Resumos
A síndrome de Vogt-Koyanagi-Harada é doença crônica, sistêmica e auto-imune, com manifestações oculares, nervosas, auditivas e tegumentares. Descrevemos aqui o caso de uma criança com início dos sintomas aos quatro anos e dois meses de idade, com positividade para o HLA DRB1*01.
Síndrome uveomeningoencefálica; Criança; Antígenos HLA-DR; Uveite; Uveite; Relatos de casos
Vogt-Koyanagi-Harada syndrome is chronic systemic autoimmune disease with ocular, nervous, auditory and tegumental manifestations. We report here the case of a child with onset of symptoms at four years and two months of age, with positive HLA DRB1*01.
Uveomeningoencephalitic syndrome; Child; HLA-DR antigens; Uveitis; Uveitis; Case reports
RELATOS DE CASOS
Síndrome de Vogt-Koyanagi-Harada incompleta associada a HLA DRB1*01 em criança de quatro anos de idade: relato de caso
Incomplete Vogt-Koyanagi-Harada syndrome associated with HLA DRB1*01 in a 4-year-old child: case report
Daniel Rocha LucenaI; Jayter Silva PaulaII; Gleilton Carlos Mendonça SilvaIII; Maria de Lourdes Veronese RodriguesIV
IMédico Assistente do Departamento de Oftalmologia, Otorrinolaringologia e Cirurgia de Cabeça e Pescoço - Faculdade de Medicina da Universidade de São Paulo - USP - Ribeirão Preto (SP) - Brasil
IIProfessor Colaborador do Departamento de Oftalmologia, Otorrinolaringologia e Cirurgia de Cabeça e Pescoço - Faculdade de Medicina da USP - Ribeirão Preto (SP) - Brasil
IIIPós-graduando do Departamento de Oftalmologia, Otorrinolaringologia e Cirurgia de Cabeça e Pescoço - Faculdade de Medicina da USP - Ribeirão Preto (SP) - Brasil
IVProfessora Associada do Departamento de Oftalmologia, Otorrinolaringologia e Cirurgia de Cabeça e Pescoço - Faculdade de Medicina da USP - Ribeirão Preto (SP) - Brasil
Endereço para correspondência Endereço para correspondência: Jayter Silva de Paula Departamento de Oftalmologia Otorrinolaringologia e Cirurgia de Cabeça e Pescoço Hospital das Clínicas de Ribeirão Preto - Campus (USP) Av. Bandeirantes, 3900 Ribeirão Preto (SP) Cep 14049-900 E-mail: jayterdepaula@yahoo.com.br
RESUMO
A síndrome de Vogt-Koyanagi-Harada é doença crônica, sistêmica e auto-imune, com manifestações oculares, nervosas, auditivas e tegumentares. Descrevemos aqui o caso de uma criança com início dos sintomas aos quatro anos e dois meses de idade, com positividade para o HLA DRB1*01.
Descritores: Síndrome uveomeningoencefálica; Criança; Antígenos HLA-DR; Uveite/etiologia; Uveite/diagnóstico; Relatos de casos [Tipo de publicação]
ABSTRACT
Vogt-Koyanagi-Harada syndrome is chronic systemic autoimmune disease with ocular, nervous, auditory and tegumental manifestations. We report here the case of a child with onset of symptoms at four years and two months of age, with positive HLA DRB1*01.
Keywords: Uveomeningoencephalitic syndrome; Child; HLA-DR antigens; Uveitis/etiology; Uveitis /diagnosis; Case reports [Publication type]
INTRODUÇÃO
A síndrome de Vogt-Koyanagi-Harada (SVKH) é uma doença sistêmica que envolve tecidos que contém melanina. É caracterizada por uma panuveíte bilateral, crônica, granulomatosa associada a manifestações variáveis de comprometimento neurológico, auditivo e cutâneo. O envolvimento ocular bilateral é necessário para caracterizar o diagnóstico, necessariamente sem história prévia de cirurgia ou trauma ocular. Os sinais mais comuns incluem iridociclite, vitreíte, edema ou hiperemia de disco óptico, espessamento coroideano e descolamento de retina neuro-sensorial(1-2).
Os critérios diagnósticos, atualmente utilizados, foram publicados a partir do I "Workshop" Internacional da SVKH e incluem: (1) ausência de história prévia de trauma ou cirurgia ocular; (2) ausência de evidência de doença ocular concomitante; (3) envolvimento ocular bilateral precoce (com áreas focais de fluido sub-retiniano ou descolamento seroso de retina) ou tardio (despigmentação, fundo de olho em pôr-do-sol, cicatrizes coriorretinianas despigmentadas de Dalen-Fuchs, e migração ou acúmulo de epitélio pigmentar da retina); (4) história ou sinais auditivos e/ou neurológicos; (5) alterações cutâneas que apareçam durante ou após as manifestações neurológicas e oculares(3).
Baseado nesses critérios, a SVKH pode ser classificada em completa, quando todos os critérios são preenchidos, incompleta, quando estão presentes os critérios 1, 2, 3 e 4 ou 5, ou provável, quando apenas os critérios 1, 2 e 3 são preenchidos(3). A idade usual de início dos sintomas é entre 20 e 50 anos, sendo que apenas 5% dos casos foram descritos com idade inferior aos 16 anos(4).
Este artigo descreve um caso raro de SVKH incompleta complicada com neovascularização sub-retiniana bilateral e associada à expressão do HLA DRB1*01 em uma criança com 4 anos e 2 meses de idade, o qual deve ser o mais jovem caso relatado com esta síndrome.
RELATO DE CASO
Criança branca, sexo masculino, foi admitida no Ambulatório de Oftalmologia do Hospital das Clínicas da Faculdade de Medicina de Ribeirão Preto - Universidade de São Paulo, com história de hiperemia, dor ocular e fotofobia em ambos os olhos (AO) há 4 meses. Queixava-se ainda de cefaléia freqüente, dislalia e possível hipoacusia no período.
Ao exame oftalmológico apresentava boa fixação em ambos olhos, porém o paciente não informava acuidade visual, por não entender optotipos. À biomicroscopia evidenciou-se intensa reação em câmara anterior, incluindo "Tyndall" celular de 3+/4+ e finos precipitados ceráticos. Havia também extensas sinéquias posteriores em AO. Ao exame fundoscópico o corpo vítreo apresentava-se sem "haze", e lesões amareladas compatíveis com áreas de coroidite podiam ser observadas inferiormente no pólo posterior e na região nasal AO. Optou-se pelo tratamento tópico com colírios de dexametasona 1% e atropina 1%, até a conclusão dos exames complementares solicitados.
Exames de rotina e hemograma estavam dentro dos limites da normalidade, assim como a pesquisa de anticorpos anti-núcleo, anti-cardiolipina, fator reumatóide, componentes do complemento sérico, RIF-toxoplasmose, VDRL, FTA-ABS, enzima conversora de angiotensina e raios-x de tórax. A velocidade de hemossedimentação era de 5 mm/1 hora.
No retorno em 2 meses, a angiofluoresceinografia evidenciou alguns pontos de vazamento no epitélio pigmentar da retina ("pin-points"), com acúmulo de contraste no espaço sub-retiniano em AO (figura não disponível, de baixa qualidade, dada pela não colaboração do paciente). O exame audiométrico evidenciou perda auditiva leve a partir de 6 kHz, bilateralmente. Concluiu-se o diagnóstico de síndrome de Vogt-Koyanagi-Harada incompleta e se instituiu o seguinte tratamento: albendazol 400 mg/dia por 3 dias (profilático), colírios de dexametasona 1% e atropina 1%, e prednisona 1 mg/kg/dia (via oral). Após 1 mês, a acuidade visual corrigida era 1,0 (paciente informando adequadamente os optotipos), com controle da inflamação e ausência de coroidite ativa em AO.
Apesar da retirada gradual do corticóide sistêmico, houve recorrência da doença após 7 meses de seguimento, com recrudescimento da baixa acuidade visual (0,2 no olho direito, e 0,4 no olho esquerdo) e da uveíte anterior AO. Optou-se novamente pelo tratamento com a dose inicial de corticosteróide sistêmico, associado a metotrexate 7,5 mg/semana (esquema indicado pelo setor de imunopediatria). Apesar deste esquema de tratamento, observou-se o aparecimento de uma membrana neovascular sub-retiniana na região macular de AO. Conseqüentemente, indicou-se ciclosporina-A (50 mg de 12/12 horas), com a suspensão abrupta do metotrexate e progressiva da corticoterapia oral.
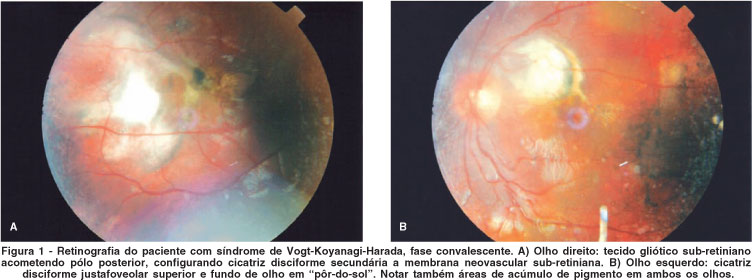
Após 3 meses de tratamento, a fundoscopia revelava a presença de tecido sub-retiniano de aspecto gliótico compatível com cicatriz disciforme, rarefação generalizada de epitélio pigmentar da retina, cicatrizes do tipo Dalen-Fuchs e fundo de olho em pôr-do-sol bilateralmente (Figura 1). A tipificação dos alelos de histocompatibilidade (HLA) evidenciou os subtipos DRB1*01, DRB1*14, DRB3 e DQB1*05.
Atualmente o paciente se apresenta assintomático, em uso de ciclosporina A. Não há atividade inflamatória intra-ocular, porém mantém acuidade visual corrigida de 0,2 e lesões cicatriciais acometendo a região macular AO. Acrescenta-se ainda que, em nenhum momento da evolução clínica, o paciente apresentou sinais ou sintomas de artrite.
DISCUSSÃO
A SVKH é mais freqüente entre negros, orientais, índios americanos e hispânicos(5). Os principais diagnósticos diferenciais desta síndrome incluem: esclerites posteriores, oftalmia simpática, coroidite multifocal, dentre outras(6). Sua etiologia é obscura, sendo que a predileção por raças pode condizer com uma predisposição genética(5). Há indícios de que uma infecção, provavelmente viral, nestes pacientes poderia induzir a formação de novos antígenos na superfície dos melanócitos, os quais desencadeariam uma reação imune antígeno-específica(7).
No caso relatado, o paciente apresentou um quadro de uveíte difusa, com iridociclite altamente sintomática e descolamento seroso da retina bilateralmente. Houve, posteriormente, recorrência da inflamação em segmento anterior e formação de membrana neovascular sub-retiniana AO. Também apresentou hipoacusia documentada, que associada aos achados citados acima, confirma a hipótese de SVKH incompleta.
Há relatos na literatura do diagnóstico de SVKH em crianças aos 6(8) e 7(9) anos de idade. O caso mais jovem publicado até o momento é o de uma criança de 4 anos e 6 meses de idade por ocasião do início dos sintomas(10). No presente caso, a criança apresentava 4 anos e 2 meses, o mais precoce dentre aqueles revisados na literatura. Embora haja disparidade nos achados entre os casos citados, todos têm em comum o fato de não preencherem os 5 critérios para a SVKH completa segundo a classificação atualmente aceita(3). Além disso, esses trabalhos foram publicados anteriormente à revisão de alguns desses critérios.
Parece haver associação da SVKH com os subtipos dos alelos de histocompatibilidade HLA-DRB1 e -DQB1(11-15). Dentre os subtipos apresentados pelo paciente, os alelos HLA-DRB1*01 e - DQB1*05, os quais apresentam baixa prevalência na população local(16), já foram previamente associados de forma direta a essa doença(15).
Algumas outras doenças auto-imunes também se associam a alguns subtipos dos alelos DRB1*01, tais como a artrite reumatóide juvenil(17). No entanto, muitos deles, incluindo também o DRB1*14, se apresentam como fatores ditos "de proteção" de algumas formas dessa doença(18-20).
A presença da associação de expressão dos alelos DRB1*01 e DQB1*05, juntamente com a apresentação clínica de inflamação do segmento anterior altamente sintomática e achados típicos de segmento posterior, fortalece a hipótese diagnóstica levantada, afastando outras possíveis na infância(15).
Embora muito incomum em crianças(6,8-10), a SVKH deve ser considerada no diagnóstico diferencial de quadros de panuveíte idiopática com resposta terapêutica rápida com corticoterapia sistêmica. Nesses casos, a família do paciente deve ser informada quanto ao curso da doença e seu possível prognóstico visual reservado.
Recebido para publicação em 19.07.2005
Última versão recebida em 21.08.2006
Aprovação em 12.09.2006
Nota Editorial: Depois de concluída a análise do artigo sob sigilo editorial e com a anuência da Dra. Luciana Peixoto Finamor sobre a divulgação de seu nome como revisora, agradecemos sua participação neste processo.
Trabalho realizado no Departamento de Oftalmologia, Otorrinolaringologia e Cirurgia de Cabeça e Pescoço - Faculdade de Medicina da Universidade de São Paulo - USP - Ribeirão Preto (SP) - Brasil.
- 01. Touitou V, Escande C, Boddaghi B, Cassoux N, Wechsler B, Lemaitre C, et al. [Diagnostic and therapeutic management of Vogt-Koyanagi-Harada syndrome]. J Fr Ophtalmol. 2005;28(1):9-16. French.
- 02. Bezerra HL, Santos LP, Carvalho AM, Muccioli C, Belfort Junior R. Síndrome de Vogt-Koyanagi-Harada: revisão de 89 casos. Arq Bras Oftalmol. 1998; 61(3):331-4.
- 03. Read RW, Holland GN, Rao NA, Tabbara KF, Ohno S, Arellanes-Garcia L, et al. Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: report of an international committee on nomenclature. Am J Ophthalmol. 2001;131(5): 647-52.
- 04. Laghmari M, Karim A, Ibrahimy W, Essakalli NH, Mohcine Z. [Vogt-Koyanagi-Harada syndrome in children]. J Fr Ophtalmol. 2002;25(6):636-40. French.
- 05. Beniz J, Forster DJ, Lean JS, Smith RE, Rao NA. Variations in clinical features of the Vogt-Koyanagi-Harada syndrome. Retina. 1991;11(3):275-80.
- 06. Moorthy RS, Inomata H, Rao NA. Vogt-Koyanagi-Harada syndrome. Surv Ophthalmol. 1995;39(4):265-92.
- 07. Matsuda H, Sugiura S. Ultrastructural changes of the melanocyte in Vogt-Koyanagi-Harada syndrome and sympathetic ophthalmia. Jpn J Ophthalmol. 1971;15:69-80.
- 08. Perkins ES. Pattern of uveitis in children. Br J Ophthalmol. 1966;50(4):169-85.
- 09. Forster DJ, Green RL, Rao NA. Unilateral manifestation of the Vogt-Koyanagi-Harada syndrome in a 7-year-old child. Am J Ophthalmol. 1991;111(3):380-2.
- 10. Gruich MJ, Evans OB, Storey JM, Bradley ST, Chen CJ. Vogt-Koyanagi-Harada syndrome in a 4-year-old child. Pediatr Neurol. 1995;13(1):50-1.
- 11. Zhang M, Oiu C, Hu T. [Association of HLA-DRB genes with Vogt-Koyanagi-Harada syndrome in a Chinese Han population]. Zhongguo YI Xue Ke Xue Yuan Xue Bao. 2000;22(1):36-40. Chinese.
- 12. Goldberg AC, Yamamoto JH, Chiarella JM, Marin ML, Sibinelli M, Neufeld R, et al. HLA-DRB1*0405 is the predominant allele in Brazilian patients with Vogt-Koyanagi-Harada disease. Hum Immunol. 1998;59(3):183-8.
- 13. Weisz JM, Holland GN, Roer LN, Park MS, Yuge AJ, Moorthy RS, et al. Association between Vogt-Koyanagi-Harada syndrome and HLA-DR1 and -DR4 in Hispanic patients living in southern California. Ophthalmology. 1995; 102(7):1012-5.
- 14. Arellanes-Garcia L, Bautista N, Mora P, Ortega-Larrocea G, Burguet A, Gorodezky C. HLA-DR is strongly associated with Vogt-Koyanagi-Harada disease in Mexican Mestizo patients. Ocul Immunol Inflamm. 1998;6(2):93-100.
- 15. Levinson RD, See RF, Rajalingam R, Reed EF, Park MS, Rao NA, Holland GN. HLA-DRB1 and -DQB1 alleles in mestizo patients with Vogt-Koyanagi-Harada's disease in Southern California. Hum Immunol. 2004;65(12):1477-82.
- 16. Louzada-Junior P, Smith AG, Hansen JA, Donadi EA. HLA-DRB1 and - DQB1 alleles in the Brazilian population of the northeastern region of the state of Sao Paulo. Tissue Antigens. 2001;57(2):158-62.
- 17. Miterski B, Drynda S, Boschow G, Klein W, Oppermann J, Kekow J, Epplen JT. Complex genetic predisposition in adult and juvenile rheumatoid arthritis. BMC Genet. 2004;5:2.
- 18. Malagon C, Van Kerckhove C, Giannini EH, Taylor J, Lovell DJ, Levinson JE, et al. The iridocyclitis of early onset pauciarticular juvenile rheumatoid arthritis: outcome in immunogenetically characterized patients. J Rheumatol. 1992;19(1):160-3.
- 19. Haas JP, Truckenbrodt H, Paul C, Hoza J, Scholz S, Albert ED. Subtypes of HLA-DRB1*03, *08, *11, *12, *13 and *14 in early onset pauciarticular juvenile chronic arthritis (EOPA) with and without iridocyclitis. Clin Exp Rheumatol. 1994;12 Suppl 10:S7-14.
- 20. Garavito G, Yunis EJ, Egea E, Ramirez LA, Malagon C, Iglesias A, et al. HLA-DRB1 alleles and HLA-DRB1 shared epitopes are markers for juvenile rheumatoid arthritis subgroups in Colombian mestizos. Hum Immunol. 2004;65(4):359-65.
Endereço para correspondência:
Datas de Publicação
-
Publicação nesta coleção
19 Jun 2007 -
Data do Fascículo
Mar 2007
Histórico
-
Aceito
12 Set 2006 -
Recebido
19 Jul 2005 -
Revisado
21 Ago 2006