Abstracts
Angelman syndrome (AS) and Prader-Willi syndrome (PWS) are distinct human neurogenetic disorders; however, a clinical overlap between AS and PWS has been identified. We report on a further case of a patient showing the PWS phenotype with the AS molecular defect. Despite the PWS phenotype, the DNA methylation analysis of SNRPN revealed an AS pattern. Cytogenetic and FISH analysis showed normal chromosomes 15 and microsatellite analysis showed heterozygous loci inside and outside the 15q11-13 region. The presence of these atypical cases could be more frequent than previously expected and we reinforce that the DNA methylation analysis is important for the correct diagnosis of severe mental deficiency, congenital hypotonia and obesity.
Angelman syndrome; Prader-Willi syndrome; imprinting defect
A síndrome de Angelman (SA) e a síndrome de Prader-Willi (SPW) são doenças neurogenéticas distintas; entretanto, já foi observada sobreposição clínica entre essas duas patologias. Descrevemos mais um caso de um paciente apresentando o fenótipo da SPW e exames moleculares compatíveis com a SA. Apesar do fenótipo da SPW, o teste da metilação do DNA no gene SNRPN revelou padrão compatível com a SA. A análise citogenética e análise por FISH mostraram ambos os cromossomos 15 normais e a análise de polimorfismo de microssatélite mostrou heterozigozidade para marcadores dentro e fora da região 15q11-13. A presença destes casos atípicos pode ser mais freqüente que o esperado e salientamos que a análise da metilação do DNA é importante para o diagnóstico correto de deficiência mental, hipotonia congênita e obesidade.
síndrome de Angelman; síndrome de Prader-Willi; defeito no centro de imprinting
A FURTHER CASE OF A PRADER-WILLI SYNDROME PHENOTYPE IN A PATIENT WITH ANGELMAN SYNDROME MOLECULAR DEFECT
Greice Andreotti De Molfetta1 1 Genetics Department, School of Medicine from Ribeirão Preto, University of São Paulo (USP), Ribeirão Preto SP, Brazil; 2 Medical Genetics Service, Hospital de Clinicas de Porto Alegre, Porto Alegre RS, Brazil. This study was supported by grants from FAPESP (98/02378-9). , Temis Maria Felix2 1 Genetics Department, School of Medicine from Ribeirão Preto, University of São Paulo (USP), Ribeirão Preto SP, Brazil; 2 Medical Genetics Service, Hospital de Clinicas de Porto Alegre, Porto Alegre RS, Brazil. This study was supported by grants from FAPESP (98/02378-9). , Mariluce Riegel2 1 Genetics Department, School of Medicine from Ribeirão Preto, University of São Paulo (USP), Ribeirão Preto SP, Brazil; 2 Medical Genetics Service, Hospital de Clinicas de Porto Alegre, Porto Alegre RS, Brazil. This study was supported by grants from FAPESP (98/02378-9). ,
Victor Evangelista de Faria Ferraz1 1 Genetics Department, School of Medicine from Ribeirão Preto, University of São Paulo (USP), Ribeirão Preto SP, Brazil; 2 Medical Genetics Service, Hospital de Clinicas de Porto Alegre, Porto Alegre RS, Brazil. This study was supported by grants from FAPESP (98/02378-9). , João Monteiro de Pina Neto1 1 Genetics Department, School of Medicine from Ribeirão Preto, University of São Paulo (USP), Ribeirão Preto SP, Brazil; 2 Medical Genetics Service, Hospital de Clinicas de Porto Alegre, Porto Alegre RS, Brazil. This study was supported by grants from FAPESP (98/02378-9).
ABSTRACT - Angelman syndrome (AS) and Prader-Willi syndrome (PWS) are distinct human neurogenetic disorders; however, a clinical overlap between AS and PWS has been identified. We report on a further case of a patient showing the PWS phenotype with the AS molecular defect. Despite the PWS phenotype, the DNA methylation analysis of SNRPN revealed an AS pattern. Cytogenetic and FISH analysis showed normal chromosomes 15 and microsatellite analysis showed heterozygous loci inside and outside the 15q11-13 region. The presence of these atypical cases could be more frequent than previously expected and we reinforce that the DNA methylation analysis is important for the correct diagnosis of severe mental deficiency, congenital hypotonia and obesity.
KEY WORDS: Angelman syndrome, Prader-Willi syndrome, imprinting defect.
Outro caso de fenótipo da síndrome de Prader-Willi em um paciente com defeito molecular da síndrome de Angelman
RESUMO - A síndrome de Angelman (SA) e a síndrome de Prader-Willi (SPW) são doenças neurogenéticas distintas; entretanto, já foi observada sobreposição clínica entre essas duas patologias. Descrevemos mais um caso de um paciente apresentando o fenótipo da SPW e exames moleculares compatíveis com a SA. Apesar do fenótipo da SPW, o teste da metilação do DNA no gene SNRPN revelou padrão compatível com a SA. A análise citogenética e análise por FISH mostraram ambos os cromossomos 15 normais e a análise de polimorfismo de microssatélite mostrou heterozigozidade para marcadores dentro e fora da região 15q11-13. A presença destes casos atípicos pode ser mais freqüente que o esperado e salientamos que a análise da metilação do DNA é importante para o diagnóstico correto de deficiência mental, hipotonia congênita e obesidade.
PALAVRAS-CHAVE: síndrome de Angelman, síndrome de Prader-Willi, defeito no centro de imprinting.
Angelman syndrome (AS) and Prader-Willi syndrome (PWS) are distinct human neurogenetic disorders involving the imprinting mechanism at the 15q11-13 region. The predominant genetic defects in PWS are 15q11-13 deletions of paternal origin and maternal chromosome 15 uniparental disomy1,2. In contrast, maternal deletions and paternal chromosome 15 uniparental disomy are associated with AS3,4. A small number of patients with PWS and AS were found to have an imprinting defect. Such an imprinting defect can be result of an imprinting center (IC) mutation or occur spontaneously5. Mutations at the UBE3A gene account for approximately 5% of AS6,7. However, 10-15% of the AS patients have an unknown etiology8.
AS is clinically characterized by central congenital hypotonia, severe mental deficiency, microcephaly with occipital flattening, profound speech delay, jerky voluntary movements, a happy disposition with paroxysms of laughter and a characteristic facial appearance which includes a proeminent jaw wide mouth and midfacial hypoplasia9. PWS is clinically characterized by central hypotonia, hyperfagia and obesity starting after the first year of life, delayed neuromotor developmental and later, mental deficiency, hypogenitalism, hypogonadotrofic hypogonadism and some dysmorphisms10.
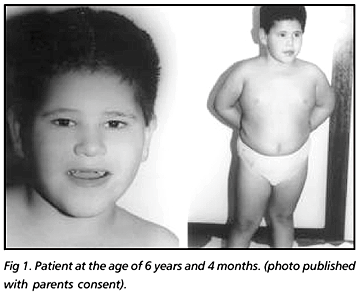
We report on a 6 year and 4 month old boy who was referred to our laboratory in order to investigate a clinical suspicion of PWS and, unexpectedly, the molecular diagnosis based on the analysis of the differential parental specific DNA methylation within the 15q11-13 region was compatible with AS.
CASE
The patient (Fig 1), an adopted child at the first days of life, was born to non-consanguineous and presumed healthy biological parents. Informed consent for publication was obtained from the adopted parents. After a normal pregnancy the patient was delivered by caesarian section at full term, with a birth weight of 2950 g (between 50-75 centile). He presented with neonatal hypotonia but feeding problems were not noted in the newborn period. His developmental progress was delayed. He walked at 2 years and did not develop any speech until 4 years; now he is able to speak some words. He now attends a school for children with severe learning difficulties. At the age of 1 year and 6 months he had one episode of seizures and has been on carbamazepine with good control. He had an abnormal EEG and a normal MRI. After the molecular result a sleep EEG was performed and showed the typical slow wave bursts.
When he was examined at the age of 6 years and 4 months his head circumference was 51.7 cm (50th centile), height 115 cm (between 25-50 centile) and weight 35000 g (above the 97 centile).
The cytogenetic analysis with GTG-banding revealed a normal 46,XY karyotype. Fluorescence in situ hybridisation (FISH) using probes for SNRPN and D15S21 loci (Vysis), which map inside the chromosomal region 15q11-13 and a control probe for the PML locus (Vysis) which maps to 15q22, a commom large deletion of 15q11-13 could be excluded (Fig 2). Methylation analysis at the SNRPN exon 1 carried out by Southern blot analysis revealed an abnormal methylation pattern with the presence of a 0.9kb paternal band and the absence of the 4.2kb maternal band (Fig 2). Microsatellites analysis of the patient showed the presence of two bands using markers for the 15q11-13 region and markers which map outside the 15q11-13 region (Fig 2). DNA of the parents could not be studied because the child is adopted. Quantitative Southern blot analysis for the IC region showed a normal dosage in the patient's DNA and therefore an IC deletion could be excluded (data not shown).
DISCUSSION
To our knowledge, 11 phenotypically atypical AS patients have been reported11-15. Three patients11-13 have a typical AS phenotype with additional PWS features, specially obesit. Cytogenetic analysis using GTG banding technique revelead a deletion in the 15q11-13 region in two of these cases. The third one had a paternal UPD(15) due a 45,XY,t(15q15q). Another atypical AS patient14 showed early onset of obesity, muscular hypotonia and mental deficiency leading to the clinical suspicion of SPW. This patient had a paternal UPD(15) case due a mosaic 47,XX, +mar/48,XX,+2mar. In addition, seven AS patients15 first suspected to have PWS showed early onset of obesity, muscular hypotonia and mental deficiency. All these patients were found to have an imprinting defect with no detectable IC deletion.
Our patient is quite similar to the seven patients reported by Gillessen-Kaesbach et al.15. He lacks the major signs of AS including movement or balance disorder, inappropriate happiness and most of the associated clinical features (Table 1). The main PWS typical features in our patient are neonatal hypotonia and hyperfagia with obesity. Although initially PWS was suspected, a molecular diagnosis of AS was made by methylation analysis, which detects PWS and AS in the same test. Unfortunately we were not able to define the exact genetic cause of AS in this patient because the parental DNA samples were not avaiable. We could only rule out the presence of a commom large deletion of the 15q11-13 as well as an IC deletion, but were not able to determine if the patient has a paternal UPD or an imprinting defect. However, the fact that our patient shares exactly the same developmental and clinical histories showed with the seven imprinting defect AS patients who display a "PWS-like" phenotype15 make us to believe that our patient has an IC molecular defect. Indeed, our patient has a normal karyotype also shown by all the IC molecular defect patients while the AS in patients with a cytogenetical abnormality was caused by a 15q11-13 deletion or by paternal UPD(15). Another factor reinforcing the presence of a imprinting defect in this patient is that he is heterozygous by the microsatellite analysis at the 15q11-13 region and outside the 15q11-13 region. Althought this do not allow us to definitely rule out the possibility of paternal heterodisomy, we should consider that the majority of AS uniparental disomy cases are paternal isodisomy instead of paternal heterodisomy.
We describe an additional case with overlapping features of AS and PWS, suggesting that the presence of these atypical could be more frequent than previously expected. With regard to genetic counselling, clinicians should be aware of the existence of this form of atypical AS. It is important that new cases be identified as it might help to explain how the imprinting mechanism works within the 15q11-13 region. We would like to reinforce that the analysis of the DNA methylation within the 15q11-13 region is an important tool for the correct diagnosis among children who presents with severe mental deficiency, congenital hypotonia and obesity.
Acknowledgements - We wish to thank Dr Gabriele Gillessen-Kaesbach and Dr Karin Buiting from Essen, Germany, for the IC quantitative Southern blot analysis as well as for their helpful suggestions on the manuscript.
Received 9 March 2002, received in final form 17 June 2002. Accepted 25 June 2002.
Dr. Greice Andreotti De Molfetta - Departamento de Genética, Faculdade de Medicina de Ribeirão Preto USP - Avenida Bandeirantes 3900 - 14049-900 Ribeirão Preto SP - Brasil. E-mail: gamolf@rge.fmrp.usp.br
- 1. Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawford JD. Deletions of chromosome 15 as cause of the Prader-Willi syndrome. N Eng J Med 1981;304:325-329.
- 2. Nicholls RD, Knoll JH, Butler MG, Karam S, Lalande M. Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willi syndrome. Nature 1989;342:281-285.
- 3. Knoll JH, Nicholls RD, Magenis RE, Graham JM Jr, Lalande M, Latt SA. Angelman and Prader-Willi syndromes share a common chromosome 15 deletion but differ in parental origin of the deletion. Am J Med Genet 1989;32:285-290.
- 4. Malcolm S, Clayton-Smith J, Nichols M, et al. Uniparental paternal disomy in Angelman's syndrome. Lancet 1991;337:694-697.
- 5. Buiting K, Saitoh S, Gross S, et al. Inherited microdeletions in Angelman and Prader-Willi syndromes define na imprinting center on human chromosome 15. Nat Genet 1995;9:395-400.
- 6. Kishino T, Lalande M, Wagstaff J.UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet 1997;15:70-73.
- 7. Matsuura T, Sutcliffe JS, Fang P, et al.De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat Genet 1997;15:74-77.
- 8. Chan CT, Clayton-Smith J, Cheng X-J, et al. Molecular mechanisms in Angelman syndrome: a survey of 93 patients. J Med Genet 1993;30:895-902.
- 9. Williams CA, Angelman H, Clayton-Smith J, et al. Angelman syndrome: consensus for diagnostic criteria. Am J Med Genet 1995;56:237-238.
- 10. Holm VA, Cassidy SB, Butler MG, et al. Prader-Willi syndrome: consensus for diagnostic criteria. Pediatrics 1993;91:398-402.
- 11. Magenis RE, Toth-Fejel S, Allen LJ, et al. Comparison of the 15q deletions in Prader-Willi and Angelman syndromes: specific regions, extent of deletions, parental origin and clinical consequences. Am J Med Genet 1990;35:333-349.
- 12. Kirkilionis AJ, Chudley AE, Gregory CA, Hamerton JL. Molecular and clinical overlap of Angelman and Prader-Willi syndrome phenotypes. Am J Med Genet 1991;40: 454-459.
- 13. Fridman C, Varela MC, Nicholls RD, Koiffmann CP. Unusual clinical feature in an Angelman syndrome patient with uniparental disomy due to a translocation 15q15q. Clin Genet 1998;54:303-308.
- 14. Dupont J-M, Le Tessier D, Rabineau D, et al. Unexpected Angelman syndrome molecular defect in a girl displaying clinical features of Prader-Willi syndrome. J Med Genet 1999;36:652-654.
- 15. Gillessen-Kaesbach G, Demuth S, Thiele H, et al. A previously unrecognized phenotype characterized by obesity, muscular hypotonia, and ability to speak in patients with Angelman syndrome caused by an imprinting defect. Eur J Hum Genet 1999;7:638-644.
Publication Dates
-
Publication in this collection
14 Jan 2003 -
Date of issue
Dec 2002
History
-
Accepted
25 June 2002 -
Reviewed
17 June 2002 -
Received
09 Mar 2002