Resumos
Apresenta-se um caso de síndrome de Cockayne, definem-se os critérios diagnósticos e identificam-se as complicações desta síndrome. Os critérios para diagnóstico deste quadro são: crescimento pobre, observado desde a vida intra-uterina, anormalidades neurológicas (desde o nascimento), perda auditiva neurossensorial, catarata (aparecimento nos primeiros 3 anos), anomalias estruturais congênitas do olho, retinopatia pigmentosa, fotossensibilidade cutânea, cáries dentárias, vida média de 12-13 anos (pode chegar aos 20 anos). O largo espectro de sintomas e sua severidade indicam distúrbio bioquímico e genético raro, de herança autossômica recessiva.
síndrome de Cockayne; retardo de desenvolvimento; herança autossômica recessiva; microcefalia
We describe a girl with Cockayne syndrome (CS), the diagnostic criteria and the complications of this syndrome. The required criteria for the diagnosis include: prenatal poor growth failure, congenital structural eye anomalies, cataracts, pigmentary retinopathy, severe neurologic dysfunction from birth, sensorineural hearing loss, cutaneous photosensitivity and dental caries. CS is a rare autosomal recessive and biochemical disorder.
Cockayne syndrome; developmental delay; autosomal recessive inheritance; microcephaly
SÍNDROME DE COCKAYNE
RELATO DE CASO
ANA GUARDIOLA* * Professora Adjunta do Departamento de Neurologia da Fundação Faculdade Federal de Ciências Médicas de Porto Alegre (FFFCMPA); ** Professor Adjunto do Departamento de Neurologia da FFFCMPA,; *** Residente de Neurologia da FFFCMPA; **** Residente de Genética Médica da FFFCMPA. Aceite: 30-outubro-1998. , CLÉBER RIBEIRO ÁLVARES-DA-SILVA** * Professora Adjunta do Departamento de Neurologia da Fundação Faculdade Federal de Ciências Médicas de Porto Alegre (FFFCMPA); ** Professor Adjunto do Departamento de Neurologia da FFFCMPA,; *** Residente de Neurologia da FFFCMPA; **** Residente de Genética Médica da FFFCMPA. Aceite: 30-outubro-1998. , JOSÉ RENATO GUIMARÃES GRISOLIA*** * Professora Adjunta do Departamento de Neurologia da Fundação Faculdade Federal de Ciências Médicas de Porto Alegre (FFFCMPA); ** Professor Adjunto do Departamento de Neurologia da FFFCMPA,; *** Residente de Neurologia da FFFCMPA; **** Residente de Genética Médica da FFFCMPA. Aceite: 30-outubro-1998. , ROGÉRIO SILBERMANN**** * Professora Adjunta do Departamento de Neurologia da Fundação Faculdade Federal de Ciências Médicas de Porto Alegre (FFFCMPA); ** Professor Adjunto do Departamento de Neurologia da FFFCMPA,; *** Residente de Neurologia da FFFCMPA; **** Residente de Genética Médica da FFFCMPA. Aceite: 30-outubro-1998.
RESUMO - Apresenta-se um caso de síndrome de Cockayne, definem-se os critérios diagnósticos e identificam-se as complicações desta síndrome. Os critérios para diagnóstico deste quadro são: crescimento pobre, observado desde a vida intra-uterina, anormalidades neurológicas (desde o nascimento), perda auditiva neurossensorial, catarata (aparecimento nos primeiros 3 anos), anomalias estruturais congênitas do olho, retinopatia pigmentosa, fotossensibilidade cutânea, cáries dentárias, vida média de 12-13 anos (pode chegar aos 20 anos). O largo espectro de sintomas e sua severidade indicam distúrbio bioquímico e genético raro, de herança autossômica recessiva.
PALAVRAS-CHAVE: síndrome de Cockayne, retardo de desenvolvimento, herança autossômica recessiva, microcefalia.
Cockayne syndrome: case report
ABSTRACT - We describe a girl with Cockayne syndrome (CS), the diagnostic criteria and the complications of this syndrome. The required criteria for the diagnosis include: prenatal poor growth failure, congenital structural eye anomalies, cataracts, pigmentary retinopathy, severe neurologic dysfunction from birth, sensorineural hearing loss, cutaneous photosensitivity and dental caries. CS is a rare autosomal recessive and biochemical disorder.
KEY WORDS: Cockayne syndrome, developmental delay, autosomal recessive inheritance, microcephaly.
A síndrome de Cockayne é um distúrbio raro, autossômico recessivo, de patogênese desconhecida com prejuízo no crescimento e disfunção progressiva neurológica. Cockayne em 1936 descreveu esta entidade na observação de duas crianças com nanismo, atrofia e surdez1.
Moossy2 fazendo uma revisão desta síndrome salienta que as características mais importantes são: 1) gestação e parto sem particularidades; 2) peso de nascimento normal; 3) dermatite por fotossensibilidade que começa em torno dos 6 meses de idade; 4) retardo de crescimento com nanismo e cifose; 5) alterações neurológicas como microcefalia, retardo mental, retinite pigmentosa, atrofia óptica e sinais de comprometimento do neurônio motor superior e do cerebelo de progressão lenta; 6) aspecto característico do rosto com nariz em bico de "papagaio" e dermatite seborréica na face em forma de asa de borboleta; 7) deterioração progressiva com cegueira, surdez e morte por inanição e complicações infecciosas na adolescência ou na terceira década de vida como máximo; 8) dados genéticos compatíveis com herança autossômica recessiva. Esta entidade tem se associado a diversos transtornos, como hiperbetalipoproteinemia, hiperinsulinemia, nefropatia e níveis baixos de glicose no sangue, anormalidades na regulação da glicose sanguinea, neuropatia periférica com diminuição da velocidade de condução nervosa e desmielinização segmentar3-5.
O propósito de apresentar este relato é chamar a atenção para essa entidade rara, hereditária, que cursa com alterações neurológicas, oculares, dermatológicas e odontológicas, evoluindo com deterioração progressiva e inexoravelmente para morte.
RELATO DE CASO
DS, 3 anos e 10 meses, feminina, branca, natural e procedente de Eldorado do Sul (Figs 1 e 2), que consulta por retardo de desenvolvimento. Nos antecedentes gineco-obstétricos destaca-se que a mãe é Gesta 8 Para 6 Aborto 1 natimorto1, sendo esta a 7ª gestação (1 aborto e 1 natimorto prévios), parto domiciliar e sem atendimento pré-natal, peso ao nascimento 3030g. Na revisão de sistemas foi referida cabeça pequena (não tem certeza se já tinha esta característica ao nascimento), alergia na face quando é exposta ao sol, cáries nos dentes e retardo no crescimento. Firmou a cabeça com 5 meses, sentou com apoio com 8 meses, sentou sem apoio aos 12 meses , engatinhou aos 16 meses, ficou em pé aos 18 meses, primeiras palavras aos 18 meses, dissílabas aos 24 meses. Tem história de hepatite com 1 mês e meio de vida, internada em hospital para avaliação e tratamento (realizou exames: IgM positivo para citomegalovírus e ecografia abdominal mostrando esplenomegalia). Seus são pais consanguíneos, tem 3 irmãos homens (18 anos, 10 anos e 2 anos), 2 irmãs mulheres (17 anos e 16 anos) normais, não havendo história familiar semelhante na família.
No exame físico geral constata-se regular estado geral, mucosas úmidas e coradas, acianótica, anictérica, afebril, eupnéica, sinais vitais estáveis, retardo de crescimento (peso 8250g / estatura 78cm), microcefalia visível, vários dentes sépticos, micrognatia, clinodactilia do 5º dedo da mão esquerda, lesão hiperemiada e crostosa extensa na face.
Exame neurológico - Psiquismo: contactua pobre e inadequadamente com o examinador.Crânio: perímetro cefálico 39cm, distância anteroposterior 22,5cm, distância biauricular 21cm, fontanelas fechadas.Fácies: aspecto de "pássaro".Atitude: deitada em decúbito dorsal com os quatro membros em extensão (membros inferiores com sinal de Little).Emissão de sons: choro/lalação.Movimentação voluntária: escassa.Movimentação involuntária: ausente.Tonos: hipertonia elástica dos quatro membros.Reflexos miotáticos fásicos: hiperreflexia global simétrica.Reflexos superficiais: cutâneoplantar esquerdo indiferente e direito em flexão, cutâneo-abdominais ausentes.Força: tetraparesia grau IV.Sensibilidade: álgica e vibratória presentes.Reflexos arcaicos: ausentes.Nervos cranianos: fotomotor presente, oculocefálico ausente.Fundo de olho com papilas pálidas e retinopatia pigmentar.Trofismo da pele e muscular: hipotrofia muscular generalizada
Desenvolvimento neuropsicomotor - Postura: mantém-se sentada sem apoio durante um período curto; deitada em decúbito ventral, desfaz-se do pano colocado na sua cabeça.Coordenação visomanual: segue com o olhar uma pessoa que se desloca no quarto; segue um objeto num ângulo de 180º; mantém firmemente o chocalho e o sacode com movimentos bruscos.Linguagem: emite algumas vocalizações.Sociabilidade: imobiliza-se ou fica quieta quando se lhe fala, sorri aos rostos familiares.
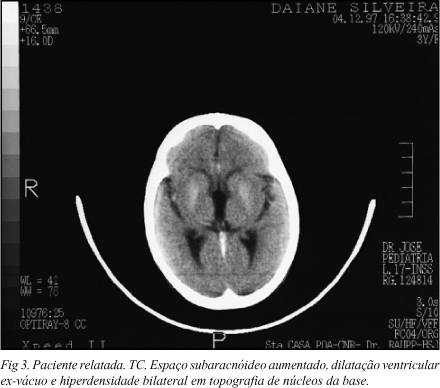
Investigação - STORCH: negativo.Anti-HIV: negativo.Alteração de função hepática com aumento de transaminases (TGO, TGP) e desidrogenase lática (DHL).Marcadores para hepatite negativos (A, B e C). Ecografia abdominal: esplenomegalia.Raio X de crânio: microcefalia.Tomografia computadorizada do crânio (TC): espaço subaracnóideo aumentado, dilatação ventricular ex-vácuo, hiperdensidade bilateral em topografia de núcleos da base (Fig 3).Avaliação oftalmológica: microftalmia, papilas pálidas bilateral em ambos olhos com embainhamento dos vasos, principalmente no olho esquerdo, retina pálida e com padrão salpicado acinzentado (sal e pimenta); exame oftalmológico compatível com síndrome de Cockayne.Avaliação genética: quadro clínico compatível com síndrome de Cockayne.
DISCUSSÃO
Cantani e cols6 fazem revisão em 129 casos relatados na literatura e Nance e Berry7 apresentam 5 pacientes por eles diagnosticados e 135 casos da literatura, definindo as características clínicas, discutem casos atípicos e comentam a importância dos ensaios bioquímicos do metabolismo do ácido nucléico, ajudando na definição deste quadro.
Os Quadros 1,2, 3 e 4 mostram as manifestações neurológicas, oftalmológicas, otorrinolaringológicas e dentárias observadas na SC.
A avaliação do paciente com suspeita de SC inclui cuidadosa história e detalhado exame físico focalizando particularmente as áreas neurológica, oftalmológica, cutânea e odontológica8. Devem ser feitos exames que confirmem ou não a disfunção do sistema nervoso central (substância branca) e periférico: TC e ressonância magnética do encéfalo, potenciais evocados auditivo e visual, pesquisa de erros inatos do metabolismo e líquor.
A análise da sensibilidade dos fibroblastos à luz ultravioleta em combinação com estudos do metabolismo do DNA representa o teste específico no diagnóstico da SC7. Também pode-se realizar diagnóstico pela medida da síntese de RNA após a irradiação com ultravioleta9.
O manejo da SC é puramente sintomático, não existe restrição na dieta, nem tratamento farmacológico. Preconiza-se avaliação periódica, fisioterapia para prevenir contraturas e manter a deambulação, aconselha-se a não exposição solar.
Impõe-se avaliação e aconselhamento genético nas famílias de portadores desta afecção, a fim de prevenir o nascimento de mais acometidos.
Nosso caso apresentava as características clínicas neurológicas, oftalmológicas, dermatológicas e odontológicas da síndrome de Cockayne. Na TC, observou-se atrofia cortical e hiperdensidade dos núcleos da base e o raio X simples de crânio mostrou microcefalia. As provas de função hepática (TGO, TGP e DHL) mostraram alterações. A avaliação laboratorial para hepatite foi negativa observação comentada na literatura7.
Dra. Ana Guardiola - Rua Sarmento Leite 245 - 90050-170 Porto Alegre RS - Brasil.
- 1. Cockayne EA. Dwarfism with retinal atrophy and deafness. Arch Dis Child 1936;11:1-8.
- 2. Moossy J. The neuropathology of Cockayne's syndrome. J Neuropathol Exp Neurol 1967;26:654-660.
- 3. Fujimoto WY, Greene ML, Seegmiller JE. Cockayne's syndrome: report of a case with hyperlipoproteinemia, hyperinsulinemia, renal disease, and abnormal growth hormone. J Pediatr 1969;75:881-884.
- 4. Cotton RB, Keats TE, McCoy EE. Abnormal blood glucose regulation in Cockayne's syndrome. Pediatrics 1970;46:54-60.
- 5. Moosa A, Dubowitz V. Peripheral neuropathy in Cockayne's syndrome. Arch Dis Child 1970;45:674-677.
- 6. Cantani A, Bamonte G, Belioni P, Tucci Bamonte M, Ceccoli D, Tacconi ML. Cockayne syndrome: a review of 129 cases so far reported in the literature. Eur Rev Med Pharm Sci 1987;9:9-17.
- 7. Nance MA, Berry SA. Cockayne syndrome: review of 140 cases. Am J Med Genet 1992;42:68-84.
- 8. Jones KL. Smith´s recognizable patterns of human malformation. 5Ed. Philadelphia: W. B. Saunders 1997:144-147.
- 9. Lehmann AR, Thompson AF, Harcourt SA, Stefanini M, Norris PG. Cockayne's syndrome: correlation of clinical features with cellular sensitivity of RNA synthesis to UV irradiation. J Med Genet 1993;30:679-682.
****
Datas de Publicação
-
Publicação nesta coleção
06 Nov 2000 -
Data do Fascículo
Mar 1999
Histórico
-
Aceito
30 Out 1998