ABSTRACT
Purpose:
To analyze the clinical features, visual acuity, and full-field electroretinogram (ERG) findings of 15 patients with the neuronal ceroid lipofuscinosis (NCL) phenotype and to establish the role of ERG testing in NCL diagnosis.
Methods:
The medical records of five patients with infantile NCL, five with Jansky-Bielschowsky disease, and five with juvenile NCL who underwent full-field ERG testing were retrospectively analyzed.
Results:
Progressive vision loss was the initial symptom in 66.7% of patients and was isolated or associated with ataxia, epilepsy, and neurodevelopmental involution. Epilepsy was present in 93.3% of patients, of whom 86.6% presented with neurodevelopmental involution. Fundus findings ranged from normal to pigmentary/atrophic abnormalities. Cone-rod, rod-cone, and both types of dysfunction were observed in six, one, and eight patients, respectively.
Conclusion:
In our study, all patients with the NCL phenotype had abnormal ERG findings, and the majority exhibited both cone-rod and rod-cone dysfunction. We conclude that ERG is a valuable tool for the characterization of visual dysfunction in patients with the NCL phenotype and is useful for diagnosis.
Keywords:
Neuronal ceroid lipofuscinoses; Membrane proteins/genetics; Retina/physiopathology; Electroretinography; Retinal dystrophies; Visual acuity
RESUMO
Objetivo:
Analisar o quadro clínico, a acuidade visual e o eletrorretinograma de campo total (ERG) de 15 pacientes com o fenótipo da lipofuscinose ceróide neuronal (LCN), estabelecendo o papel do eletrorretinograma no seu diagnóstico.
Métodos:
Eletrorretinograma foi realizado em 5 pacientes com lipofuscinose ceróide neuronal infantil, 5 com doença de Jansky-Bielschowsky e 5 com lipofuscinose ceróide neuronal juvenil sendo feita uma análise retrospectiva dos registros médicos.
Resultados:
A perda progressiva da acuidade visual foi o sintoma inicial em 66,7%; isolada ou associada à ataxia, epilepsia e involução do desenvolvimento neuropsico motor. Epilepsia foi o sintoma inicial em 93,3% e 86,6% apresentaram involução do desenvolvimento neuropsicomotor. Achados fundoscópicos variaram de normal a alterações pigmentares/atróficas. Disfunção de cone-bastonete foi constatada em 6 pacientes, bastonete-cone em 1 e em 8 pacientes observou-se disfunção proporcional de ambos os sistemas.
Conclusão:
O eletrorretinograma foi alterado em todos os pacientes, e o achado mais frequente foi o comprometimento de cones e bastonetes. O eletrorretinograma constitui, portanto, uma ferramenta valiosa para caracterizar a disfunção visual em pacientes com o fenótipo da lipofuscinose ceróide neuronal, contribuindo para seu diagnóstico.
Descritores:
Lipofuscinose ceróides neuronais; Proteínas de membrana/genética; Retina/fisiopatologia; Eletrorretinografia; Distrofias retinianas; Acuidade visual
INTRODUCTION
Many childhood neurodegenerative disorders affect vision, and therefore, diagnostic, prognostic, treatment, and rehabilitation pro grams involve visual evaluations. Neuronal ceroid lipofuscinoses (NCLs), which comprise the most common pediatric neurodegenerative disorders, are lysosomal storage diseases. Pathophysiologically, NCLs have been attributed to lysosomal lipofuscin ceroid deposits11 Kohan R, Cismondi IA, Oller-Ramirez AM, Guelbert N, Anzolini TV, Alonso G, et al. Therapeutic approaches to the challenge of neuronal ceroid lipofuscinoses. Curr Pharm Biotechnol. 2011;12(6):867-83. and are caused by enzymatic dysfunction22 Miller JN, Pearce DA. A novel c.776_777insA mutation in CLN1 leads to infantile neuronal ceroid lipofuscinosis. J Child Neurol. 2013;28(9):1106-11.,33 Mink JW, Augustine EF, Adams HR, Marshall FJ, Kwon JM. Classification and natural history of the neuronal ceroid lipofuscinoses. J Child Neurol. 2013;28(9):1101-5. consequent to autosomal recessive genetic mutations. To date, 14 genes have been linked to NCL.
Clinically, although NCL is the main cause of childhood pro gressi ve encephalopathy, it can manifest at varying ages, from early in childhood to during adulthood11 Kohan R, Cismondi IA, Oller-Ramirez AM, Guelbert N, Anzolini TV, Alonso G, et al. Therapeutic approaches to the challenge of neuronal ceroid lipofuscinoses. Curr Pharm Biotechnol. 2011;12(6):867-83.,44 Rider JA, Rider DL. Thirty years of Batten disease research: present status and future goals. Mol Genet Metab. 1999;66(4):231-3.,55 Santavuori P, Lauronen L, Kirveskari E, Åberg L, Sainio K, Autti T. Neuronal ceroid lipofuscinoses in childhood. Neurol Sci. 2000;21(3 Suppl):S35-41.. Worldwide, the re ported NCL incidence is one in 12,500 live newborn infants66 Birch DG, Weleber RG. Diseases of fatty acid storage and metabolism: Lipofuccinoses and the long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. In: Heckenlively JR, Arden GB, editors. Principles and practice of clinical electrophysiology of vision. 2nd ed. Cambridge, MA: MIT Press; 2006. p.889-96.. Curren tly, the three most frequently reported NCLs are infantile (INCL), Jansky-Biels chowsky disease (JB), and juvenile NCL (JNCL), also called Batten-Spielmeyer-Vogt-Sjogren disease.
NCLs present with various symptoms, including cognitive and motor decline, movement disorders, seizures, and retinopathy. In cases of INCL, the initial symptom onset occurs during the first 2 years of life, and the disease progresses quickly. INCL is characterized by severe developmental milestone losses, blindness, and microcephaly, and affected individuals have a life expectancy of 8-13 years11 Kohan R, Cismondi IA, Oller-Ramirez AM, Guelbert N, Anzolini TV, Alonso G, et al. Therapeutic approaches to the challenge of neuronal ceroid lipofuscinoses. Curr Pharm Biotechnol. 2011;12(6):867-83.,22 Miller JN, Pearce DA. A novel c.776_777insA mutation in CLN1 leads to infantile neuronal ceroid lipofuscinosis. J Child Neurol. 2013;28(9):1106-11.. JB disease first manifests as ataxia, progressive developmental milestone losses, epilepsy, and posterior vision losses in children of 2-4 years of age. JNCL, which is caused by a mutation in CLN3 (16p12), is the most common type of N CL. The onset of this form occurs during 4-8 years of age and is characterized by vision loss and progression to total blindness within 2 years, epilepsy, and motor and cognitive declines77 Bozorg S, Ramirez-Montealegre D, Chung M, Pearce DA. Juvenile neuronal ceroid lipofuscinosis (JNCL) and the eye. Surv Ophthalmol. 2009;54(4):463-71.
8 Puri PK, Leilani Valdes C, Angelica Selim M, Bentley RC. Neuronal ceroid lipofuscinosis diagnosed via skin biopsy. J Clin Neurosc. 2010;17(12):1585-7.
9 Kwon JM, Adams H, Rothberg PG, Augustine EF, Marshall FJ, Deblieck EA, et al. Quantifying physical decline in juvenile neuronal ceroid lipofuscinosis (Batten disease). Neurology. 2011;77(20):1801-7. Comment in: Neurology. 2011;77(20):1779-80.
10 Cialone J, Adams H, Augustine EF, Marshall FJ, Kwon JM, Newhouse N, et al. Females experience a more severe disease course in Batten disease. J Inherit Metab Dis. 2012; 35(3):549-55.
11 Dolisca SB, Mehta M, Pearce DA, Mink JW, Maria BL. Batten disease: clinical aspects, molecular mechanisms, translational science, and future directions. J Child Neurol. 2013;28(9):1074-100.-1212 Drack AV, Miller JN, Pearce DA. A novel c.1135_1138delCTGT mutation in CLN3 leads to juvenile neuronal ceroid lipofuscinosis. J Child Neurol. 2013;28(9):1112-6..
NCL is an important ophthalmologic concern. Eye symptoms are usually the first sign of disease and can facilitate an early diagnosis. The disease course is mainly determined by central nervous system degradation and corresponding motor and intellectual deficits, and most patients die before reaching 30 years of age1313 Seeliger M, Rüther K, Apfelstedt-Sylla E, Schlote W, Wohlrab M, Zrenner E. [Juvenile neuronal ceroid lipofuscinosis (Batten-Mayou) disease. Ophthalmologic diagnosis and findings]. Ophthalmologe. 1997;94(8):557-62. German.. Retinal degeneration is an early lysosomal storage disease event, although the eye fundus appearance might remain normal or exhibit nonspecific abnormalities during the initial disease phase. However, a full-field electroretinogram (ERG) evaluation can detect changes that might precede fundus abnormalities, even during the initial NCL phase, as affected patients will not exhibit detectable ERG responses during progression1414 Weleber RG. The dystrophic retina in multisystem disorders: the electroretinogram in neuronal ceroid lipofuscinoses. Eye (Lond). 1998;12(Pt 3b):580-90.,1515 Weleber RG, Gupta N, Trzupek KM, Wepner MS, Kurz DE, Milam AH. Electroretinographic and clinicopathologic correlations of retinal dysfunction in infantile neuronal ceroid lipofuscinosis (infantile Batten disease). Mol Genet Metab. 2004;83(1-2):128-37.. The 2009 Hamburg-Germany NCL consensus therefore considered neuro-ophthalmologic evaluation as the first step toward a NCL diagnosis11 Kohan R, Cismondi IA, Oller-Ramirez AM, Guelbert N, Anzolini TV, Alonso G, et al. Therapeutic approaches to the challenge of neuronal ceroid lipofuscinoses. Curr Pharm Biotechnol. 2011;12(6):867-83.. The present study therefore aimed to ascertain the characteristics of clinical and retinal dysfunction in patients with the NCL phenotype and to establish the role of ERG in early NCL diagnosis.
METHODS
Subjects
This retrospective study evaluated the case notes of 15 patients with the NCL phenotype who were referred to the Clinical Electrophysiology of Vision Laboratory, Department of Ophthalmology and Visual Sciences, Paulista School of Medicine, Federal University of São Paulo, São Paulo, Brazil, for full-field ERG between July 2001 and December 2008. All patients, who were identified retrospectively, had undergone a comprehensive ophthalmologic examination before full-field ERG. Five patients each with INCL, JB disease, and JNCL were included. Neurologic and clinical abnormalities, consanguinity, and familial NCL history were determined from anamnesis and/or available medical records. The inclusion criterion was the presence of the NCL phenotype, which comprised visual complaints, neurodevelopmental involution, ataxia, and intractable epileptic seizures.
Ophthalmic examination
All patients underwent comprehensive ophthalmic exams, including slit-lamp, refraction, and dilated indirect ophthalmoscopy, performed by one of the authors (SSW), a retinal specialist. Presenting visual acuity (PVA) was measured in each eye using a retro-illuminated Early Treatment Diabetic Retinopathy Study Chart of with Tumble "E" optotypes presented at a 4-m distance, when possible. Ocular motility was assessed using cover/uncover and alternate cover testing for distance and near vision testing, with and without glasses, with emphasis on the diagnosis of strabismus and nystagmus.
Full-field electroretinography
Each patient was administered a drop of tropicamide 1% with a drop of phenylephrine 10% to achieve a minimum pupil diameter of 6 mm and allowed to adapt to the dark for 30 min. Under dim red illumination, a bipolar contact lens electrode (Burian-Allen bipolar electrode; Hansen Ophthalmic Development Lab, Coralville, IA, USA) was placed on the corneal surface of one or both eyes, depending on the patient's cooperation. The corneal surface was anesthetized with two drops of tetracaine 1.0%, and a drop of methylcellulose 2% was placed on the inside surface of the contact lens electrode for protection and to ensure good electrical contact. A gold cup ground electrode was applied to the earlobe. All stimuli were presented in a Ganzfeld dome (LKC Technologies Inc., Gaithersburg, MD, USA). Signals were amplified (gain, 910,000; 0.3-500 Hz), digitized, averaged, saved, and displayed using a digital plotter (UTAS E-3000 System, LKC Technologies Inc., Gaithersburg, MD, USA). ERGs were recorded according to the standard International Society for Clinical Electrophysiology of Vision (ISCEV) protocol1616 Marmor MF, Fulton AB, Holder GE, Miyake Y, Brigell M, Bach H; International Society for Clinical Electrophysiology of Vision. ISCEV standard for full-field clinical electroretinography (2008 update). Doc Ophthalmol. 2009;118(1):69-77.. The stimulus and recording details have been described previously1717 Berezovsky A, Moraes NS, Nusinowitz S, Salomão SR. Standard full-field electroretinography in healthy preterm infants. Doc Ophthalmol. 2003;107(3):243-9.. The peak-to-peak amplitude (µV) and implicit time (ms) from each step of the ISCEV standard protocol were determined, and amplitudes ≤2 µV were considered non-detectable responses. Retinal dysfunction was classified as cone only, rod only, rod-cone, or cone-rod dysfunction according to standard clinical criteria, with consideration of history, fundus examination findings, and previous full-field ERG responses (recorded from one or both eyes with bipolar contact lens electrodes). ERG peak-to-peak amplitude and implicit time data from each step according to the ISCEV standard protocol were measured and compared with normative data from our own laboratory1818 Pereira JM, Mendieta L, Sacai PY, Salomão SR, Berezovsky A. Estudo normativo do eletrorretinograma de campo total em adultos jovens. Arq Bras Oftalmol. 2003;66(1): 137-44..
Ethics committee approval
This study was approved by the Committee of Ethics in Research of the Federal University of São Paulo (expert's report no. 1185.562) and conducted according to the tenets of the Declaration of Helsinki.
RESULTS
The 15 enrolled patients (three males) belonged to 13 families and ranged in age from 1 to 12 years (mean=5.9; median=5). The 12 females ranged in age from 3 to 12 years (mean=6.6; median=6), whereas the males ranged in age from 1 to 4 years (mean=3; median=4). Symptom onset ranged from 0.2 to 7 years (mean=3.3; median=3) among females and from 1 to 3 years (mean=2.3; median=3) among males. Seven (46.7%) patients had a history of consanguinity, and five (33.3%) reported a similar familial case. The self-reported skin color was white for 13 (86.7%) patients, and black for two (13.3%) patients.
Visual symptoms
The PVA results of the 15 patients are shown in table 1. Visual acuity in the better-seeing eye could be measured in only two patients, who received scores of 20/25 and 20/250. Of the remaining 13 patients, seven (46.7%) could not fixate on nor follow lights and objects, and six (40.0%) could fixate on and follow light and objects. The most frequent visual symptom was progressive visual acuity loss, reported by 10 (66.7%) patients. One patient reported only visual loss as the initial complaint, whereas this symptom was associated with other neurologic features in the other nine patients (60%). Five (33.3%) patients reported no visual symptoms.
Clinical assessment
Epilepsy was the initial presenting neurological symptom in 14 (93.3%) patients. Thirteen (86.6%) patients presented with neurodevelopmental involution, five exhibited ataxia, and one developed cardiopathy. Most patients (n=13) had multiple neurologic symptoms. The patients were then classified into three groups based on their clinical characteristics and age at initial symptom onset. All five JB disease patients presented with epilepsy and neuro psycho-motor involution (NPMI). Among the five patients with INCL, all had epilepsy and four presented with NPMI. All five JNCL patients presented with epilepsy; four presented with NPMI and reduced visual acuity, whereas one patient did not report these symptoms. The major clinical characteristics are shown in table 1.
Ophthalmic assessment
The fundus examination yielded normal results in seven (46.7%) patients, including two with JB disease and all with INCL disease. In contrast, all patients with JNCL exhibited fundus changes. The observed fundus changes included optic disk pallor in seven (46.7%) patients, bull's eye maculopathy in three (20%, all JNCL patients), peri-macular pigmentary deposits in three (20%), retinal pigmentary epithelium atrophy in two (13.3%), severe arteriolar narrowing in one, and neurosensory retinal atrophy in one patient. Six (40%) patients presented with two or more retinal abnormalities. Furthermore, two (13.3%) patients exhibited exotropia, and one patient had nystagmus (Table 1).
Full-field ERG and its correlations
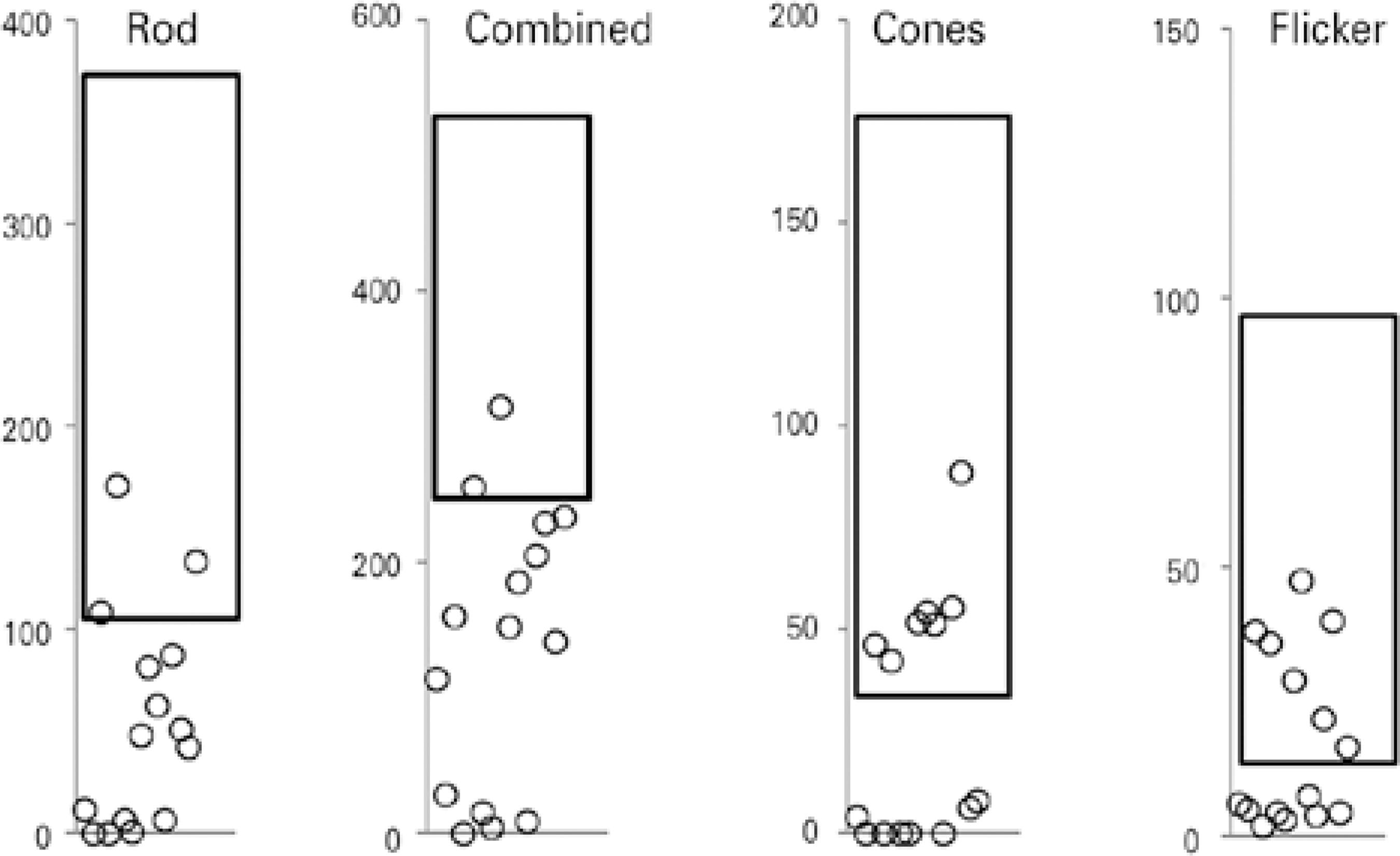
ERG abnormalities were found in all 15 patients. Figure 1 summarizes the ERG parameters for scotopic (rod and mixed) and photopic (cone and flicker) retinal function obtained from NCL patients, compared with normative data from our own laboratory. Scotopic (rod and mixed) and photopic (cone and flicker) a and b wave amplitudes and implicit times are shown in table 2, where abnormal values are indicated in bold. Cone-rod dysfunction was observed in six patients (cases 1, 3, 5, 6, 9, 10), rod-cone dysfunction in one patient (case 2), and both types in eight patients (cases 4, 7, 8, 11, 12, 13, 14, 15) (Table 2).
Electroretinogram (ERG) amplitudes (μV) for rod, combined, cone, and 30-Hz flicker responses recorded from one eye in each of 15 patients with the neuronal ceroid lipofuscinosis phenotype, compared with normative data from our own laboratory (rectangle=mean ± two standard deviations).
Amplitudes (mV) and implicit times (ms) from full-field electroretinogram (ERG) recordings of 15 patients with the NCL phenotype
Patients with JB disease
Two JB disease patients with normal fundoscopy findings and mild visual impairment presented with cone-rod dysfunction (cases 1, 5). Two other patients (cases 2, 4) with the worst visual acuity (did not fixate/follow objects/light) exhibited more fundoscopic abnormalities compared to those with better acuity, severe retinal changes, undetectable rod responses, and very low-amplitude cone responses, characteristic of diffuse retinal dysfunction affecting both systems (rods and cones). One patient (case 3) presented with optic disk pallor and cone-rod ERG dysfunction. Generally, those with the lowest visual acuity had more severe ophthalmic changes.
Patients with INCL
All five patients had normal fundus examinations (case 6 had nystagmus) and abnormal ERG findings. Three patients exhibited cone-rod dysfunction (cases 6, 9, 10), and two presented with diffuse retinal dysfunction affecting both systems (cases 7, 8). There was no correlation between visual acuity and ERG dysfunction.
Patients with JNCL
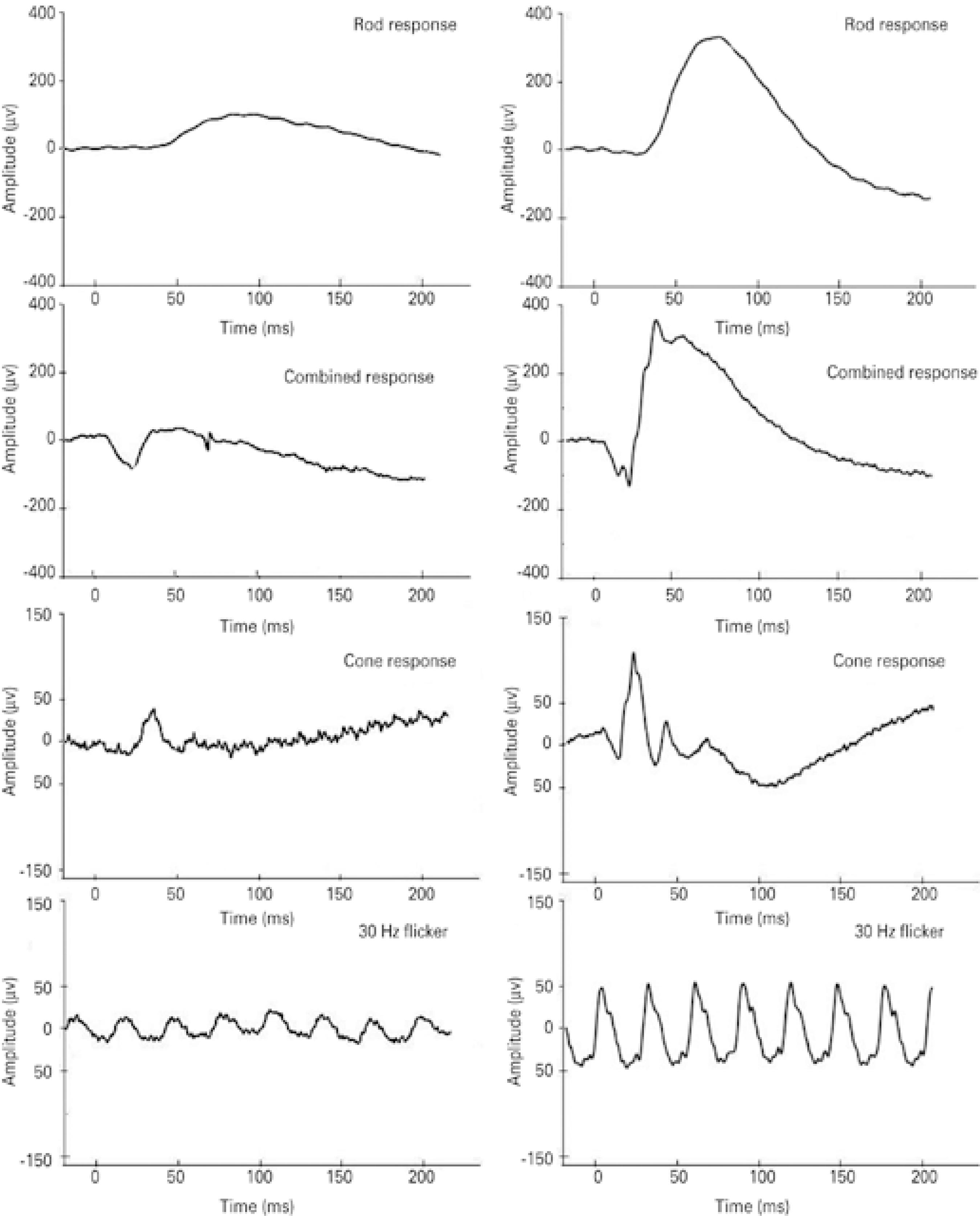
All five patients exhibited fundus changes, with ERG results indi cative of diffuse retinal dysfunction affecting both systems (rods and cones). Figure 2 presents representative standard full-field ERG waveforms (rod, maximal, cone, and 30 HZ-flicker responses) from a patient with the NCL phenotype (left) and a healthy subject (sex- and age-matched).
Representative standard full-field electroretinogram (ERG) waveforms (rod, combined, cone, and 30 Hz-flicker responses). Amplitudes (μV) and time (ms) are presented on the Y and X axes, respectively. Data are shown from case #12, a 6-year old female patient (left panels) with the neuronal ceroid lipofuscinosis phenotype, and a 6-year old female healthy subject (right panels).
DISCUSSION
This retrospective case series of the three most common forms of the NCL phenotype demonstrated that all 15 included patients exhibited phenotypic NCL characteristics, including progressive visual impairment, epilepsy, neurodevelopmental milestone losses, and ataxia. In addition, almost half of the patients reported a family history of consanguinity, as would be expected with recessive genetic transmission.
In our series, the most frequent presenting visual symptom was progressive visual acuity loss, a hallmark of NCL, in 10 (66.7%) patients44 Rider JA, Rider DL. Thirty years of Batten disease research: present status and future goals. Mol Genet Metab. 1999;66(4):231-3.
5 Santavuori P, Lauronen L, Kirveskari E, Åberg L, Sainio K, Autti T. Neuronal ceroid lipofuscinoses in childhood. Neurol Sci. 2000;21(3 Suppl):S35-41.-66 Birch DG, Weleber RG. Diseases of fatty acid storage and metabolism: Lipofuccinoses and the long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. In: Heckenlively JR, Arden GB, editors. Principles and practice of clinical electrophysiology of vision. 2nd ed. Cambridge, MA: MIT Press; 2006. p.889-96.. The five (33.3%) patients with no visual symptoms at the initial disease phase (three, one, and one with JB disease, INCL, and JNCL, respectively), were the youngest among our subjects, and will likely develop these symptoms during disease progression1919 Horiguchi M, Miyake Y. Batten disease - deteriorating course of ocular findings. Jpn J Ophthalmol. 1992;36(1):91-6.. Among the five patients with JNCL (all female), four initially presented with progressive visual acuity loss at a mean age of 5.7 years, thus corroborating previous studies1010 Cialone J, Adams H, Augustine EF, Marshall FJ, Kwon JM, Newhouse N, et al. Females experience a more severe disease course in Batten disease. J Inherit Metab Dis. 2012; 35(3):549-55..
Seven (46%) patients had a normal fundus, including two and five classified as having JB disease and INCL, respectively. Those findings corroborated previous reports of normal fundus findings during the initial NCL phase1515 Weleber RG, Gupta N, Trzupek KM, Wepner MS, Kurz DE, Milam AH. Electroretinographic and clinicopathologic correlations of retinal dysfunction in infantile neuronal ceroid lipofuscinosis (infantile Batten disease). Mol Genet Metab. 2004;83(1-2):128-37.
16 Marmor MF, Fulton AB, Holder GE, Miyake Y, Brigell M, Bach H; International Society for Clinical Electrophysiology of Vision. ISCEV standard for full-field clinical electroretinography (2008 update). Doc Ophthalmol. 2009;118(1):69-77.
17 Berezovsky A, Moraes NS, Nusinowitz S, Salomão SR. Standard full-field electroretinography in healthy preterm infants. Doc Ophthalmol. 2003;107(3):243-9.
18 Pereira JM, Mendieta L, Sacai PY, Salomão SR, Berezovsky A. Estudo normativo do eletrorretinograma de campo total em adultos jovens. Arq Bras Oftalmol. 2003;66(1): 137-44.-1919 Horiguchi M, Miyake Y. Batten disease - deteriorating course of ocular findings. Jpn J Ophthalmol. 1992;36(1):91-6.. On the other hand, all JNCL patients (i.e., older patients) presented with detectable fundoscopic abnormalities affecting the central retina, med-peripheral retina, retinal vasculature, and optic disk. Three of 5 JNCL patients, or 60%, presented with bull's eye maculopathy, a higher frequency than the 22% reported by Collins et al.2020 Collins J, Holder GE, Herbert H, Adams GG. Batten disease: features to facilitate early diagnosis. Br J Ophthalmol. 2006;90(9):1119-24..
Regarding ERG, full-field abnormalities were observed in all 15 patients to varying degrees, even those with no visual complaints and/or normal fundus findings. These findings agree with previous reports in which abnormal ERG findings were observed during the early stages of all three NCLs, with progression to a non-recordable status1515 Weleber RG, Gupta N, Trzupek KM, Wepner MS, Kurz DE, Milam AH. Electroretinographic and clinicopathologic correlations of retinal dysfunction in infantile neuronal ceroid lipofuscinosis (infantile Batten disease). Mol Genet Metab. 2004;83(1-2):128-37.,2020 Collins J, Holder GE, Herbert H, Adams GG. Batten disease: features to facilitate early diagnosis. Br J Ophthalmol. 2006;90(9):1119-24.. As per previous reports, the earliest ERG manifestations of INCL and JNCL include a marked loss and increased implicit latency of the scotopic and photopic b-wave, with relative preservation of the a-wave. This defect, which is evident for both rods and cones, suggests preservation of the photoreceptor outer segment function, with severely disturbed transmission of signals to bipolar cells. These ERG findings support the early existence of a relative pre- or post-synaptic block of effective neurotransmission from the photoreceptor inner segments to second-order bipolar neurons in affected patients1414 Weleber RG. The dystrophic retina in multisystem disorders: the electroretinogram in neuronal ceroid lipofuscinoses. Eye (Lond). 1998;12(Pt 3b):580-90.,1515 Weleber RG, Gupta N, Trzupek KM, Wepner MS, Kurz DE, Milam AH. Electroretinographic and clinicopathologic correlations of retinal dysfunction in infantile neuronal ceroid lipofuscinosis (infantile Batten disease). Mol Genet Metab. 2004;83(1-2):128-37..
Patients with JB disease exhibited decreases in cone function of 25% to 95%, whereas rod function ranged from normal to non-de tectable. Among patients with INCL, the ERG cone and rod functions ranged from normal to a decrease of 95%. Those with JNCL exhibited decreases in cone and rod function from 8% to 95% and from 15% to 95%, respectively. Under conditions of dark adaptation, only three and two patients respectively exhibited rod and combined responses with normal amplitudes. During the light adaptation phase, seven patients each exhibited cone and flicker responses with normal amplitudes (Figure 1). Our finding suggests a more pronounced impairment of rod function and relative preservation of cone function among this series of patients with NCL. Previous histopathologic evaluations of retinas affected by NCL revealed reduced cell numbers and auto-fluorescent lipofuscin granules in all retinal layers, with a central epi-retinal membrane. The periphery was better preserved but featured short photoreceptor outer segments. Further immunofluorescence analysis revealed degenerate rods and cones throughout the retina, with better preservation in peripheral areas1515 Weleber RG, Gupta N, Trzupek KM, Wepner MS, Kurz DE, Milam AH. Electroretinographic and clinicopathologic correlations of retinal dysfunction in infantile neuronal ceroid lipofuscinosis (infantile Batten disease). Mol Genet Metab. 2004;83(1-2):128-37..
Despite research into experimental therapies, NCL is currently untreatable, and early diagnosis is needed to implement appropriate counseling and support2121 Selden NR, Al-Uzri A, Huhn SL, Koch TK, Sikora DM, Nguyen-Driver MD, et al. Central nervous system stem cell transplantation for children with neuronal ceroid lipofuscinosis. J Neurosurg Pediatr. 2013;11(6):643-52.. Our above results confirm the early deleterious effects of NCL on retinal function and visual acuity, and indicate that full-field ERG may be useful for NCL diagnosis, particularly in patients who do not have access to genotyping.
-
Funding: No specific financial support was available for this study.
-
Approved by the following research ethics committee: Universidade Federal de São Paulo (CAAE: 46327515.1.0000.5505).
REFERENCES
-
1Kohan R, Cismondi IA, Oller-Ramirez AM, Guelbert N, Anzolini TV, Alonso G, et al. Therapeutic approaches to the challenge of neuronal ceroid lipofuscinoses. Curr Pharm Biotechnol. 2011;12(6):867-83.
-
2Miller JN, Pearce DA. A novel c.776_777insA mutation in CLN1 leads to infantile neuronal ceroid lipofuscinosis. J Child Neurol. 2013;28(9):1106-11.
-
3Mink JW, Augustine EF, Adams HR, Marshall FJ, Kwon JM. Classification and natural history of the neuronal ceroid lipofuscinoses. J Child Neurol. 2013;28(9):1101-5.
-
4Rider JA, Rider DL. Thirty years of Batten disease research: present status and future goals. Mol Genet Metab. 1999;66(4):231-3.
-
5Santavuori P, Lauronen L, Kirveskari E, Åberg L, Sainio K, Autti T. Neuronal ceroid lipofuscinoses in childhood. Neurol Sci. 2000;21(3 Suppl):S35-41.
-
6Birch DG, Weleber RG. Diseases of fatty acid storage and metabolism: Lipofuccinoses and the long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. In: Heckenlively JR, Arden GB, editors. Principles and practice of clinical electrophysiology of vision. 2nd ed. Cambridge, MA: MIT Press; 2006. p.889-96.
-
7Bozorg S, Ramirez-Montealegre D, Chung M, Pearce DA. Juvenile neuronal ceroid lipofuscinosis (JNCL) and the eye. Surv Ophthalmol. 2009;54(4):463-71.
-
8Puri PK, Leilani Valdes C, Angelica Selim M, Bentley RC. Neuronal ceroid lipofuscinosis diagnosed via skin biopsy. J Clin Neurosc. 2010;17(12):1585-7.
-
9Kwon JM, Adams H, Rothberg PG, Augustine EF, Marshall FJ, Deblieck EA, et al. Quantifying physical decline in juvenile neuronal ceroid lipofuscinosis (Batten disease). Neurology. 2011;77(20):1801-7. Comment in: Neurology. 2011;77(20):1779-80.
-
10Cialone J, Adams H, Augustine EF, Marshall FJ, Kwon JM, Newhouse N, et al. Females experience a more severe disease course in Batten disease. J Inherit Metab Dis. 2012; 35(3):549-55.
-
11Dolisca SB, Mehta M, Pearce DA, Mink JW, Maria BL. Batten disease: clinical aspects, molecular mechanisms, translational science, and future directions. J Child Neurol. 2013;28(9):1074-100.
-
12Drack AV, Miller JN, Pearce DA. A novel c.1135_1138delCTGT mutation in CLN3 leads to juvenile neuronal ceroid lipofuscinosis. J Child Neurol. 2013;28(9):1112-6.
-
13Seeliger M, Rüther K, Apfelstedt-Sylla E, Schlote W, Wohlrab M, Zrenner E. [Juvenile neuronal ceroid lipofuscinosis (Batten-Mayou) disease. Ophthalmologic diagnosis and findings]. Ophthalmologe. 1997;94(8):557-62. German.
-
14Weleber RG. The dystrophic retina in multisystem disorders: the electroretinogram in neuronal ceroid lipofuscinoses. Eye (Lond). 1998;12(Pt 3b):580-90.
-
15Weleber RG, Gupta N, Trzupek KM, Wepner MS, Kurz DE, Milam AH. Electroretinographic and clinicopathologic correlations of retinal dysfunction in infantile neuronal ceroid lipofuscinosis (infantile Batten disease). Mol Genet Metab. 2004;83(1-2):128-37.
-
16Marmor MF, Fulton AB, Holder GE, Miyake Y, Brigell M, Bach H; International Society for Clinical Electrophysiology of Vision. ISCEV standard for full-field clinical electroretinography (2008 update). Doc Ophthalmol. 2009;118(1):69-77.
-
17Berezovsky A, Moraes NS, Nusinowitz S, Salomão SR. Standard full-field electroretinography in healthy preterm infants. Doc Ophthalmol. 2003;107(3):243-9.
-
18Pereira JM, Mendieta L, Sacai PY, Salomão SR, Berezovsky A. Estudo normativo do eletrorretinograma de campo total em adultos jovens. Arq Bras Oftalmol. 2003;66(1): 137-44.
-
19Horiguchi M, Miyake Y. Batten disease - deteriorating course of ocular findings. Jpn J Ophthalmol. 1992;36(1):91-6.
-
20Collins J, Holder GE, Herbert H, Adams GG. Batten disease: features to facilitate early diagnosis. Br J Ophthalmol. 2006;90(9):1119-24.
-
21Selden NR, Al-Uzri A, Huhn SL, Koch TK, Sikora DM, Nguyen-Driver MD, et al. Central nervous system stem cell transplantation for children with neuronal ceroid lipofuscinosis. J Neurosurg Pediatr. 2013;11(6):643-52.
Publication Dates
-
Publication in this collection
Jul-Aug 2017
History
-
Received
01 Apr 2016 -
Accepted
18 Jan 2017