação de resistência insulínica (HOMA-IR) foi calculado pela fórmula: insulina sérica (μU/mL) x glicose plasmática (mmol/L)/22,5. O índice da função da célula β (HOMA-B) foi calculado pela fórmula: 20 x insulina sérica (μU/mL)/glicose plasmática (mmol/L) 3,5. Foram excluídos dessa estimativa os indivíduos diabéticos em uso de insulina.
Adicionalmente, nos indivíduos propósitos, foram dosados LH, FSH, E2, progesterona, prolactina, S-DHEA, testosterona total e livre, GH, cortisol sérico após supressão noturna com 1 mg de dexametasona e cortisol livre urinário (ensaio imunoenzimático); IGF-1 (ensaio imunorradiométrico); androstenediona, DHEA, diidrotestosterona, testosterona livre e 17OHP (RIE/DPC).
Os indivíduos afetados foram divididos em diabéticos (n = 5) e não diabéticos (n = 12). Excluiu-se da amostragem um indivíduo do grupo afetado não diabético por ser criança. Visando à comparação dos valores de glicemia, HbA1c, insulina, peptídeo C, leptina, perfil lipídico, HOMA-IR e HOMA-B entre os grupos afetado diabético (GAD), afetado não diabético (GAND) e não afetado (GNA), foi necessário selecionar, entre os familiares não afetados, um grupo equivalente (n = 17) em termos de IMC, relação cintura/quadril (C/Q), idade e relação feminino/masculino, uma vez que essas variáveis têm influência nas dosagens de insulina e de leptina.
Análise estatística
As variáveis categoriais foram apresentadas como proporções e as variáveis quantitativas foram expressas como média e desvio-padrão. O teste de Fisher foi utilizado para cálculo de significância na comparação de proporções e o método paramétrico de análise de variância (ANOVA), complementado pelo teste de Tukey-Kramer, foi utilizado para as médias. Empregou-se o teste de Kolmogorov-Smirnov para avaliação da distribuição normal dos dados e, quando apropriado, estes foram normalizados por transformação logarítmica. Foi adotado o nível de significância de 5% (p < 0,05).
RESULTADOS
Propósito da família I
GLV, 18 anos, feminino, branca, relatava irregularidade menstrual desde a menarca aos 11 anos e estava em uso de terapia estrogênica desde os 16 anos. Notou-se desenvolvimento progressivo da musculatura dos membros superiores e inferiores após a puberdade, com acentuação aos 14 anos, quando também refere início de acantosis nigricans em regiões axilar e cervical, bem como de deposição de gordura localizada em face e pescoço.
Apresentava, ao exame físico, aparência masculinizada decorrente de importante diminuição do tecido adiposo subcutâneo de braços, pernas e tórax (mais acentuada na face anterior), com aparente hipertrofia muscular e vasos proeminentes, e acentuada acantosis nigricans, com hipertrofia de papilas em regiões axilar e cervical. O exame da genitália mostrava uma importante hipertrofia de grandes lábios, sem clitoromegalia. Apresentava também hirsutismo leve em linha alba ínfero-umbilical e raiz de coxa (+/4) e submentoniano (++/4), e face em "lua cheia", em virtude de deposição de gordura em face e pescoço, esboçando duplo-queixo, sem giba (Figura 1). Altura = 173 cm; peso = 72,5 kg; cintura = 74 cm; quadril = 96 cm; C/Q = 0,77; IMC = 24,5 kg/m²; PA = 130 x 90 mmHg.
Os exames complementares não evidenciaram hipercortisolismo. Os demais exames, após suspensão da terapia estrogênica, mostravam a função tireoidiana normal; colesterol e triglicérides elevados; hiperinsulinemia; glicemia de jejum e TTOG normais. A avaliação hormonal mostrou níveis séricos de androstenediona e DHEA elevados na primeira fase do ciclo sem inversão LH/FSH (Quadro 1). A ultrassonografia pélvica evidenciou ovários aumentados de volume com aspecto micropolicístico.
Propósito da família II
AMAS, 39 anos, feminino, branca, com história de várias investigações para síndrome de Cushing desde os 33 anos. Relatava irregularidade menstrual desde a menarca aos 10 anos e dificuldade para engravidar. Fez uso de estrógeno associado à progestágeno dos 33 aos 35 anos, necessitando suspender o uso em virtude de trombose venosa profunda. Queixava-se de deposição de gordura progressiva em face, região supraclavicular e posterior do pescoço desde a puberdade, semelhante à tia materna. Negava diabetes e dislipidemia.
Ao exame físico, apresentava discreta acantosis nigricans cervical, leve hirsutismo em linha alba ínfero-umbilical (++/4) e em região submentoniana (++/4), face em "lua cheia" com duplo queixo; acúmulo de gordura em pescoço, fossa supraclavicular e presença de giba. Apresentava também diminuição de tecido adiposo subcutâneo em membros superiores e inferiores, com aparência de hipertrofia muscular e vasos proeminentes. A região suprapúbica tinha acúmulo de tecido adiposo em grande quantidade. Não se observaram alterações na genitália (Figura 2). Altura = 153 cm; peso = 66 kg; cintura = 91 cm; quadril = 94 cm; C/Q = 0,96; IMC = 28,2 kg/m²; PA = 130 x 80 mmHg.
Os exames complementares afastaram hipercortisolismo. Os demais exames demonstraram função tireoidiana normal; colesterol e triglicérides elevados; hiperinsulinemia; glicemia de jejum e TTOG normais; inversão da relação FSH/LH com níveis séricos de DHEA e de androstenediona ligeiramente elevados na primeira fase do ciclo (Quadro 1). A ultrassonografia pélvica evidenciou moderado aumento do volume ovariano, com aspecto micropolicístico. A tomografia computadorizada de abdome superior foi normal.
Avaliação das famílias I e II
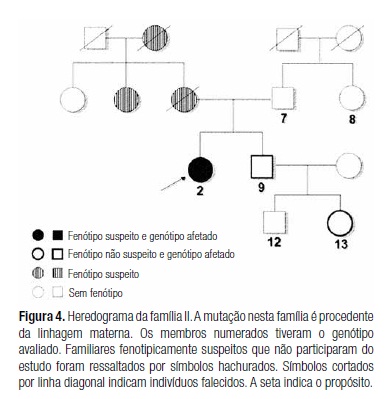
Avaliou-se um total de 50 indivíduos de duas famílias brasileiras não relacionadas. Na família I, além do propósito (01-GLV), foram estudados outros 43 familiares. Na família II, menos numerosa, avaliamos o propósito (02-AMAS) e cinco de seus familiares. Ambas as famílias têm ascendência portuguesa. A análise dos heredogramas da família I e da família II demonstra padrão de transmissão autossômico dominante (Figuras 3 e 4).
Os dados clínicos e antropométricos dos indivíduos afetados (portadores de mutação no gene LMNA) estão demonstrados no quadro 2. Designou-se como lipodistrofia parcial familiar (FPLD) o fenótipo clínico de diminuição de tecido adiposo subcutâneo em membros superiores e inferiores, com vasos aparentes e proeminentes, em associação ao acúmulo de tecido adiposo em face, pescoço e região supraclavicular. O fenótipo lipoatrófico (LA) foi caracterizado por diminuição de tecido adiposo subcutâneo em membros superiores e inferiores, com vasos aparentes e proeminentes, sem associação com acúmulo de tecido adiposo em face e região cervical.
Não foram constatadas alterações fenotípicas em cinco dos dezoito pacientes com genótipo alterado (três destes da família I e dois da família II). Excluindo-se a paciente 13-TRSS, uma menina impúbere que ainda poderia manifestar lipodistrofia, não houve alteração fenotípica em 23,5% dos indivíduos com genótipo alterado, sendo três deles do sexo masculino (09-LCS, 48-SR, 49-MRF) e um do sexo feminino (50-MDR).
Houve maior prevalência de diabetes e de SOP no grupo de afetados (risco 2,8 vezes maior de desenvolvimento dessas entidades clínicas neste grupo em relação ao GNA). No grupo feminino, a idade de diagnóstico do diabetes variou entre 30 e 45 anos. Não houve diferença significativa na prevalência de angina e hipertensão entre os dois grupos (Tabela 1) e nenhum dos indivíduos apresentou alterações na função renal e nas enzimas hepáticas.
Entre os indivíduos afetados do sexo feminino, houve relato de SOP em cinco delas, incluindo os propósitos. Todas apresentavam o fenótipo clássico de lipodistrofia, irregularidade menstrual e leve hirsutismo, com idade da menarca variando de 10 a 14 anos. Avaliou-se o eixo gonadal em duas dessas mulheres (21-LCGM e 22-SCGM), além dos propósitos. Elas eram irmãs e apresentavam DHEA elevada na primeira fase do ciclo, com os demais androgênios dentro da normalidade. Os exames de 21-LCGM são: DHEA = 10,9 ng/mL, androstenediona = 2.201 pg/mL, testosterona = 273 pg/mL, testosterona livre = 1,6 pg/mL, DHT = 147 pg/mL, S-DHEA = 178 mcg/dL, 17OHP = 138 ng/dL. Os exames de 22-SCGM são: DHEA 15 ng/mL, androstenediona = 1.396 pg/mL, testosterona = 454 pg/mL, testosterona livre = 1,4 pg/mL e 17OHP = 105 ng/dL. Os ensaios utilizados e os valores de referência são os mesmos descritos para avaliação dos propósitos. Entre os indivíduos não afetados, apenas uma das mulheres era portadora de SOP.
Para comparação de dados antropométricos e laboratoriais, foi conveniente dividir os indivíduos afetados em diabéticos e não diabéticos, tendo em vista a maior prevalência de diabetes no grupo afetado. A paciente 13-TRSS foi excluída da avaliação por ser criança. Obtiveram-se, então, três grupos para comparação: GAD (n = 5), GAND (n = 12) e GNA (n = 17).
Os resultados das comparações entre os três grupos encontram-se na tabela 2. Houve um predomínio de indivíduos do sexo feminino no GAD, sem diferenças na proporção entre os sexos nos demais grupos. A média de idade do GAD foi significativamente mais elevada.
O GAD apresentou níveis plasmáticos médios de glicose e de HbA1c mais elevados que os demais grupos. Os valores médios do índice HOMA-B foram mais elevados no GAND do que no GNA.
Os níveis séricos médios da insulina, triglicérides e VLDL e os valores médios do índice HOMA-IR foram mais elevados em ambos os grupos afetados.
Os níveis séricos médios da leptina e os valores médios da relação leptina/IMC foram menores em ambos os grupos afetados (p = 0,012 e p = 0,002, respectivamente).
Os níveis de leptina correlacionaram-se com o IMC no GNA (r = 0,53; IC95% = 0,07-0,80; p = 0,02), o que não foi observado no grupo de pacientes com lipodistrofia. Ambos os grupos apresentaram correlação significativa entre os níveis séricos de leptina e a relação leptina/IMC (r = 0,98; IC95% = 0,96-0,99; p < 0,0001 no grupo de afetados e r = 0,95; IC95% = 0,87-0,98; p < 0,0001 no GNA).
Não houve diferença nos valores médios do IMC, da C/Q, dos níveis séricos médios do peptídeo C, colesterol total, HDL-c e LDL-c entre os três grupos.
Sequenciamento do gene LMNA
Realizou-se o sequenciamento das amostras de seis indivíduos, quatro deles apresentando mutação pelo método PCR-RFLP (casos 01, 03 e 31 da família I e 02 da família II) e dois deles sem evidência de mutação pelo método PCR-RFLP (casos 04 da família I e 07 da família II) (Figuras 5 e 6).
A partir da análise do sequenciamento, identificamos mutação pontual no códon 482 no éxon 8 do gene LMNA, com padrão heterozigoto. A troca do nucleotídeo citosina por timina leva à substituição do aminoácido arginina (CGG) por triptofano (TGG) na lâmina A/C, caracterizando mutação missense, simbolizada por p.R482W, nos quatro casos cujo PCR-RFLP evidenciou mutação.
Os sequenciamentos das amostras para os éxons 8 e 11 do gene LMNA nos dois casos sem evidência de mutação pelo PCR-RFLP não mostraram alterações nas sequências dos nucleotídeos.
DISCUSSÃO
Foram estudadas duas famílias brasileiras de indivíduos portadores de FPLD2. A suspeita clínica, com base no fenótipo dos pacientes, foi um bom preditor para identificação dos portadores da mutação, especialmente em pacientes diabéticos. Entre os pacientes estudados, alterações fenotípicas foram detectadas em 100% dos afetados diabéticos, em 66,66% dos afetados não diabéticos e em nenhum dos indivíduos não afetados. Entretanto, em um estudo que rastreou o DNA genômico de 277 indivíduos adultos não relacionados portadores de fenótipos metabólicos variados, incluindo FPLD, mutações no gene LMNA só foram identificadas em aproximadamente 10% dos casos (15).
A mutação no gene LMNA foi verificada em seis indivíduos do sexo masculino, três deles com fenótipo lipoatrófico e três sem alteração fenotípica. A avaliação clínica do fenótipo lipoatrófico é comprometida nos homens, uma vez que a hipertrofia muscular já é rotineiramente identificada em indivíduos do sexo masculino.
Sinais clássicos de FLPD foram mais evidentes nas mulheres, em virtude da aparência masculinizada facilmente detectável. A evolução para o progressivo acúmulo de tecido adiposo subcutâneo em face, pescoço e região supraclavicular, com ocorrência após três a quatro anos da menarca, foi bastante previsível. O surgimento de acantosis nigricans, hirsutismo leve e SOP ocorreu precocemente. O diabetes, embora frequente, foi um evento mais tardio.
Apesar de ambas as famílias terem apresentado a mesma mutação no éxon 8 do gene LMNA, houve heterogeneidade fenotípica entre as mulheres com sinais clássicos de lipodistrofia. Achados semelhantes aos nossos, evidenciando heterogeneidade fenotípica em indivíduos de mesmo sexo e com a mesma mutação, não são os mais frequentes. A maioria dos estudos familiares que avaliaram a correlação entre genótipo e fenótipo aponta heterogeneidade fenotípica especialmente relacionada ao sexo (7-11). A lipoatrofia foi caracteristicamente associada aos homens, enquanto o padrão que envolve a diminuição de tecido adiposo no tronco foi relatado em mulheres com fenótipo lipodistrófico na maioria dos trabalhos. O fenótipo evidenciado no propósito da família II em nosso estudo é menos comum, embora já tenha sido relatado por outros autores (9,13).
Observou-se uma maior prevalência de SOP nas pacientes com FLPD2, com magnitude de risco 2,8 vezes maior nas afetadas em relação às não afetadas pela mutação. A relação inversa, entretanto, parece não ser consistente na literatura. Em um estudo visando caracterizar o papel de variações no gene LMNA na etiologia da SOP (16), 53 polimorfismos obtidos a partir do sequenciamento do gene LMNA em 43 mulheres portadoras de SOP com fenótipo similar à FPLD foram genotipados em 624 casos de SOP e em 544 mulheres controle. Os autores concluíram que nenhuma das variantes mostrou evidência significativa de associação com SOP, embora não excluam a possibilidade de variações no gene LMNA contribuírem para o fenótipo de SOP em uma pequena parcela das pacientes (2%-5%).
Além disso, os indivíduos afetados pela mutação apresentaram resistência insulínica, comprovada por níveis séricos de insulina e por índice HOMA-IR mais elevados, além de hipertrigliceridemia, quando comparados com seus familiares não afetados. Essas alterações precedem o início do diabetes, já que foram observadas tanto no GAD quanto no GAND.
Esses dados são corroborados por um estudo que comparou pacientes portadores de FPLD2 sem diabetes com indivíduos controle pareados para idade e peso (17). Foram detectados valores de glicemia em jejum similares em ambos os grupos, porém com aumento significativo dos valores de insulinemia em jejum (p = 0,004), índice HOMA-B (p = 0,01), ácidos graxos livres (p = 0,03) e triglicérides (p = 0,0004) nos pacientes com FPLD2. Após ingestão de glicose, os pacientes com FPLD2 apresentaram uma excessiva resposta insulínica em relação ao controle, embora os valores glicêmicos tenham sido similares em ambos os grupos.
Constatou-se que os indivíduos afetados apresentaram níveis séricos de leptina e relação leptina/IMC significativamente diminuídos em relação aos seus familiares não afetados. Considerando que as concentrações de leptina dependem diretamente da massa de células adiposas, a hipoleptinemia é uma anormalidade metabólica esperada nas FLPD, sendo ainda mais importante nas lipodistrofias familiares generalizadas (18). Em um estudo aberto com seis mulheres portadoras de FPLD2 recebendo doses máximas de antidiabéticos orais e hipolipemiantes, leptina recombinante foi administrada em injeções subcutâneas duas vezes ao dia na dose máxima de 0,08 mg/kg/dia por 12 meses (19). Foi observada uma redução significativa dos triglicérides em cinco pacientes (p = 0,03) e do colesterol total nas seis pacientes (p = 0,01). Houve melhora também significativa da sensibilidade à insulina e da glicemia em jejum (p < 0,01), sem contudo ter sido observada melhora na tolerância à glicose e nos valores de HbA1c (p = 0,07). Esses resultados sugerem que pacientes com FPLD2 podem se beneficiar do uso de leptina recombinante, com melhora do perfil lipídico, da glicemia em jejum e da sensibilidade à insulina.
Nesse estudo, foi identificada a mutação pontual missense no códon 482 do éxon 8 do gene LMNA. Essa mutação heterozigota, denominada p.R482W, leva à substituição do aminoácido arginina por triptofano na lâmina A/C.
Desde o estudo pioneiro de sequenciamento de DNA nos membros de uma família canadense com fenótipo de FPLD, que evidenciou a mutação heterozigótica missense p.R482Q no éxon 8 do gene LMNA (12), a lista de mutações associadas à FPLD2 foi rapidamente ampliada, sendo a esmagadora maioria dessas encontradas no éxon 8, especialmente no códon 482 (mutações missense p.R482Q, p.R482W e p.R482L) (14). Atualmente, a maioria dos novos casos de FPLD2 identificados em países norte-americanos apresenta uma dessas mutações no códon 482. Virtualmente todos os pacientes com FPLD2 por mutações no códon 482 apresentam um fenótipo "clássico" puro de lipodistrofia, com acometimento mínimo de outros tecidos, como os músculos esqueléticos e cardíacos (20).
Por fim, acredita-se que a exata compreensão das alterações clínicas, bioquímicas e genéticas da FPLD2 poderá auxiliar na elucidação de mecanismos envolvidos na deterioração metabólica associada à resistência insulínica de forma mais ampla. Isso possibilitaria o desenvolvimento de novas terapias não apenas para condições raras como as lipodistrofias, mas para outras formas epidêmicas de resistência insulínica cujos mecanismos genéticos não estão elucidados, como as presentes em pacientes com síndrome metabólica.
Agradecimentos: aos doutores Jacqueline Capeau e Olivier Lascols pelo auxílio técnico referente aos desenhos dos primers e à Ms. Daniele Costa Abreu, pelo apoio na realização da PCR nas amostras deste estudo.
Declaração de financiamento: este estudo contou com o auxílio financeiro da Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (Capes).
Recebido em 10/Jul/2012
Aceito em 15/Mai/2013
Declaração: os autores declaram não haver conflitos de interesse científico neste estudo.
- 1. Garg A. Lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab. 2011;96(11):3313-25.
- 2. Bhayana S, Hegele RA. Genetic syndromes of human lipodystrophy. Can J Diab. 2002;26(4):363-8.
- 3. Vantyghem MC, Pigny P, Maurage CA, Rouaix-Emery N, Stojkovic T, Cuisset JM, et al. Patients with familial partial lipodystrophy of the Dunnigan type due to a LMNA R482W mutation show muscular and cardiac abnormalities. J Clin Endocrinol Metab. 2004;89(11):5337-46.
- 4. Broers JLV, Ramaekers FCS, Bonne G, Yaou RB, Hutchison CJ. Nuclear lamins: laminopathies and their role in premature ageing. Physiol Rev. 2006;86:967-1008.
- 5. Goldberg M. Import and export at the nuclear envelope. Symp Soc Exp Biol. 2004;1:115-33.
- 6. Herrmann H, Bar H, Kreplak L, Strelkov SV, Aebi U. Intermediate filaments: from cell architecture to nanomechanics. Nat Rev Mol Cell Biol. 2007;8:562-73.
- 7. Hegele RA, Anderson CM, Wang J, Jones DC, Cao H. Association between nuclear lamin A/C R482Q mutation and partial lipodystrophy with hyperinsulinemia, dyslipidemia, hypertension and diabetes. Genome Res. 2000;10:652-58.
- 8. Speckman RA, Garg A, Du F, Bennett L, Veile R, Arioglu E, et al. Mutational and haplotype analyses of families with familial partial lipodystrophy (Dunnigan variety) reveal recurrent missense mutations in the globular C-terminal domain of lamin A/C. Am J Hum Genet. 2000;66:1192-8.
- 9. Vigouroux C, Magré J, Vantyghem MC, Bourut C, Lascols O, Shackleton S, et al. Lamin A/C gene: sex-determined expression of mutations in Dunnigan-type familial partial lipodystrophy and absence of coding mutations in congenital and acquired generalized lipoatrophy. Diabetes. 2000;49:1958-62.
- 10. Shackleton S, Lloyd DJ, Jackson SNJ, Evans R, Niermeijer MF, Singh BM, et al. LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat Genet. 2000;24:153-6.
- 11. Schmidt H, Genschel J, Baier P, Schmidt M, Ockenga J, Tietge UJ, et al. Dyslipemia in familial partial lipodystrophy caused by an R482W mutation in the LMNA gene. J Clin Endocrinol Metab. 2001;86:2289-95.
- 12. Cao H, Hegele RA. Nuclear lamin A/C R482Q mutation in canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum Mol Genet. 2000;9:109-12.
- 13. Garg A, Vinaitheerthan M, Weatherall PT, Bowcock AM. Phenotypic heterogeneity in patients with familial partial lipodystrophy (Dunnigan variety) related to the site of missense mutations in lamin A/C gene. J Clin Endocrinol Metab. 2001;86:50-65.
- 14. Hegele RA. LMNA mutation position predicts organ system involvement in laminopathies. Clin Genet. 2005;8:31-4.
- 15. Decaudain A, Vantyghem MC, Guerci B, Hécart AC, Auclair M, Reznik Y, et al. New metabolic phenotypes in laminopathies: LMNA mutations in patients with severe metabolic syndrome. J Clin Endocrinol Metab. 2007;92(12):4835-44.
- 16. Urbanek M, Nampiaparampil G, D'Souza J, Sefton E, Ackerman C, Legro RS, et al. The role of genetic variation in the lamin A/C gene in the etiology of polycystic ovary syndrome. J Clin Endocrinol Metab. 2009;94:2665-9.
- 17. Boschmann M, Engeli S, Moro C, Luedtke A, Adams F, Gorzelniak K, et al. LMNA mutations, skeletal muscle lipid metabolism, and insulin resistance. J Clin Endocrinol Metab. 2010;95(4):1634-43.
- 18. Haque WA, Shimomura I, Matsuzawa Y, Garg A. Serum adiponectin and leptin levels in patients with lipodystrophies. J Clin Endocrinol Metab. 2002;87:2395.
- 19. Park JY, Javor ED, Cochran EK, DePaoli AM, Gorden P. Long-term efficacy of leptin replacement in patients with Dunnigan-type familial partial lipodystrophy. Metabolism. 2007;56(4):508-16.
- 20. Hegele RA. Phenomics, lamin A/C, and metabolic disease. J Clin Endocrinol Metab. 2007;92(12):4566-8.
Correspondência para:
Datas de Publicação
-
Publicação nesta coleção
11 Dez 2013 -
Data do Fascículo
Nov 2013
Histórico
-
Recebido
10 Jul 2012 -
Aceito
15 Maio 2013