ABSTRACT
BACKGROUND:
Primary sclerosing cholangitis is a rare disease, but its prevalence has been underestimated in children and adolescents due to broad variation in clinical presentation as well as diagnostic challenges in this life period.
OBJECTIVE:
To evaluate children and adolescents with primary sclerosing cholangitis and to describe their clinical, laboratorial, histopathological, and cholangiography conditions.
METHODS:
This is an observational descriptive research that took place from 2005 to 2016 and included all the patients seen in the Outpatient Unit for Pediatric Hepatology of Hospital das Clinicas of UFMG who had been diagnosed with primary sclerosing cholangitis before the age of 18. Diagnosis was established through clinical, laboratory, radiographic and/or histopathologic criteria. Other chronic liver diseases were excluded, as well as secondary causes of cholangitis. Data analysis used statistic resources in SPSS software. Variables were expressed as averages, standard deviation, absolute frequency, and percentage.
RESULTS:
Twenty-one patients fulfilled criteria to be included in the research sample. Male patients predominated (3.2:1) and average age at diagnosis was 6.7±3.9 years. Five (23.8%) patients had associated inflammatory bowel disease, four had ulcerative colitis and one indeterminate colitis. Signs and symptoms vary and are usually discrete at presentation. The most frequent symptom was abdominal pain (47.6%) followed less frequently by jaundice (28.6%) and itching (14.3%). The reason for medical investigation was asymptomatic or oligosymptomatic enzyme alterations in 33.3% of patients. All patients presented increased hepatic enzymes: aminotransferases, gamma glutamyl transferase, and alkaline phosphatase. Twenty patients had alterations compatible to primary sclerosing cholangitis in their cholangiography exam; one patient had no alterations at magnetic resonance cholangiography, but presented histopathologic alterations that were compatible to small duct cholangitis. Hepatic fibrosis was present in 60% of 15 patients who were biopsied upon admission; cirrhosis being present in four (26.7%) patients. A total of 28.5% of patients had unfavorable outcomes, including two (9.5%) deaths and four (19%) transplants.
CONCLUSION:
Primary sclerosing cholangitis is a rare disease in childhood and adolescence and its initial diagnosis may be delayed or overlooked due to asymptomatic or unspecific clinical manifestations. The association with inflammatory bowel disease is common. Prognosis may be unfavorable as the disease progresses and hepatic transplant is the definitive treatment.
HEADINGS:
Sclerosing cholangitis; diagnosis; Child; Adolescent; Liver cirrhosis; Liver transplantation
RESUMO
CONTEXTO:
A colangite esclerosante primária é uma doença rara, mas sua prevalência tem sido subestimada em crianças e adolescentes, tanto pela variedade de apresentação clínica quanto pela dificuldade diagnóstica neste período.
OBJETIVO:
Avaliar crianças e adolescentes com colangite esclerosante primária descrevendo seu quadro clínico, laboratorial, histopatológico e colangiográfico.
MÉTODOS:
Trata-se de um estudo descritivo observacional, de 2005 a 2016, de todos os pacientes atendidos no Ambulatório de Hepatologia Pediátrica do Hospital das Clínicas da UFMG, com o diagnóstico de colangite esclerosante primária até a idade de 18 anos. O diagnóstico foi estabelecido segundo os critérios clínicos, laboratoriais, radiológicos e/ou histopatológicos. Foi realizada investigação para exclusão de outras doenças hepáticas crônicas e causas secundárias de colangite. A análise dos dados foi efetuada com os recursos estatísticos do software SPSS. As variáveis foram expressas por meio de médias, desvio-padrão, frequência absoluta e porcentagem.
RESULTADOS:
Vinte e um pacientes preencheram os critérios de inclusão. Houve predomínio no sexo masculino (3,2:1). A média de idade ao diagnóstico foi 6,7±3,9 anos. Cinco (23,8%) pacientes apresentavam doença inflamatória intestinal associada, sendo quatro casos de colite ulcerativa e um de colite indeterminada. Na apresentação, os sinais e sintomas são variados, em geral discretos. O sintoma mais frequente foi dor abdominal (47,6%), seguido menos frequentemente por icterícia (28,6%) e prurido (14,3%). Em 33,3% dos pacientes, o motivo de início da investigação foi alteração de enzimas em pacientes assintomáticos ou oligossintomáticos. Todos os pacientes exibiam aumento das enzimas hepáticas: aminotransferases, gama glutamiltransferase e fosfatase alcalina. Vinte pacientes apresentavam alterações em exame colangiográfico, compatíveis com colangite esclerosante primária; um paciente não apresentava alterações na colangiorressonância, mas apresentava alterações histopatológicas compatíveis com colangite de pequenos ductos. Fibrose hepática já estava presente em 60% dos 15 pacientes com biópsia à admissão; com cirrose estabelecida em quatro pacientes. 28,5% dos pacientes tiveram evolução desfavorável, com dois (9,5%) óbitos e quatro (19%) pacientes transplantados.
CONCLUSÃO:
Colangite esclerosante primária é uma doença rara na criança e no adolescente, cujo diagnóstico inicial pode ser atrasado ou passar despercebido, principalmente por apresentar manifestações clínicas inespecíficas ou cursar de forma assintomática. É frequente a associação com doença inflamatória intestinal. O prognóstico pode ser reservado com o avançar da doença, sendo o transplante hepático o tratamento definitivo.
DESCRITORES:
Colangite esclerosante primária; diagnóstico; Criança; Adolescente; Cirrose hepática; Transplante de fígado
INTRODUCTION
Primary sclerosing cholangitis (PSC) is a chronic liver disease of unknown etiology with an autoimmune component, characterized by chronic inflammation and fibrosis of intra and/or extra-hepatic biliary ducts, and a slow, but progressive, evolution towards cirrhosis55. Feldstein AE, Perrault J, El-Youssif M, Lindor KD, Freese DK, Angulo P. Primary Sclerosing Cholangitis in Children: A Long-Term Follow-Up Study. Hepatology . 2003;38:210-7.,77. Mieli-Vergani G, Vergani D. Unique features of primary sclerosing cholangitis in children. Curr Opin Gastroentetrol. 2010;26:265-8.,88. Mieli-Vergani G, Vergani D. Sclerosing Cholangitis in Children and Adolescents. Clin Liver Dis. 2016;20:99-111.. PSC is predominant in males (2:1) and young adults, although it can occur at any age. However, some characteristics are specific to children. Itching, for example, is less frequent in children and adolescents when compared to adults. PSC prevalence has been underestimated in children and adolescents, due to a broad range of clinical presentations as well as diagnostic challenges inherent to this age group. The initial phase of the disease is usually insidious and silent. Diagnosis may be delayed and occur only when chronic liver disease, cirrhosis with ascites or digestive hemorrhage or portal hypertension complications have already set in99. Miethke AG, Balistreri WF. Sclerosing cholangitis. In: Suchy FG, Sokol RJ, Balistreri WF. Liver Disease in Children. New York. Cambridge University Press. 2014:322-40.. Therefore, PSC should always be considered in any patient with chronic liver disease of unidentified etiology, particularly males.
Diagnosis of PSC is based on cholangiography findings (multifocal dilations and stricture in part or the entirety of the biliary tract), clinical, laboratory, and histological findings, as well as exclusion of secondary causes of cholangitis33. Deneau M, Jensen MK, Holmen J, Williams MS, Book LS, Guthery SL. Primary Sclerosing Cholangitis, Autoimmune Hepatitis, and Overlap in Utah Children: Epidemiology and Natural History. Hepatology. 2013;58:1392-400.,66. Girarda M, Franchi-Abellab S, Lacaille F, Debraya D. Specificities of sclerosing cholangitis in childhood. Clin Res Hepatol Gastroenterol. 2012;36:530-5.,77. Mieli-Vergani G, Vergani D. Unique features of primary sclerosing cholangitis in children. Curr Opin Gastroentetrol. 2010;26:265-8.,99. Miethke AG, Balistreri WF. Sclerosing cholangitis. In: Suchy FG, Sokol RJ, Balistreri WF. Liver Disease in Children. New York. Cambridge University Press. 2014:322-40.. Aminotransferases levels are slightly increased; gamma glutamyl transferase (GGT) can reach twice the normal levels and is more sensitive than alkaline phosphatase for diagnosis in children33. Deneau M, Jensen MK, Holmen J, Williams MS, Book LS, Guthery SL. Primary Sclerosing Cholangitis, Autoimmune Hepatitis, and Overlap in Utah Children: Epidemiology and Natural History. Hepatology. 2013;58:1392-400.,55. Feldstein AE, Perrault J, El-Youssif M, Lindor KD, Freese DK, Angulo P. Primary Sclerosing Cholangitis in Children: A Long-Term Follow-Up Study. Hepatology . 2003;38:210-7.,77. Mieli-Vergani G, Vergani D. Unique features of primary sclerosing cholangitis in children. Curr Opin Gastroentetrol. 2010;26:265-8.. Recently, PSC of small ducts was described, when chronic cholestasis and histopathologic findings compatible with PSC associated with normal imaging exams for the biliary tract88. Mieli-Vergani G, Vergani D. Sclerosing Cholangitis in Children and Adolescents. Clin Liver Dis. 2016;20:99-111..
Diagnosis and management of PSC is challenging in pediatric patients. Prognosis is reserved and 20% to 30% of patients will require liver transplantation88. Mieli-Vergani G, Vergani D. Sclerosing Cholangitis in Children and Adolescents. Clin Liver Dis. 2016;20:99-111..
Taking into account the limited number of studies in literature, the objective of this study is to evaluate children and adolescents with PSC, describing their clinical, laboratory, histopathologic and cholangiography findings.
METHODS
This study has an observational descriptive research design and took place from 2005 to 2016, included all patients seen in the Outpatient Unit for Pediatric Hepatology in the Pediatric Gastroenterology sector of the Clinical Hospital of the Federal University of Minas Gerais (UFMG) who had been diagnosed with PSC before the age of 18.
Diagnosis was established through clinical, laboratory, radiographic and/or histopathologic criteria: clinical evidence of chronic liver disease (hepatosplenomegaly, palmar erythema, telangiectasis) associated to alteration in laboratory exams (increased GGT, associated or not to increased aminotransferases), and/or histopathologic alterations (bile duct injury, periductular onion skin fibrosis, cholestasis), and/or imaging alterations (multifocal dilations and stricture in part or the entirety of the biliary tract compatible to diagnosis). Patients with superimposed autoimmune hepatitis or secondary causes of cholangitis (such as trauma, tumors, histiocytosis, immunodeficiency, and infections) were excluded from the sample.
All patients who are admitted to the outpatient unit undergo clinical and laboratory evaluation, including hepatic enzymes, histopathologic and imaging evaluation, among others. Other chronic liver diseases were investigated in order to exclude autoimmune hepatitis (AIH), Wilson’s disease, α1-antitripsine deficiency, viral hepatitis, and hepatitis due to drugs, as well as secondary causes of cholangitis. Follow-up was established according to clinical conditions of each patient.
Biopsies were evaluated searching for signs of PSC, such as aggression and ductal proliferation, onion skin fibrosis, reduced ducts, cholestasis, or inflammation. Other histopathologic findings were also evaluated to facilitate differential diagnosis, particularly with superimposed autoimmune hepatitis. Presence of fibrosis and cirrhosis was also taken into consideration.
Cholangiography findings were obtained through magnetic resonance cholangiopancreatography (MRCP) or endoscopic retrograde cholangiopancreatography (ERCP). Images were then reevaluated by the health team responsible for follow up. Findings suggestive of PSC include multifocal dilations and strictures in part or entirety of biliary tract.
Colonoscopy to investigate for inflammatory bowel disease (IBD) was performed in all patients with gastrointestinal symptoms. The diagnosis of IBD was confirmed on histology in conjunction with the endoscopic findings reported by the endoscopist.
Data analysis used statistic resources in SPSS software. Variables were expressed as averages, standard deviation, absolute frequency, and percentage.
The study was approved by the Research Ethics Committee of UFMG. (no. 17395313.0.0000.5149).
RESULTS
Twenty-one patients fit sample inclusion criteria. Average follow-up time was of 4.8±3.4 years, varying from 3 months to 10 years.
The main clinical findings are summarized in Table 1. Male gender was predominant (3.2:1). Age at initial presentation of symptoms and diagnosis was predominantly in the first decade of life, with a lag of 1.6 years from onset to average age at diagnosis. Five patients presented inflammatory bowel disease (IBD), four of these having ulcerative colitis and one with undetermined colitis. In four patients, diagnosis of IBD took place before or concurrently to PSC diagnosis. In one patient, ulcerative colitis became evident after diagnosis of PSC.
The most frequent symptom at disease onset was abdominal pain (47.6%), followed by jaundice (28.6%), and itching (14.3%). Hepatosplenomegaly was present in 52.4% of patients at clinical exam. Altered enzyme levels were the reason for liver disease investigation in 33.3% of patients and, in 9.5%, low platelet count secondary to hepatosplenomegaly.
Laboratory results are found in Table 2. All patients showed increased hepatic enzymes: alanine aminotransferase (ALT), aspartate aminotransferase (AST), GGT and alkaline phosphatase. Anti-nuclear factor (ANA) was positive in only three (14.2%) patients. Positive anti-neutrophil cytoplasmic antibody (ANCA) was found in 14.2% of our sample.
All 21 patients underwent cholangiogram exam - one through ERCP and the remaining through MRCP. Only one patient had no biliary tract alterations, although GGT and alkaline phosphatase were increased and the biopsy sample showed small duct anomalies.
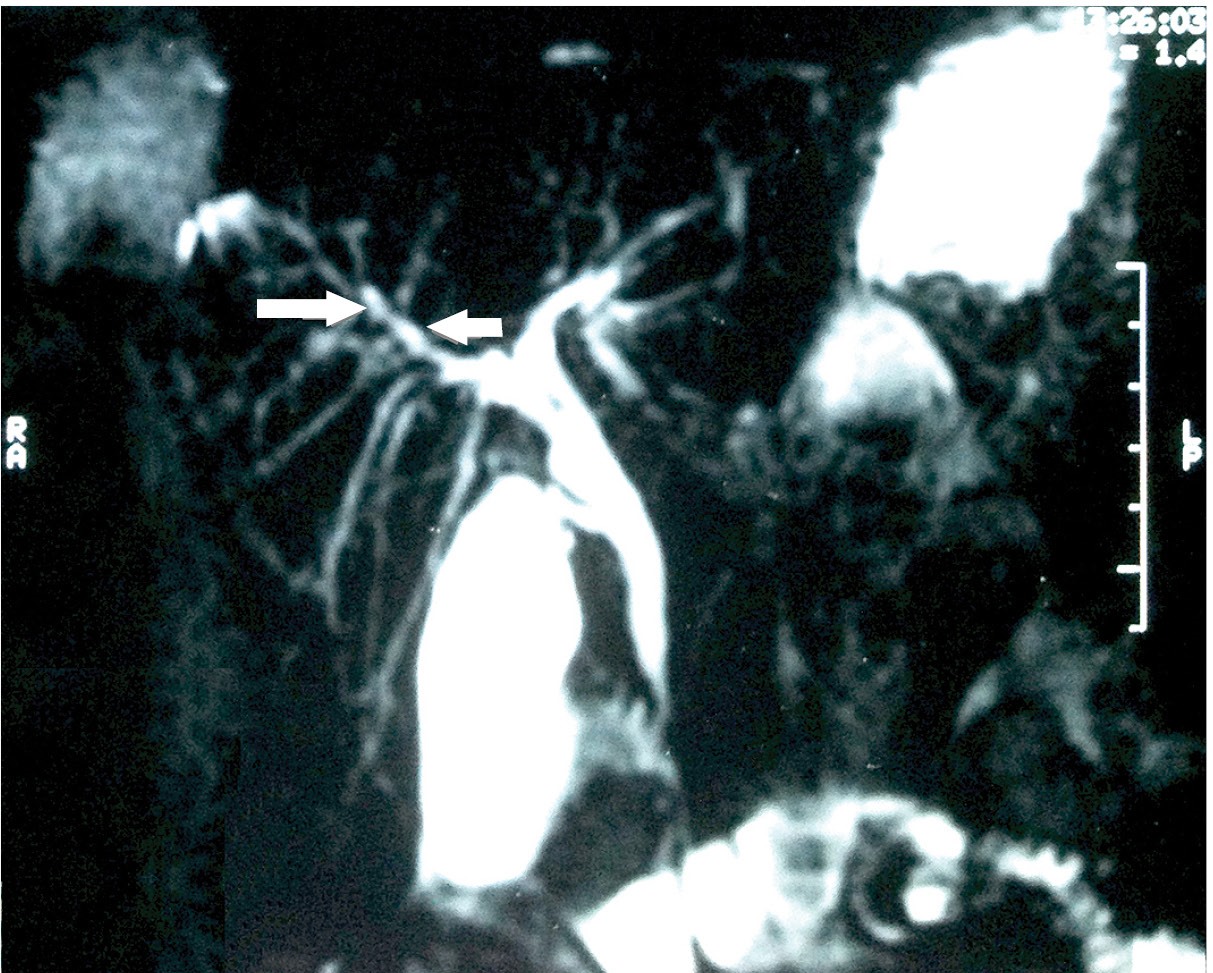
The 20 patients who presented alterations in the cholangiogram had findings compatible with PSC diagnosis (Table 3). Dilations and/or stricture of intra or extrahepatic biliary ducts (Figure 1) were the most common findings (81%). Alterations exclusive to extrahepatic sites were found in only two (9.5%) patients. No patients had exclusively intrahepatic biliary alterations. One patient evolved with dominant stenosis of the biliary tract. Following repeated episodes of cholangitis and sepsis, this last patient received a liver transplant and is currently well.

Magnetic resonance cholangiopancreatography image shows overall irregularity of the intra and extrahepatic ducts which produces the beaded appearance (arrows).
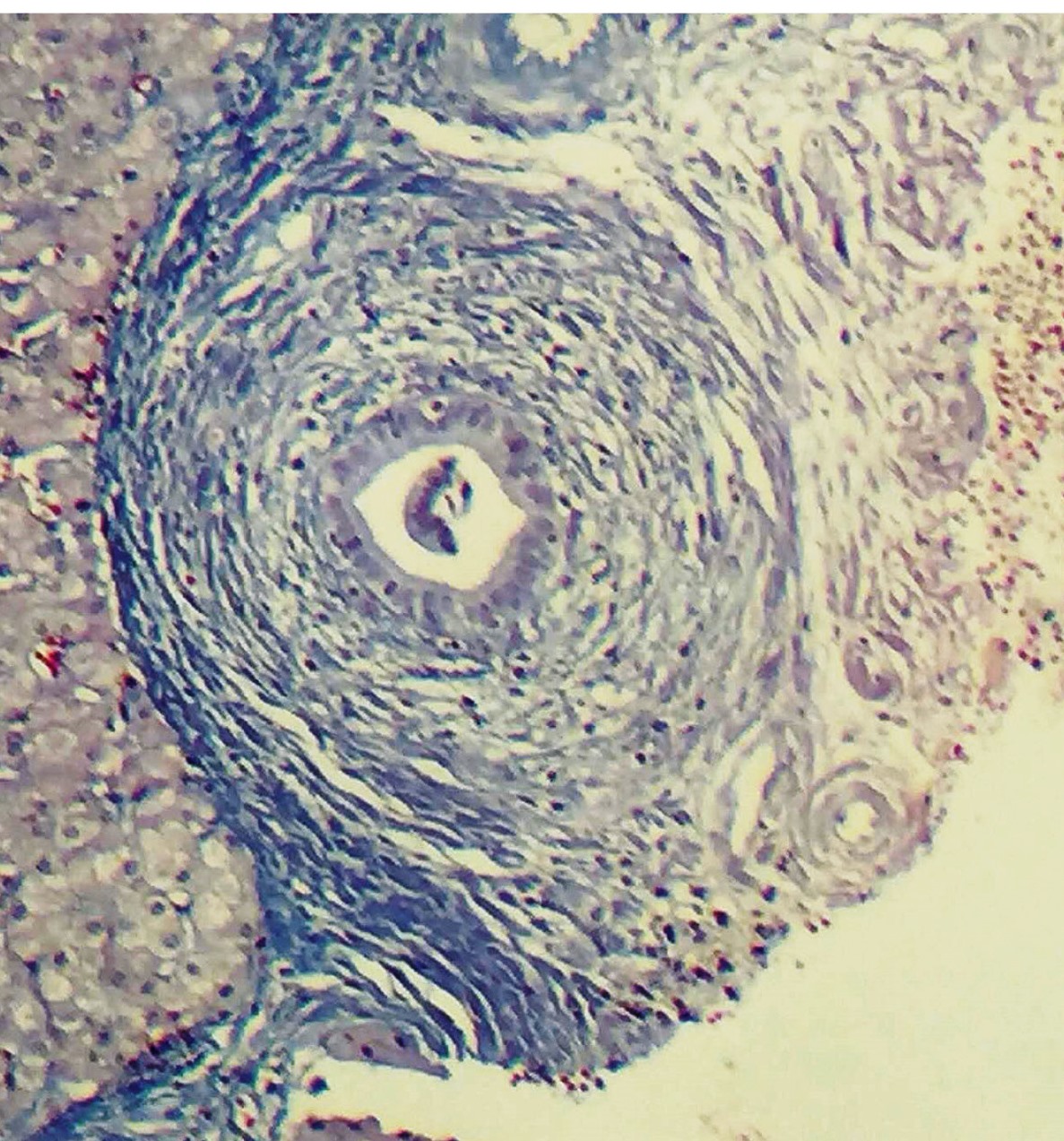
Liver biopsy was performed in 15 of the 21 patients. Biopsy results are summarized in Table 4. Fibrosis was established in 9 (60%) of the 15 biopsied patients, and cirrhosis in four (26.7%). Onion skin fibrosis (Figure 2) was found in only two (13.3%) patients. Lesions and ductal aggression were present in 33.3% of patients.

Fatal outcome occurred in two patients. The first was a 13-year-old with cholangiocarcinoma presenting with acute and sudden deterioration of liver function, jaundice, and difficult-to-manage ascites, leading to death within eight months of diagnosis. This patient did not present IBD. The second patient died at 22-years-of-age due to hepatic failure before transplant.
Four (19%) patients received transplants, one at 11, two at 13, and one at 15 years of age. All had good outcomes and have not presented disease recurrence to date (1 to 8 years following liver transplant).
Twenty patients were treated with ursodeoxycholic acid (UDCA) at an average dose of 15 mg/kg/day. One patient did not make use of this medication due to swift progression to liver failure and need for transplant. Of those who made use of UDCA, 12 (60%) presented biochemical improvement. The two patients who died and three of the four patients who received a liver transplant never reached normal enzyme concentrations while in use of UDCA. No patients presented adverse reactions significant enough to lead to suspension of medication.
Five patients also received steroids and/or azathioprine due to IBD, with good results.
DISCUSSION
Studies on PSC in pediatric patients are limited, especially in Brazil. Only one report of four patients was published in 19981313. Silva OO, Santos TE, Cardoso ES, et al. Colangite esclerosante primária em crianças e adolescentes. Arq Gastroenterol. 1998;35:267-73.. Existing literature sometimes presents diverging results depending on time of publication and the service in which the study took place, reflecting diverse research designs and diagnostic protocols. Older research studies, in particular, failed to exclude secondary causes of cholangitis. Cholangitis that is secondary to systemic diseases such as immunodeficiency may lead to poor prognosis. Besides that, it is important separate PSC-AIH overlap with features of both diseases that is termed autoimmune sclerosing cholangitis (ASC)22. Debray D, Pariente D, Urvoas E, et al. Sclerosing cholangitis in children. J Pediatr. 1994;124:49-56.,44. el-Shabrawi M, Wilkinson ML, Portmann B, et al. Primary sclerosing cholangitis in childhood. Gastroenterology. 1987;92:1226-35.,55. Feldstein AE, Perrault J, El-Youssif M, Lindor KD, Freese DK, Angulo P. Primary Sclerosing Cholangitis in Children: A Long-Term Follow-Up Study. Hepatology . 2003;38:210-7.,1515. Wilschanski M, Chait P, Wade JA, et al. Primary sclerosing cholangitis in 32 children: clinical, laboratory and radiographic features, with survival analysis. Hepatology . 1995;22:1415-22.. Separation between patients with isolated PSC and those with ASC is relevant because their prognosis is different due to response to immunosuppressants in ASC, unlike isolated PSC88. Mieli-Vergani G, Vergani D. Sclerosing Cholangitis in Children and Adolescents. Clin Liver Dis. 2016;20:99-111.,1212. Rodrigues AT, Liu PMF, Fagundes EDT, et al. Clinical Characteristics and Prognosis in Children and Adolescents With Autoimmune Hepatitis and Overlap Syndrome. JPGN. 2016;63:76-81.. Differentiation between the two isn’t always clear and progression of AIH to ASC has been documented in children in particular88. Mieli-Vergani G, Vergani D. Sclerosing Cholangitis in Children and Adolescents. Clin Liver Dis. 2016;20:99-111.. Another limitation of these studies is that most are, as is ours, retrospective.
PSC remains a rare disease in childhood. In the present study, we described 21 patients seen throughout 12 years of outpatient follow up. Cases of secondary cholangitis, immunodeficiency, and ASC were excluded from the sample. Various authors in literature report different sample sizes (Table 5), but some included patients with secondary cholangitis and ASC. Valentino et al.1414. Valentino PL, Wiggins S, Harney S, Raza R, Lee CK, Jonas MM. The natural history of primary sclerosing cholangitis in children: a large single-center longitudinal cohort study. JPGN. 2016;63:603-9. published the largest sample size to date, where they describe 120 patients, 89 of these with isolated PSC.
The disease is more predominant in males, unlike autoimmune hepatites, as reported in literature33. Deneau M, Jensen MK, Holmen J, Williams MS, Book LS, Guthery SL. Primary Sclerosing Cholangitis, Autoimmune Hepatitis, and Overlap in Utah Children: Epidemiology and Natural History. Hepatology. 2013;58:1392-400.,55. Feldstein AE, Perrault J, El-Youssif M, Lindor KD, Freese DK, Angulo P. Primary Sclerosing Cholangitis in Children: A Long-Term Follow-Up Study. Hepatology . 2003;38:210-7.,77. Mieli-Vergani G, Vergani D. Unique features of primary sclerosing cholangitis in children. Curr Opin Gastroentetrol. 2010;26:265-8.,1414. Valentino PL, Wiggins S, Harney S, Raza R, Lee CK, Jonas MM. The natural history of primary sclerosing cholangitis in children: a large single-center longitudinal cohort study. JPGN. 2016;63:603-9.. In this study, disease onset was within the first decade of life. Most population samples reported show onset in second decade of life33. Deneau M, Jensen MK, Holmen J, Williams MS, Book LS, Guthery SL. Primary Sclerosing Cholangitis, Autoimmune Hepatitis, and Overlap in Utah Children: Epidemiology and Natural History. Hepatology. 2013;58:1392-400.,55. Feldstein AE, Perrault J, El-Youssif M, Lindor KD, Freese DK, Angulo P. Primary Sclerosing Cholangitis in Children: A Long-Term Follow-Up Study. Hepatology . 2003;38:210-7.,1414. Valentino PL, Wiggins S, Harney S, Raza R, Lee CK, Jonas MM. The natural history of primary sclerosing cholangitis in children: a large single-center longitudinal cohort study. JPGN. 2016;63:603-9.,1515. Wilschanski M, Chait P, Wade JA, et al. Primary sclerosing cholangitis in 32 children: clinical, laboratory and radiographic features, with survival analysis. Hepatology . 1995;22:1415-22.. In Valentino and collaborators’1414. Valentino PL, Wiggins S, Harney S, Raza R, Lee CK, Jonas MM. The natural history of primary sclerosing cholangitis in children: a large single-center longitudinal cohort study. JPGN. 2016;63:603-9. sample, 40% of cases were diagnosed in the first decade of life, 4 of these under two years of age and the youngest at 6 months of age.
Signs and symptoms vary at onset and are generally subtle. Assymptomatic cases are also common and are evaluated due to hepatosplenomegaly or alteration of laboratory results55. Feldstein AE, Perrault J, El-Youssif M, Lindor KD, Freese DK, Angulo P. Primary Sclerosing Cholangitis in Children: A Long-Term Follow-Up Study. Hepatology . 2003;38:210-7.. Cases that present jaundice, which would facilitate the identification of liver disease, are less frequent, and consisted of 28.6% in our study and 15% to 25% in other studies55. Feldstein AE, Perrault J, El-Youssif M, Lindor KD, Freese DK, Angulo P. Primary Sclerosing Cholangitis in Children: A Long-Term Follow-Up Study. Hepatology . 2003;38:210-7.,1010. Miloh T, Arnon R, Shneider B, Suchy F, Kerkar N. A Retrospective Single-Center Review of Primary Sclerosing Cholangitis in Children. Clin Gastroenterol Hepatol. 2009;7:239-45.,1515. Wilschanski M, Chait P, Wade JA, et al. Primary sclerosing cholangitis in 32 children: clinical, laboratory and radiographic features, with survival analysis. Hepatology . 1995;22:1415-22.. Itching was reported in 15% of our patients, a rate similar to the 19% described by Feldstein et al.55. Feldstein AE, Perrault J, El-Youssif M, Lindor KD, Freese DK, Angulo P. Primary Sclerosing Cholangitis in Children: A Long-Term Follow-Up Study. Hepatology . 2003;38:210-7.) and Miloh et al.1010. Miloh T, Arnon R, Shneider B, Suchy F, Kerkar N. A Retrospective Single-Center Review of Primary Sclerosing Cholangitis in Children. Clin Gastroenterol Hepatol. 2009;7:239-45., evidence that it is a less frequent symptom in childhood when compared to adults. In Valentino and collaborators’1414. Valentino PL, Wiggins S, Harney S, Raza R, Lee CK, Jonas MM. The natural history of primary sclerosing cholangitis in children: a large single-center longitudinal cohort study. JPGN. 2016;63:603-9. sample, most patients were diagnosed due to alterations in hepatic biochemistry and only 6% of patients had jaundice, 26% abdominal pain and/or itching, while 64% were asymptomatic. Due to the lack of specificity in clinical presentations, PSC should always be taken into consideration in any patient with chronic liver disease without etiologic definition, particularly in males.
Our study found a lag of 1.6 years between onset of disease and final diagnosis. Subtle and unspecific diagnosis and a need for broad evaluations to exclude other liver diseases in differential diagnosis partially explain the noteworthy delay in reaching a final diagnosis. However, in their American sample, Valentino et al.1414. Valentino PL, Wiggins S, Harney S, Raza R, Lee CK, Jonas MM. The natural history of primary sclerosing cholangitis in children: a large single-center longitudinal cohort study. JPGN. 2016;63:603-9. found a shorter span, of only 2.5 months, between onset and diagnosis. We speculate that, due to the rarity of the disease, health professionals do not readily recognize it among differential diagnosis and this might contribute to diagnostic delay and may be the most effective area for intervention for more effective and prompt diagnosis.
As in adults, PSC is frequently associated to IBD. In this study, only 23.8% of cases had associated IBD, predominantly ulcerative colitis. In literature, the association rate is described as 53% to 85% of cases, also predominately colitis55. Feldstein AE, Perrault J, El-Youssif M, Lindor KD, Freese DK, Angulo P. Primary Sclerosing Cholangitis in Children: A Long-Term Follow-Up Study. Hepatology . 2003;38:210-7.,1414. Valentino PL, Wiggins S, Harney S, Raza R, Lee CK, Jonas MM. The natural history of primary sclerosing cholangitis in children: a large single-center longitudinal cohort study. JPGN. 2016;63:603-9.,1515. Wilschanski M, Chait P, Wade JA, et al. Primary sclerosing cholangitis in 32 children: clinical, laboratory and radiographic features, with survival analysis. Hepatology . 1995;22:1415-22., but lower rates of 10.7% were found by Debray et al.22. Debray D, Pariente D, Urvoas E, et al. Sclerosing cholangitis in children. J Pediatr. 1994;124:49-56., although their sample included cases of secondary cholangitis. This difference may owe itself to the fact that not all our patients were submitted to colonoscopy, only those with symptoms, which may have lead to a lower prevalence. Therefore, colonoscopy might be recommended in all patients with PSC, regardless of symptoms, as recommended by English King’s College88. Mieli-Vergani G, Vergani D. Sclerosing Cholangitis in Children and Adolescents. Clin Liver Dis. 2016;20:99-111.. Most cases occurred previous to or concurrent with diagnosis of PSC, as described in literature55. Feldstein AE, Perrault J, El-Youssif M, Lindor KD, Freese DK, Angulo P. Primary Sclerosing Cholangitis in Children: A Long-Term Follow-Up Study. Hepatology . 2003;38:210-7.,1414. Valentino PL, Wiggins S, Harney S, Raza R, Lee CK, Jonas MM. The natural history of primary sclerosing cholangitis in children: a large single-center longitudinal cohort study. JPGN. 2016;63:603-9..
No other associated autoimmune diseases were found among our patients. Regarding autoimmune hepatitis, overlap with sclerosing cholangitis was an exclusion criterion. Valentino et al.1414. Valentino PL, Wiggins S, Harney S, Raza R, Lee CK, Jonas MM. The natural history of primary sclerosing cholangitis in children: a large single-center longitudinal cohort study. JPGN. 2016;63:603-9. found low association (7%) of PSC to other autoimmune diseases such as diabetes and celiac disease.
All our patients had alterations in blood concentrations of hepatic enzymes, aminotransferases and GGT and alkaline phosphatase. Feildstein et al.55. Feldstein AE, Perrault J, El-Youssif M, Lindor KD, Freese DK, Angulo P. Primary Sclerosing Cholangitis in Children: A Long-Term Follow-Up Study. Hepatology . 2003;38:210-7. found altered alkaline phosphatase in 25% of their sample; however, their study included patients with autoimmune cholangitis, which may explain some discrepancies between results. These authors also found positive ANA and AML in 69% of cases while our sample had a 14.3% rate of positivity for ANA. These differences highlight the importance of analyzing patients with sclerosing cholangitis separately from those with ASC since both clinical presentation and disease progression appear to be different.
Regarding imaging of biliary tract, MRCP has the advantage of not being invasive and not contributing to risk of radiation or associated pancreatitis, unlike ERCP. It has also been shown to be accurate in diagnosis of PSC1010. Miloh T, Arnon R, Shneider B, Suchy F, Kerkar N. A Retrospective Single-Center Review of Primary Sclerosing Cholangitis in Children. Clin Gastroenterol Hepatol. 2009;7:239-45.. The findings are those classically described in studies of children and adults: predominance of alterations in both intra and extrahepatic ducts as found in this study55. Feldstein AE, Perrault J, El-Youssif M, Lindor KD, Freese DK, Angulo P. Primary Sclerosing Cholangitis in Children: A Long-Term Follow-Up Study. Hepatology . 2003;38:210-7.,1010. Miloh T, Arnon R, Shneider B, Suchy F, Kerkar N. A Retrospective Single-Center Review of Primary Sclerosing Cholangitis in Children. Clin Gastroenterol Hepatol. 2009;7:239-45.. In the study by Miloh et al., 16 (32.7%) of 49 patients had normal cholangiograms, with abnormalities only in histopathology, characterizing cholangitis of small ducts1010. Miloh T, Arnon R, Shneider B, Suchy F, Kerkar N. A Retrospective Single-Center Review of Primary Sclerosing Cholangitis in Children. Clin Gastroenterol Hepatol. 2009;7:239-45..
Presence of histopathologic fibrosis in 60% of patients, 26.7% of these with established cirrhosis might explain patient outcomes. Onion skin fibrosis was found in only two patients and is reported to be rare88. Mieli-Vergani G, Vergani D. Sclerosing Cholangitis in Children and Adolescents. Clin Liver Dis. 2016;20:99-111.. Patients with advanced fibrosis (METAVIR 3 or 4) at diagnosis composed 41% of Valentino and collaborators’ sample and 65% of Batres et al.11. Batres LA, Russo P, Mathews M, Piccoli DA, Chuang E, Ruchelli E. Primary sclerosing cholangitis in children: a histologic follow-up study. Pediatr Dev Pathol. 2005;8:568-76.. Miloh et al.1010. Miloh T, Arnon R, Shneider B, Suchy F, Kerkar N. A Retrospective Single-Center Review of Primary Sclerosing Cholangitis in Children. Clin Gastroenterol Hepatol. 2009;7:239-45. found that all patients had abnormalities that suggested PSC, with some degree of fibrosis, cirrhosis in 9%, and 12 (26.7%) of 45 patients with onion skin fibrosis. Small duct PSC was observed in 34% of Miloh’s cases, a higher rate than that found by other authors11. Batres LA, Russo P, Mathews M, Piccoli DA, Chuang E, Ruchelli E. Primary sclerosing cholangitis in children: a histologic follow-up study. Pediatr Dev Pathol. 2005;8:568-76.,88. Mieli-Vergani G, Vergani D. Sclerosing Cholangitis in Children and Adolescents. Clin Liver Dis. 2016;20:99-111.,1010. Miloh T, Arnon R, Shneider B, Suchy F, Kerkar N. A Retrospective Single-Center Review of Primary Sclerosing Cholangitis in Children. Clin Gastroenterol Hepatol. 2009;7:239-45.. In our study only one patient presented small duct cholangitis.
Prognosis of PSC is poor as evidenced by results of follow up showing high rates of transplants and complications throughout disease progression. Approximately 20% to 30% of patients will require liver transplants11. Batres LA, Russo P, Mathews M, Piccoli DA, Chuang E, Ruchelli E. Primary sclerosing cholangitis in children: a histologic follow-up study. Pediatr Dev Pathol. 2005;8:568-76.,88. Mieli-Vergani G, Vergani D. Sclerosing Cholangitis in Children and Adolescents. Clin Liver Dis. 2016;20:99-111.. In this study it is noteworthy that 9.5% of patients had a fatal outcome and 19% required transplant; therefore 28.5% of patients had a poor prognosis, a rate similar to that described by Wilschanski et al.1515. Wilschanski M, Chait P, Wade JA, et al. Primary sclerosing cholangitis in 32 children: clinical, laboratory and radiographic features, with survival analysis. Hepatology . 1995;22:1415-22.. Miloh et al.1010. Miloh T, Arnon R, Shneider B, Suchy F, Kerkar N. A Retrospective Single-Center Review of Primary Sclerosing Cholangitis in Children. Clin Gastroenterol Hepatol. 2009;7:239-45. found that 17% of patients required transplants; one patient progressed to cholangiocarcinoma and death eight months after diagnosis. Deneau et al.33. Deneau M, Jensen MK, Holmen J, Williams MS, Book LS, Guthery SL. Primary Sclerosing Cholangitis, Autoimmune Hepatitis, and Overlap in Utah Children: Epidemiology and Natural History. Hepatology. 2013;58:1392-400. also reported two cases of cholangiocarcinoma among 29 patients aged 17.9 to 18 years of age. Our patient, at 13-years-old, was younger and had been diagnosed with PSC at 6.5 years of age. In Feldstein et al.55. Feldstein AE, Perrault J, El-Youssif M, Lindor KD, Freese DK, Angulo P. Primary Sclerosing Cholangitis in Children: A Long-Term Follow-Up Study. Hepatology . 2003;38:210-7. study, average survival with native liver was of 12.7 years, further evidencing the poor prognosis of the disease. Deneau et al.33. Deneau M, Jensen MK, Holmen J, Williams MS, Book LS, Guthery SL. Primary Sclerosing Cholangitis, Autoimmune Hepatitis, and Overlap in Utah Children: Epidemiology and Natural History. Hepatology. 2013;58:1392-400.) reported that 11 (38%) of 29 patients developed complications such as portal hypertension, cholangiocarcinoma, liver transplant, and death. In Valentino et al.1414. Valentino PL, Wiggins S, Harney S, Raza R, Lee CK, Jonas MM. The natural history of primary sclerosing cholangitis in children: a large single-center longitudinal cohort study. JPGN. 2016;63:603-9., in a sample of 89 patients, none progressed to neoplasia and fatal outcome was of 2%, less than previous studies. One of our patients presented dominant stenosis of biliary tract and underwent dilation of biliary tracts (4.8%), a rate compatible with previous publications which vary from 2% to 12%33. Deneau M, Jensen MK, Holmen J, Williams MS, Book LS, Guthery SL. Primary Sclerosing Cholangitis, Autoimmune Hepatitis, and Overlap in Utah Children: Epidemiology and Natural History. Hepatology. 2013;58:1392-400.,55. Feldstein AE, Perrault J, El-Youssif M, Lindor KD, Freese DK, Angulo P. Primary Sclerosing Cholangitis in Children: A Long-Term Follow-Up Study. Hepatology . 2003;38:210-7.,1414. Valentino PL, Wiggins S, Harney S, Raza R, Lee CK, Jonas MM. The natural history of primary sclerosing cholangitis in children: a large single-center longitudinal cohort study. JPGN. 2016;63:603-9..
Regarding treatment, 20 patients made use of UDCA, with no significant adverse effects. Patients who progressed to fatal outcome or transplants did not reach normal enzyme values despite medication. UDCA improves itchiness, may improve biochemistry, but studies in adults have failed to show improvement in disease progression or increased survival rates1010. Miloh T, Arnon R, Shneider B, Suchy F, Kerkar N. A Retrospective Single-Center Review of Primary Sclerosing Cholangitis in Children. Clin Gastroenterol Hepatol. 2009;7:239-45.,1111. Nayagam JS, Pereira SP, Devlin J, Harrison PM, Joshi D. Controversies in the management of primary sclerosing cholangitis. World J Hepatol. 2016;8:265-72.,1616. Wunsch E, Trottier J, Milkiewicz M, et al. Prospective Evaluation of UrsodeoxycholicAcid Withdrawal in Patients With Primary Sclerosing Cholangitis. Hepatology . 2014;60:931-40.. Use of UDCA remains controversial in literature and the evaluation of its efficacy in changing disease prognosis is beyond the scope of this paper. Well designed and controlled studies with pediatric samples are desirable, despite the difficulties to execute due to the disease’s rarity. Therefore, current treatment is supportive and directed more towards management of complications (dominant stenosis, itchiness, cholangitis, and nutritional deficiencies) than the cause of PSC. Liver transplant is, therefore, the only alternative for most patients. Recurrence following transplant is reported for 11% to 27% of patients55. Feldstein AE, Perrault J, El-Youssif M, Lindor KD, Freese DK, Angulo P. Primary Sclerosing Cholangitis in Children: A Long-Term Follow-Up Study. Hepatology . 2003;38:210-7.,1010. Miloh T, Arnon R, Shneider B, Suchy F, Kerkar N. A Retrospective Single-Center Review of Primary Sclerosing Cholangitis in Children. Clin Gastroenterol Hepatol. 2009;7:239-45.,1414. Valentino PL, Wiggins S, Harney S, Raza R, Lee CK, Jonas MM. The natural history of primary sclerosing cholangitis in children: a large single-center longitudinal cohort study. JPGN. 2016;63:603-9., but our four transplanted patients have yet to show signs of disease recurrence.
PSC is concluded to be a rare disease, with a difficult diagnosis and poor prognosis. Therefore, diagnosis of PSC should be taken into account in any patient with etiologically undefined chronic hepatitis, particularly in males. Further studies are necessary to better understand physiopathology and improve approach and management of patients in order to improve prognosis of this devastating disease.
ACKNOWLEDGEMENTS
Dra. Paula Vieira Teixeira Vidigal (Universidade Federal de Minas Gerais) for the figure of hepatic biopsy.
REFERENCES
-
1Batres LA, Russo P, Mathews M, Piccoli DA, Chuang E, Ruchelli E. Primary sclerosing cholangitis in children: a histologic follow-up study. Pediatr Dev Pathol. 2005;8:568-76.
-
2Debray D, Pariente D, Urvoas E, et al. Sclerosing cholangitis in children. J Pediatr. 1994;124:49-56.
-
3Deneau M, Jensen MK, Holmen J, Williams MS, Book LS, Guthery SL. Primary Sclerosing Cholangitis, Autoimmune Hepatitis, and Overlap in Utah Children: Epidemiology and Natural History. Hepatology. 2013;58:1392-400.
-
4el-Shabrawi M, Wilkinson ML, Portmann B, et al. Primary sclerosing cholangitis in childhood. Gastroenterology. 1987;92:1226-35.
-
5Feldstein AE, Perrault J, El-Youssif M, Lindor KD, Freese DK, Angulo P. Primary Sclerosing Cholangitis in Children: A Long-Term Follow-Up Study. Hepatology . 2003;38:210-7.
-
6Girarda M, Franchi-Abellab S, Lacaille F, Debraya D. Specificities of sclerosing cholangitis in childhood. Clin Res Hepatol Gastroenterol. 2012;36:530-5.
-
7Mieli-Vergani G, Vergani D. Unique features of primary sclerosing cholangitis in children. Curr Opin Gastroentetrol. 2010;26:265-8.
-
8Mieli-Vergani G, Vergani D. Sclerosing Cholangitis in Children and Adolescents. Clin Liver Dis. 2016;20:99-111.
-
9Miethke AG, Balistreri WF. Sclerosing cholangitis. In: Suchy FG, Sokol RJ, Balistreri WF. Liver Disease in Children. New York. Cambridge University Press. 2014:322-40.
-
10Miloh T, Arnon R, Shneider B, Suchy F, Kerkar N. A Retrospective Single-Center Review of Primary Sclerosing Cholangitis in Children. Clin Gastroenterol Hepatol. 2009;7:239-45.
-
11Nayagam JS, Pereira SP, Devlin J, Harrison PM, Joshi D. Controversies in the management of primary sclerosing cholangitis. World J Hepatol. 2016;8:265-72.
-
12Rodrigues AT, Liu PMF, Fagundes EDT, et al. Clinical Characteristics and Prognosis in Children and Adolescents With Autoimmune Hepatitis and Overlap Syndrome. JPGN. 2016;63:76-81.
-
13Silva OO, Santos TE, Cardoso ES, et al. Colangite esclerosante primária em crianças e adolescentes. Arq Gastroenterol. 1998;35:267-73.
-
14Valentino PL, Wiggins S, Harney S, Raza R, Lee CK, Jonas MM. The natural history of primary sclerosing cholangitis in children: a large single-center longitudinal cohort study. JPGN. 2016;63:603-9.
-
15Wilschanski M, Chait P, Wade JA, et al. Primary sclerosing cholangitis in 32 children: clinical, laboratory and radiographic features, with survival analysis. Hepatology . 1995;22:1415-22.
-
16Wunsch E, Trottier J, Milkiewicz M, et al. Prospective Evaluation of UrsodeoxycholicAcid Withdrawal in Patients With Primary Sclerosing Cholangitis. Hepatology . 2014;60:931-40.
-
Disclosure of funding: no funding received
Publication Dates
-
Publication in this collection
02 Oct 2017 -
Date of issue
2017
History
-
Received
12 June 2017 -
Accepted
16 Aug 2017