Resumos
A doença de Fabry é erro inato do metabolismo dos glicoesfingolipídeos (GL), resultante da atividade deficiente da enzima alfa-galactosidase A (a-Gal), com herança ligada ao cromossomo X. O acúmulo progressivo de GL nos tecidos resulta nas manifestações clínicas da doença, mais evidentes em homens hemizigotos, e incluem angioqueratomas, acroparestesias, córnea verticilata, hipo-hidrose, envolvimento cardíaco, renal e manifestações cerebrovasculares. Foi realizada avaliação em família acometida pela doença, sendo dois pacientes do sexo feminino e três do sexo masculino. Todos os pacientes foram submetidos a anamnese, exame oftalmológico completo e dosagem da atividade da enzima a-Gal. O único achado clínico presente em todos foi a córnea verticilata. Isto demonstra o importante papel que o exame oftalmológico apresenta no diagnóstico da doença, já que as alterações oculares são tão características.
Opacidade da córnea; Doença de Fabry; Doenças por armazenamento dos lisossomos
Fabry's disease is a rare X-linked lisosomal storage disorder of glycosphingolipid (GL) metabolism, caused by a deficiency of alpha-galactosidase A activity. The progressive accumulation of GL in tissues results in the clinical manifestations of the disease, that are more evident in hemizygous males, and include angiokeratomas, acroparesthesia, cornea verticillata, cardiac and kidney involvement, cerebrovascular manifestations. A family with Fabry's disease including 2 female patients and 3 male patients is reported. The patients were submitted to complete medical history, ophthalmological examination and alpha-galactosidase activity test. Cornea verticillata was a constant finding in all patients. This demonstrates the important role of the ophtalmological examination for the diagnosis of Fabry's disease since the eye findings are so characteristic of the disease.
Corneal opacity; Fabry disease; Lysosomal storage diseases
RELATOS DE CASOS
Córnea verticilata - marcador clínico da doença de Fabry: relato de caso
Cornea verticillata - a clinical marker of Fabry disease: case report
Cynthia Azeredo CordeiroI; Fernando OréficeII; Euler Pace LasmarIII; Helena Hollanda SantosIV; Eugênia Ribeiro ValadaresV
IPós-graduanda em Oftalmologia da Universidade Federal de Minas Gerais - UFMG - Belo Horizonte (MG) - Brasil
IIProfessor Titular de Oftalmologia da UFMG - Belo Horizonte (MG) - Brasil
IIIProfessor Titular de Nefrologia da Faculdade de Ciências Médicas de Minas Gerais - UFMG - Belo Horizonte (MG) - Brasil
IVResidente de Genética da UFMG - Belo Horizonte (MG) - Brasil
VProfessora Adjunta de Pediatria da UFMG - Belo Horizonte (MG) - Brasil
Endereço para correspondência Endereço para correspondência: Cynthia Azeredo Cordeiro Rua Gilberto Siqueira, 87/502 Campos (RJ) - CEP 28010-400 E-mail: cordeiro.cy@gmail.com
RESUMO
A doença de Fabry é erro inato do metabolismo dos glicoesfingolipídeos (GL), resultante da atividade deficiente da enzima alfa-galactosidase A (a-Gal), com herança ligada ao cromossomo X. O acúmulo progressivo de GL nos tecidos resulta nas manifestações clínicas da doença, mais evidentes em homens hemizigotos, e incluem angioqueratomas, acroparestesias, córnea verticilata, hipo-hidrose, envolvimento cardíaco, renal e manifestações cerebrovasculares. Foi realizada avaliação em família acometida pela doença, sendo dois pacientes do sexo feminino e três do sexo masculino. Todos os pacientes foram submetidos a anamnese, exame oftalmológico completo e dosagem da atividade da enzima a-Gal. O único achado clínico presente em todos foi a córnea verticilata. Isto demonstra o importante papel que o exame oftalmológico apresenta no diagnóstico da doença, já que as alterações oculares são tão características.
Descritores: Opacidade da córnea; Doença de Fabry; Doenças por armazenamento dos lisossomos
ABSTRACT
Fabry's disease is a rare X-linked lisosomal storage disorder of glycosphingolipid (GL) metabolism, caused by a deficiency of a-galactosidase A activity. The progressive accumulation of GL in tissues results in the clinical manifestations of the disease, that are more evident in hemizygous males, and include angiokeratomas, acroparesthesia, cornea verticillata, cardiac and kidney involvement, cerebrovascular manifestations. A family with Fabry's disease including 2 female patients and 3 male patients is reported. The patients were submitted to complete medical history, ophthalmological examination and alpha-galactosidase activity test. Cornea verticillata was a constant finding in all patients. This demonstrates the important role of the ophtalmological examination for the diagnosis of Fabry's disease since the eye findings are so characteristic of the disease.
Keywords: Corneal opacity; Fabry disease; Lysosomal storage diseases
INTRODUÇÃO
A doença de Fabry é um erro inato do metabolismo dos glicoesfingolipídeos (GL), em especial de globotriasilceramida, resultante da atividade deficiente da enzima alfa-galactosidaseA (a-Gal). Herdada de forma ligada ao cromossomo X, acomete principalmente homens e apresenta uma prevalência estimada de 1:40000 homens(1). A expressão da doença, em mulheres heterozigotas, pode variar de estado assintomático a doença tão grave quanto a que ocorre em homens. O gene que codifica a enzima encontra-se no braço longo do cromossomo X (Xq22.1)(2-3). Atualmente, foram identificadas 300 mutações, que podem ser de diversos tipos (missence, nonsense, defeitos no splicing) e que são praticamente específicas para cada família. A maioria dos casos ocorre de forma herdada, sendo raros os casos de mutações de novo(3).
Devido à deficiência da atividade desta enzima, ocorre um acúmulo progressivo de GL no interior de endotélios e músculos lisos de vasos sanguíneos em diversos órgãos, podendo acometer: coração, rins, pele, sistema nervoso, olhos e trato gastrintestinal. É comum, ainda, a ocorrência de febre sem causa aparente(4).
A forma clássica da doença ocorre geralmente em homens hemizigotos. Os sintomas surgem na infância e adolescência, com acroparestesias, angioqueratomas, córnea verticilata e hipo-hidrose. Entre a 3ª e a 4ª décadas, ocorre aumento dos sintomas, com comprometimento progressivo do sistema vascular, levando a alterações cardíacas, renais e cerebrais(5-6). Os achados cardiovasculares incluem hipertensão arterial, hipertrofia ventricular esquerda, angina de peito, isquemia miocárdica, insuficiência mitral, anormalidades eletrocardiográficas e dilatação da aorta. A doença renal leva a um quadro de hipertensão arterial, proteinúria e hematúria podendo evoluir para insuficiência renal crônica. A ocorrência de acidente vascular cerebral se deve à oclusão da microvasculatura ou por embolia(1,5). Distúrbios psiquiátricos, diarréia, fraqueza e edema em membros inferiores são freqüentemente relatados(3,7). Doença pulmonar, hipoacusia, vertigem e zumbido também podem ocorrer(3).
Em geral, mulheres heterozigotas são assintomáticas ou exibem manifestações leves. Mesmo nas mulheres portadoras (sem qualquer sintoma), observa-se freqüentemente, ao exame oftalmológico a córnea verticilata(4,8).
O diagnóstico é difícil quando a história familiar é negativa para a doença. Com freqüência os pacientes passam por diversos médicos, têm diagnósticos errôneos e só tardiamente o diagnóstico é estabelecido. Em homens afetados, o diagnóstico definitivo é feito pela determinação da atividade da enzima a-Gal, que pode ser dosada no plasma, em leucócitos e em fibroblastos(9). A atividade de a-Gal, na maioria dos homens afetados, é inferior a 3% do normal, mas atividade menor que 20% do normal já foi descrita(10). Nas mulheres heterozigotas, é comum ocorrer resultados de dosagem da atividade da enzima dentro dos valores de referência(9). A triagem clínica das mulheres portadoras da doença pode ser realizada por meio do achado da córnea verticilata(1).
Anteriormente, quando a diálise e o transplante renal não eram realizados, a insuficiência renal era a principal causa de mortalidade e a expectativa de vida era de 41 anos. Atualmente, as doenças cardiovascular e cerebrovascular são as maiores responsáveis pelo óbito. Após o transplante renal, a atividade da enzima do enxerto consegue metabolizar os GB3 evitando acometimento do rim transplantado. As portadoras não devem ser candidatas a doação(3).
A terapia de reposição enzimática foi aprovada pelo FDA (Food and Drug Administration) em 2003(3). Estudos têm mostrado que a infusão de a-Gal recombinante em acometidos reduz o acúmulo de GB3 no endotélio dos rins, coração e pele. O tratamento é bem tolerado pelos pacientes, com poucas reações adversas(11-12).
Uma vez firmado o diagnóstico de DF o aconselhamento genético deve ser oferecido ao paciente e seus familiares. O geneticista deve explicar os exames disponíveis e as implicações de um diagnóstico positivo, levando em consideração fatores culturais, religiosos ou sociais que possam influenciar o entendimento dos pacientes(13-14). Todas as filhas de um afetado serão portadoras, uma vez que elas herdarão o X que tem a mutação de seu pai; nenhum dos seus filhos herdará a doença, já que ele vai herdar o cromossomo Y de seu pai e não o X. Metade dos filhos de uma portadora será acometida, uma vez que ela tem um X com a mutação e outro sem; metade de suas filhas será portadora pelo mesmo motivo(3,15).
ACHADOS OFTALMOLÓGICOS
Os achados oculares têm se tornado um dos marcadores da doença e, em alguns pacientes, têm contribuído para o diagnóstico.
Alterações conjuntivais consistem em dilatação e tortuosidade vasculares, irregularidade no calibre e formação de aneurisma. Alterações retinianas são similares e podem ser exarcebadas pela presença de doença renal e hipertensão arterial.
Alterações cristalinianas consistem em deposição de material granular e catarata subcapsular anterior(8).
Verticilata é definida como uma estrutura em forma de buquê, com vários filamentos reunidos em torno de um mesmo eixo ao qual estariam inseridos. Então, córnea verticilata é descrita como opacidades amareladas caracterizadas por uma ou mais linhas irradiando de um ponto próximo ao centro da córnea. É o achado ocular mais comum, e está presente em quase todos os homens hemizigotos e em cerca de 70 a 90% das mulheres heterozigotas(3,8,16). Não há relato de comprometimento da acuidade visual e nenhuma correlação com a gravidade da doença foi estabelecida. Córnea verticilata também está associada com o uso de fenotiazina, cloroquina, indometacina, clorfazimina e amiodarona. No caso de doença de Fabry, esta alteração não desaparece com o tratamento(8), ao contrário do que ocorre nos casos associados ao uso crônico de amiodarona(17).
O exame oftalmológico é importante ferramenta no diagnóstico de portadoras, já que a dosagem da atividade da enzima a-Gal nem sempre está diminuída nestas mulheres e o exame molecular é de difícil acesso(3).
RELATO DOS CASOS
Foi realizada avaliação em uma família acometida pela doença, sendo duas mulheres heterozigotas e três homens hemizigotos (Figura 1, Tabela 1).
Todos os pacientes foram submetidos à anamnese, exame oftalmológico completo e dosagem da atividade da enzima alfa-galactosidase (Tabela 2). As atividades enzimáticas foram realizadas no laboratório de Erros Inatos do Metabolismo da Universidade Federal de São Paulo, custeadas pela empresa Genzyme do Brasil.
O exame oftalmológico incluiu acuidade visual, motilidade ocular, ectoscopia, biomicroscopia, tonometria e oftalmoscopia binocular indireta.
Foi realizado o heredograma da família (Figura 1).
Mãe
GMF, 70a, leucoderma, casada, 4 filhos, com quadro de insuficiência renal crônica de longa data (não sabe precisar). Paralisia de membros inferiores devido ao acidente vascular cerebral há 30 anos, implante de marcapasso devido à bradicardia há 15 anos e diagnóstico de doença de Alzheimer há 4 anos. A dosagem da atividade da enzima a-Gal no plasma foi de 0,54 µmol/L/h (VR.:>2,7 µmol/L/h) e em leucocitos foi de 18,4 µmol/mg proteína/h (VR:>1,4 µmol/mg proteína/h). O exame oftalmológico foi dificultado pelo estado avançado da doença de Alzheimer. Ao exame biomicroscópico, apresentava pseudofacia e córnea verticilata.
Irmão 1
GMA, masc, 46 anos, leucodermo, solteiro, sem filhos, com história de acroparestesia desde a infância. Apresentou isquemia miocárdica há 3 anos e refere episódios de diarréia, hipo-hidrose e intolerância ao calor e frio. A dosagem da atividade da enzima a-Gal no plasma foi de 0,16 µmol/L/h (VR.:>2,7 µmol/L/h) e em leucocitos não foi detectável (VR:>1,4 µmol/mg proteína/h). Ao exame oftalmológico, observou-se córnea verticilata, sem nenhuma outra alteração.
Irmão 2
GMAG, fem, 45anos, leucoderma, casada, sem filhos. Na infância, foi inadvertidamente diagnosticada febre reumática devido às alterações cardíacas e acroparestesias. Há 15 anos evoluiu com insuficiência renal, sendo submetida ao transplante em 1997. Apresenta angioqueratomas (mãos, abdome e tórax), regurgitação mitral mínima, hipertensão arterial e hipercolesterolemia. A dosagem da atividade da enzima a-Gal no plasma foi de 0,96 µmol/L/h(VR.:>2,7 µmol/L/h) e em leucocitos foi de 20,1µmol/mg proteína/h (VR:>1,4 µmol/mg proteína/h). Ao exame oftalmológico, observou-se córnea verticilata, sem nenhuma outra alteração.
Irmão 3
AMA, masc, 44 anos, casado. Também tratado para febre reumática na infância devido a acroparestesias, encontra-se em tratamento insuficiência renal há 6 anos. Apresenta ainda anidrose, hipertrofia ventricular esquerda, episódios febris vespertinos, diarréia ocasional e intolerância ao calor e frio. A dosagem da atividade da enzima a-Gal no plasma foi de 0,53 µmol/L/h (VR.:>2,7 µmol/L/h) e em leucocitos não foi detectável (VR:>1,4 µmol/mg proteína/h). Ao exame oftalmológico, observou-se córnea verticilata, sem nenhuma outra alteração.
Sua filha, CGMA, 3 anos, até o momento, não apresenta alterações da doença.
Irmão 4 - Probando (caso índice)
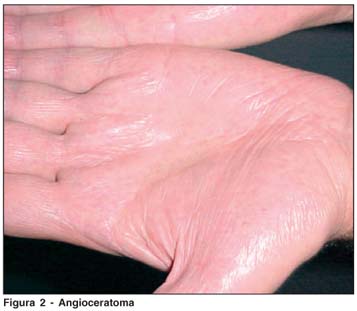
RMA, 42a, masc, casado, 2 filhos. Foi tratado na infância para febre reumática devido às acroparestesias. Apresentou insuficiência renal em 2001, quando foi submetido ao transplante renal (doadora: esposa). Como alterações cardíacas, apresenta degeneração mixomatosa, prolapso da válvula mitral e dilatação da raiz da aorta torácica. Refere hipo-hidrose, alterações da memória, intolerância ao frio e calor e apresenta angioqueratomas (mãos, quadris e genitais) (Figura 2). A dosagem da atividade da enzima a-Gal no plasma foi de 0,03 µmol/L/h (VR.:>2,7 µmol/L/h) e em leucocitos não foi detectável (VR:>1,4 µmol/mg proteína/h). Ao exame oftalmológico, observou-se córnea verticilata (Figura 3), sem nenhuma outra alteração.
Seus filhos, ambos do sexo masculino, RMAF, 11anos, tem diagnóstico de lúpus eritematoso sistêmico e ERMA, 7anos, é saudável.
Todos os pacientes negaram uso de: fenotiazina, cloroquina, indometacina, clorfazimina e amiodarona.
DISCUSSÃO
Esta série de casos de DF em uma família reforça o caráter hereditário da mesma. Como é uma doença relacionada ao cromossomo X, os homens apresentam as formas graves da doença e transmitem para todas as filhas, mas não aos filhos. Segundo a literatura, cerca de 10% das heterozigotas desenvolve insuficiência renal, o que pode ser explicado pela hipótese de inativação do X(15,18). Segundo essa hipótese, um dos cromossomos X nas mulheres é aleatoriamente inativado nas células, numa tentativa de igualar a expressão de genes ligados ao X nos sexos masculino e feminino(15). Cerca de 70 a 90% das portadoras podem ser identificadas através do exame oftalmológico mostrando a córnea verticilata(3).
Nesta série de casos a mãe, sua filha do sexo feminino e três filhos do sexo masculino apresentam sintomas graves (Tabela 1). Importante ressaltar que todos com a doença apresentaram alteração na dosagem de a-Gal no plasma mas somente os homens apresentaram alteração na dosagem em leucócitos.
O único achado clínico comum a homens e mulheres nesta família foi a córnea verticilata (Figura 3). Este tipo de alteração pode ocorrer também em uso de fenotiazina, cloroquina, indometacina, clorfazimina e amiodarona(15), mas não havia história de uso destas medicações. É um achado que pode ser encontrado tanto em afetados quanto em portadoras(13,15). O exame oftalmológico apresentou um importante papel no diagnóstico, já que esta alteração é bastante sugestiva da doença e foi observada em todos os pacientes.
Recebido para publicação em 24.05.2006
Última versão recebida em 02.11.2006
Aprovação em 06.12.2006
Trabalho realizado no Departamento de Oftalmologia do Hospital São Geraldo, Universidade Federal de Minas Gerais - UFMG.
- 1. Masson C, Cissé I, Simon V, Insalaco P, Audran M. Fabry disease: a review. Joint Bone Spine. 2004;71(5):381-3.
-
2Online Mendelian Inheritance in Man - OMIN [homepage on the Internet]. Baltimore, Maryland: Johns Hopkins University; c2001. [cited 2006 Apr 22] Available from: http://www3.ncbi.nlm.nih.gov/omim
- 3. Gene Tests [homepage on the Internet]. Seattle: University of Washington; c 1993-2007. [cited 2006 Apr 22]. Available from: http://www3.geneclinics.org
- 4. Ritter M, Dittrich R, Droste DW. Microembolus detection in four patients with Fabry's disease: further support for a primarily microangiopathic origin of early cerebrovascular symptoms. Eur Neurol. 2003;50(3):141-5.
- 5. Bloom D, Speijer D, Linthorst GE, Donker-Koopman WG, Strijland A, Aerts JM. Recombinant enzyme therapy for Fabry disease: absense of editing of human alpha-galactosidase A mRNA. Am J Hum Genet. 2003;72(1):23-31.
- 6. Garman SC, Garboczi DN. The molecular defect leading to Fabry disease: structure of human alpha-galactosidase. J Mol Biol. 2004;337(2):319-35.
- 7. López Rodríguez M, Gómez Cerezo J, Barbado Hernández FJ. Enfermedad de Fabry: una entidad multidisciplinar con nuevas perspectivas terapéuticas. Rev Clin Esp. 2003;203(6):292-5.
- 8. Nguyen TT, Gin T, Nicholls K, Low M, Galanos J, Crawford A. Ophthalmological manifestations of Fabry disease: a survey of patients at the Royal Melbourne Fabry Disease Treatment Centre. Clin Experiment Ophthalmol. 2005;33(2):164-8.
- 9. Zschocke J, Hoffmann GF, editors. Vademecum Metabolicum. Manual of metabolic paediatrics. 2nd ed. Germany: Milupa GGmbH & Co. KG; 2004.
- 10. Romeo G, D'Urso M, Pisacane A, Blum E, De Falco A, Ruffilli A. Residual activity of alpha-galactosidase A in Fabry's disease. Biochem Genet. 1975; 13(9-10):615-28.
- 11. Eng CM, Guffon N, Wilcox WR, Germain DP, Lee P, Waldek S, Caplan L, Linthorst GE, Desnick RJ; International Collaborative Fabry Disease Study Group. Safety and efficacy of recombinant human alpha-galactosidase A - replacement therapy in Fabry's disease. N Engl J Med. 2001;345(1):9-16.
- 12. Wilcox WR, Banikazemi M, Guffon N, Waldek S, Lee P, Linthorst GE, Desnick RJ, Germain DP; International Fabry Disease Study Group. Long-term safety and efficacy of enzyme replacement therapy for Fabry disease. Am J Hum Genet. 2004;75(1):65-74.
- 13. Facher JJ, Robin NH. Genetic counseling in primary care. What questions are patients likely to ask, and how should they be answered? Postgrad Med. 2000;107(3):59-60, 63-6.
- 14. Johnson KA, Brensinger JD. Genetic counseling and testing: implications for clinical practice. Nurs Clin North Am. 2000;35(3):615-26. Review.
- 15. Nussbaum RL McInnes RR, Willard HF. Thompson & Thompson genetics in medicine. 6th ed. Philadelphia: W.B. Saunders; c2001. 444p.
- 16. Hirano K, Murata K, Miyagawa A, Terasaki H, Saigusa J, Nagasaka T, Kobayashi M. Histopathologic findings of cornea verticillata in a woman heterozygous for Fabry's disease. Cornea. 2001;20(2):233-6.
- 17. Kaplan LJ, Cappaert WE. Amiodarone keratopathy. Correlation to dosage and duration. Arch Ophthalmol. 1982;100(4):601-2.
- 18. Inagaki S, Migita M, Hayakawa M, Fujita A, Yoshida J, Ishizaki M, et al. An asymptomatic heterozygous female with fabry disease: implications for enzyme replacement therapy. J Nippon Med Sch. 2005;72(6):387-90.
Datas de Publicação
-
Publicação nesta coleção
20 Set 2007 -
Data do Fascículo
Ago 2007
Histórico
-
Aceito
06 Dez 2006 -
Revisado
02 Nov 2006 -
Recebido
24 Maio 2006