Abstracts
With the aim of analyzing which complementary tests are relevant in the diagnostic evaluation of individuals with pervasive developmental disorders, a protocol of clinical and laboratory evaluation was applied in 103 outpatients. The protocol included chromosomal analysis, screening for inborn errors of metabolism, cytogenetic and molecular study of the FRAXA, FRAXE, and FRAXF mutations, EEG, SPECT, and magnetic resonance imaging study. Eighty-four subjects concluded the complementary tests and were classified either as having autism, atypical autism or Asperger syndrome according to the DSM-IV criteria. Sixteen individuals, all bellonging to the two autistic groups, presented genetic or enviromental factors that may have lead to the behavioral disorders, showing the importance of diagnostic evaluation in this group of conditions. Neuroimaging and EEG findings were non-specific and occurred in similar proportion among the groups, being considered of relative low significance in the diagnostic evaluation of individuals with pervasive developmental disorders.
autism; Asperger syndrome; pervasive developmental disorders; fragile X syndrome; SPECT; magnetic resonance imaging
Visando analisar quais exames complementares são relevantes na avaliação diagnóstica de uma amostra de indivíduos com transtornos globais do desenvolvimento, 103 pacientes atendidos em nível ambulatorial foram submetidos a um protocolo composto por avaliação clínica e exames complementares, os quais incluíam cariótipo, estudo molecular da síndrome do cromossomo X frágil, cromatografia de aminoácidos, EEG, SPECT e ressonância magnética. Foram selecionados 84 indivíduos que completaram a investigação laboratorial e apresentavam diagnóstico de autismo, autismo atípico ou síndrome de Asperger, de acordo com os critérios do DSM-IV. Em 16 indivíduos foram identificados distúrbios ambientais ou geneticamente determinados que podem ter causado o quadro comportamental, ressaltando a importância de uma avaliação diagnóstica meticulosa em tais casos. Os achados de neuroimagem e EEG foram inespecíficos e estiveram presentes em proporções semelhantes entre os três grupos, sendo considerados pouco elucidativos na avaliação diagnóstica de indivíduos com transtornos globais do desenvolvimento.
autismo; síndrome de Asperger; transtornos globais do desenvolvimento; síndrome do cromossomo X frágil; SPECT; ressonância magnética
Genetic and neurological evaluation in a sample of individuals with pervasive developmental disorders
Avaliação genética e neurológica em uma amostra de indivíduos com transtornos globais do desenvolvimento
Carlos Eduardo SteinerI; Marilisa Mantovani GuerreiroII; Antonia Paula Marques-de-FariaIII
Departamento de Genética Médica, Faculdade de Ciências Médicas (FAC), Universidade Estadual de Campinas FCM-Unicamp (UNICAMP) Campinas SP, Brasil
IMD, MSc, Departamento de Genética Médica
Departamento de Genética Médica, Faculdade de Ciências Médicas (FAC), Universidade Estadual de Campinas FCM-Unicamp (UNICAMP) Campinas SP, Brasil
IIMD, PhD, Departamento de Neurologia
Departamento de Genética Médica, Faculdade de Ciências Médicas (FAC), Universidade Estadual de Campinas FCM-Unicamp (UNICAMP) Campinas SP, Brasil
IIIMD, PhD, This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP # 97/13525-0).
ABSTRACT
With the aim of analyzing which complementary tests are relevant in the diagnostic evaluation of individuals with pervasive developmental disorders, a protocol of clinical and laboratory evaluation was applied in 103 outpatients. The protocol included chromosomal analysis, screening for inborn errors of metabolism, cytogenetic and molecular study of the FRAXA, FRAXE, and FRAXF mutations, EEG, SPECT, and magnetic resonance imaging study. Eighty-four subjects concluded the complementary tests and were classified either as having autism, atypical autism or Asperger syndrome according to the DSM-IV criteria. Sixteen individuals, all bellonging to the two autistic groups, presented genetic or enviromental factors that may have lead to the behavioral disorders, showing the importance of diagnostic evaluation in this group of conditions. Neuroimaging and EEG findings were non-specific and occurred in similar proportion among the groups, being considered of relative low significance in the diagnostic evaluation of individuals with pervasive developmental disorders.
Key words: autism, Asperger syndrome, pervasive developmental disorders, fragile X syndrome, SPECT, magnetic resonance imaging.
RESUMO
Visando analisar quais exames complementares são relevantes na avaliação diagnóstica de uma amostra de indivíduos com transtornos globais do desenvolvimento, 103 pacientes atendidos em nível ambulatorial foram submetidos a um protocolo composto por avaliação clínica e exames complementares, os quais incluíam cariótipo, estudo molecular da síndrome do cromossomo X frágil, cromatografia de aminoácidos, EEG, SPECT e ressonância magnética. Foram selecionados 84 indivíduos que completaram a investigação laboratorial e apresentavam diagnóstico de autismo, autismo atípico ou síndrome de Asperger, de acordo com os critérios do DSM-IV. Em 16 indivíduos foram identificados distúrbios ambientais ou geneticamente determinados que podem ter causado o quadro comportamental, ressaltando a importância de uma avaliação diagnóstica meticulosa em tais casos. Os achados de neuroimagem e EEG foram inespecíficos e estiveram presentes em proporções semelhantes entre os três grupos, sendo considerados pouco elucidativos na avaliação diagnóstica de indivíduos com transtornos globais do desenvolvimento.
Palavras-chave: autismo, síndrome de Asperger, transtornos globais do desenvolvimento, síndrome do cromossomo X frágil, SPECT, ressonância magnética.
Pervasive developmental disorders (PDDs) are a heterogeneous group of neurobehavioral disorders of childhood, comprising autism (the most common form of PDD), atypical autism, Asperger syndrome, Rett syndrome, and PDD not otherwise specified. All these entities are clinically characterized by abnormalities in three main areas: social interaction, language and communication, and interests and activities1. The clinical picture is quite variable among affected individuals2. Up to 75% present mental retardation, and epilepsy is manifested in more than 30%, suggesting the occurence of extensive brain damages by the action of neurobiological factors3, 4. Initially thought to have psychogenic origin, PDDs are now considered neuropsychiatrical conditions determined by still less understood factors acting in a multifactorial model. The more recent advances in this field have been generated by neuroanatomical and neurophysiological researchs on the central nervous system, as well as molecular studies with the aim of determining gene(s) involved in their etiology. In at least 10% of the cases a specific etiology can be identified such as environmental factors, chromosomal abnormalities or single gene disorders4-6, with special attention to the association of fragile X syndrome and autism.
In the absence of specific laboratory findings, several diagnostic scales and manuals were created on the basis of the clinical aspects, in order to define the diagnosis of PDDs. The most useful are the criteria listed in the Diagnostic and Statistical Manual of Mental Disorders (DSM) from the American Psychiatric Association1. Considering the heterogeneity of the PDDs and the high prevalence of associated conditions, individuals with a preliminary diagnosis of autism or related disorders should be evaluated in a systematic and interdisciplinary way, including a detailed clinical evaluation and complementary tests.
The aim of this study was to identify and analyse genetic and neurological aspects in a sample of individuals presenting PDDs by using a protocol of clinical and laboratory assessment7,8 and define which ones are relevant in the diagnostic evaluation of these conditions.
METHOD
A total of 103 individuals from the ambulatories of Genetics and Neurology were referred with a preliminary diagnosis of autism. Parents or legal guardians were invited to join the study by signing an informed consent term approved by our institutional Ethics Committee.
A comprehensive protocol comprising clinical interview, physical and neurologic examination with attention to dysmorphologic evaluation was applied. Functional diagnosis was based on the DSM-IV criteria for the PDDs1. Etiological diagnosis comprised cytogenetic analysis including techniques for fragile X chromosome9. Molecular studies of the FRAXA mutation using the PCR technique10 and the Southern blotting technique11 were also performed, as well as analysis for the FRAXE and FRAXF mutations following proper protocols12,13. Other complementary tests were screening for inborn errors of metabolism and aminoacid chromatography on urine and plasma samples14.
Neurological assesment included EEG, SPECT, and magnetic resonance imaging. EEG followed the international 10-20 system15 in a 16 channel Berger (BergerÒ, Brazil) equipment. SPECT images were acquired under general anaesthesia in a gamma-chamber capting endovenous Tc99m-HMPAO contrast16. Magnetic resonance imaging (MRI) was also obtained under general anaesthesia in a 2.0 Tesla Elscint PrestigeÒ (Haifa, Israel) equipment17.
The chi-square test of significance was used to give a measure of the significance (p < 0.05) of an observed deviation from the expected value. Comparasion was made among the groups in the sample and with a control group that included individuals presenting mental retardation without Down syndrome18, besides data from the literature.
RESULTS
The 103 outpatients initially evaluated comprised 84 males and 19 females (sex ratio 4.4:1) with ages ranging from 2.6 to 28.6 years (mean 9.9 years). Functional diagnosis based on DSM-IV criteria revealed that 68 individuals presented autism while 16 had atypical autism and 11 received the diagnosis of Asperger syndrome. Eight patients were excluded due to other neurobehavior diagnosis than autism: 2 manifested Rett syndrome, 3 actually had infantile psychosis, and 3 presented mental retardation but no autism. Besides these eight patients and despite the diagnosis of one of the PDDs, other 11 subjects did not conclude their clinical and laboratory evaluation and were also excluded. The remaining 84 individuals that composed the final sample were classified in three groups: autism, atypical autism, and Asperger syndrome. Characteristics of each group are shown on Table 1.
Following the protocol, a detailed clinical history revealed that 3 individuals presented significant environmental factors that may have contributed to the autistic features: one girl had sequelae from prematurity associated to severe neonatal hypoxia, other girl had a post-vaccinal (MMR) encephalitis at age 9 months, and a boy had neonatal meningitis . Clinical evaluation identified three patients with Down syndrome, as well as six individuals with other dysmorphic genetic conditions.
Concerning the complementary tests, structural chromosomal abnormalities were found in three individuals besides the three patients with Down syndrome. Other cytogenetic finding was the presence of the fragile site at Xq27.3 in four patients, three of them showing a very low positivity rate (1 to 4%) and another with a consistent positivity (17%). Molecular analysis revealed that the later individual had a mosaic expansion of CGG from 100 to 900 repetitions in the FMR1 gene while the other three patients were negative for the FRAXA, FRAXE, and FRAXF mutations. In two subjects the biochemical tests detected the presence of hyperphenylalaninemia and phenylketonuria leading to the diagnosis of untreated phenylketonuria.
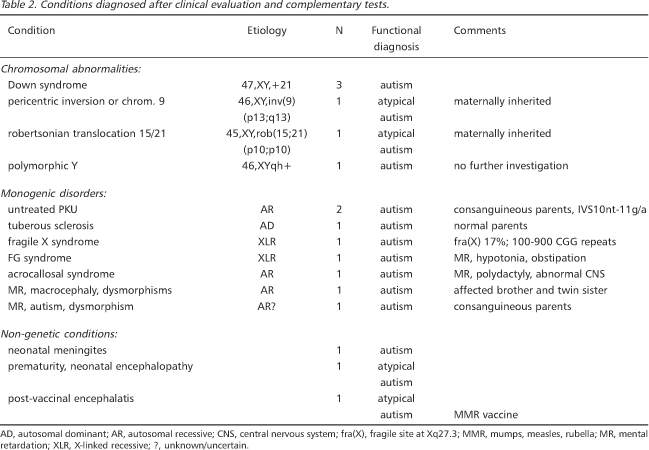
Details of the clinical and complementary tests are shown on Table 2.
Due to technical problems, some individuals did not conclude the neurological complementary evaluation.
A total of 70 EEGs were performed with 49 (71%) normal results and 21 (29%) abnormal results. The autistic group presented two cases of generalized epileptiform activity, three of unspecified generalized desorganization, and two of unspecified hemispheric desorganization (one in the right and the other in the left hemisphere). Unspecified focal desorganization was detected in six individuals presenting autism, four presenting atypical autism, and five with Asperger syndrome.
Fifty-eight subjects underwent SPECT imaging resulting in 26 (45%) normal, one (2%) inconclusive, and 31 (53%) abnormal results with similar proportion among the groups. Nine individuals with autism and one with Asperger syndrome presented hypoperfusion of the frontal lobes, while 10 with autism, two with atypical autism and one with Asperger syndrome showed hypoperfusion of the cerebelum. Hypoperfusion of the basal ganglia was seen in two patients with autism, one with atypical autism, and one with Asperger syndrome. Abnormal unilateral or bilateral temporal lobe perfusion occured in nine subjects with autism, six with atypical autism, and four with Asperger syndrome. One individual with Asperger syndrome had abnormal occipital lobe perfusion. Abnormal left hemisphere perfusion, abnormal cingulate girus, and the inconclusive test were found in one individual each of the autistic group.
Finally, 63 patients concluded the neuroanatomical evaluation which revealed 44 (70%) normal exams and 19 (30%) abnormal results, two of them showing specific abnormalites related to the syndromic diagnosis (acrocallosal syndrome and tuberous sclerosis, one case each), and the remaining revealing variable findings. Thinning or partial agenesis of the corpus callosum was seen in two individuals with autism. Hypoplasia of the cerebelar vermis was present in one subject with autism and other with atypical autism. One individual with atypical autism showed ectopic cerebelar tonsilae and reduction of the superior right temporal lobe with enlarged silvyan fissure. Perivascular enlargment was detected in one individual with autism and in other with Asperger syndrome. Gliosis was found in three individuals, one of each group. Non-hypertensive enlargment or assimetry of the lateral ventricules was seen in four patients with autism, two with atypical autism, and one with Asperger syndrome.
DISCUSSION
Several conditions with genetic etiologies have been described in association with autism19. Some deserve mention due to a consistent association, such as tuberous sclerosis or fragile X syndrome, and just a few seem to be strictly associated with autistic features, like the entity described by Orstavick et al.20 as a dysmorphic condition comprising macrocephaly, epilepsy, mental retardation, and autism. However, most of these descriptions represent anecdotal reports indicating a probable casual relationship, like the association of acrocallosal syndrome with autism which is, to our knowledge, the first report in the literature. Cytogenetic analysis showed that three individuals with Down syndrome had trisomy21. Autism is frequently seen in persons with Down syndrome in spite of a pleasant and sociable personality usually described in this condition21. Other 3 subjects presented structural abnormalities including one case each of Yqh+, pericentric inversion of chromosome 9, and Robertsonian translocation 15/21, the two latter being maternally inherited. Previously described in autism22, Yqh+ is considered a polymorphism without clinical significance and does not justify the neurological picture seen in this patient. On the other hand, pericentric inversion of chromosome 9 is common in the general population and usually causes no abnormal phenotype, although recent data suggest that it may be involved in genetic susceptibility to psychiatric disorders such as schizophrenia or even autism23. The roberstonian translocation 15/21 deserves mention because abnormalities of chromosome 15 have a higher prevalence in autism than in the general population24 and frequently have maternal origin25.
Abnormalities in the FMR1 gene cause the fragile X syndrome, an X-linked condition with a wide range of deviant behaviours, from mental impairment in variables degrees to hyperactivity and autistic features. Studies in the 80's suggested that fragile X syndrome was present in a great number of autistic subjects, and it was even proposed that it would be the cause of autism. However, recent data showed that it can be identified in less than 3% of autistic individuals, a value similar to its prevalence in samples based in other developmental disorders like mental retardation, learning disabilities or speech delay26.
Phenylketonuria is a well known autosomal recessive condition that has also been described in association with autism4, although its frequency is decreasing in industrialised countries after the introduction of neonatal screening and early dietary treatment. Molecular analysis showed that both patients present the same mutation (IVS10nt-11g/a) in the phenylalanine hydroxilase gene, a genotype that leads to severe biochemical defect and consequent worse clinical picture27.
Concerning the neurological tests, abnormalities in the EEG were found in 30% of the total sample, similar to previous reports in the literature28. A slight predominance of electroencephalografic abnormalities in the temporal regions was detected but statistical analysis revealed no difference in relation to other cerebral regions or among the three groups.
Neuroimaging (MRI) abnormalities occurred in 30% of the three groups, but no specific structural abnormality was found. Cerebellar hypoplasia has been described as a possible neuroimaging marker for autism29, however in the present sample it occurred in equal proportions among the autistic and the Asperger groups, and was present either in idiopathic autism as in autism due to a specific condition. Statistical analysis revelead a higher incidence of abnormalities in the group formed by autistic individuals with a specific syndromic diagnosis (P2(4) = 15.67; p = 0.003), which is probably correlated with the associated diagnosis but not with autism itself.
Although SPECT abnormalities were detected in more than 50% of the individuals in the present sample, showing the higher sensibility among the three tests, it revealed no specificity in autism30. In spite of cerebellum and frontal lobe(s) seemed to be more affected, no statistical difference was observed in relation to the other areas.
Finally, there was no correlation in anatomical areas or clinical severity when results from the three neurological tests were compared.
CONCLUSION
The association of autism with an environmental or genetic condition must be considered in any individual presenting abnormalities in the socialization, communication, and/or activities, despite the level of intellectual functioning. Clinical evaluation of such cases should comprise, besides careful information about behavior patterns, a dysmorphologic examination to search for suggestive signs of neurogenetic disorders. Concerning the laboratory tests, karyotype and molecular tests for the fragile X syndrome should be included in the investigation of autism. If molecular tests are not available, cytogenetic analysis in folic acid deficient medium is an alternative. On the other hand, EEG or neuroimaging exams may bring few benefits for the patient or his family and should be performed depending of individual indication, but not as a routine. We believe that the identification of typical and "pure" cases of autism will be usefull for neuroimaging researchs involving new technologies that may be available in a near future.
Acknowledgements - The authors wish to thank all the patients and their families who participated in this research, as well as the following institutions: Associação para o Desenvolvimento do Autista de Campinas (ADACAMP), Centro de Estudos e Desenvolvimento do Autismo e Patologias Associadas de Pirassununga (CEDAP), Associação dos Pais e Amigos dos Autistas de Itu (AMAI), and many Associações de Pais e Amigos dos Excepcionais (APAEs). A special thank to the colleagues and employees from Hospital de Clínicas and Faculdade de Ciências Médicas, UNICAMP. The authors are also grateful to Dr. Angelina Xavier Acosta for the molecular studies in the phenylketonuric patients.
Received 12 June 2002, received in final form 4 October 2002
Accepted 26 October 2002
Dra. Antonia Paula Marques-de-Faria - Departamento de Genética Médica, FCM, UNICAMP - Caixa Postal 6111- 13081-970 Campinas SP - Brasil - FAX: 55 19 3788 8909. E-mail: dgmfcm@unicamp.br
-
1American Psychiatric Association (APA). American Psychiatric Press, Diagnostic and statistical manual of mental disorders, 4.Ed. Washington: 1994.
- 2. Rapin I. Autistic children: diagnosis and clinical features. Pediatrics 1991;87(Suppl):751-760.
- 3. Sigman M, Arbelle S, Dissanayake C. Current research findings on childhood autism. Can J Psychiatry 1995;40:289-294.
- 4. Gillberg C. The neurobiology of infantile autism. J Child Psychol Psychiatry 1988;29:257-266.
- 5. Fommbonne E, du Mazaubrun C, Cans C, Grandjean H. Autism and associated medical disorders in a French epidemiological survey. J Am Acad Child Adolesc Psychiatry 1997;36:1561-1569.
- 6. Skjedal OH, Sponheim E, Ganes T, Jellum E, Bakke S. Childhood autism: the need for physical investigations. Brain Dev 1998;20:227-233.
- 7. Steiner CE. Aspectos genéticos e neurológicos do autismo; proposta de abordagem interdisciplinar na avaliação diagnóstica do autismo e distúrbios correlatos. Campinas, 1998. Dissertação de mestrado; Universidade Estadual de Campinas. Campinas, Brasil 1998.
- 8. Steiner CE. Genetic and neurologic aspects of autism; proposal of an interdisciplinary approach in the diagnostic evaluation of autism and related disorders. Genet Mol Biol 1999;22:457.
- 9. Jacky PB, Ahuja YR, Anyane-Yeboa K, et al. Guidelines for the preparation and analysis of the fragile X chromosome in lymphocytes. Am J Med Genet 1991;38:400-403.
- 10. Fu YK, Kuhl DPA, Pizzuti A, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell 1991;67:1047-1058.
- 11. Yu S, Mulley J, Loesch D, et al. Fragile-X syndrome: unique genetics of the heritable unstable element. Am J Hum Genet 1992;50:968-980.
- 12. Knight SJL, Flannery AV, Hirst MC, et al. Trinucleotide repeat amplifications and hypermethylation on a CpG island in FRAXE mental retardation. Cell 1993;74:127-134.
- 13. Ritchie RJ, Knight SJL, Hirst MC, et al. The cloning of FRAXF: trinucleotide repeat expansion and methylation at a third fragile site in distal Xqter. Hum Mol Genet 1994;3:2115-2121.
- 14. Davidsohn I, Henry JB. Clinical diagnosis by laboratory methods. Philadelphia: Saunders 1974.
- 15. American Electroencephalographic Society. Guideline seven: a proposal for standard montages to be used in clinical EEG. J Clin Neurophysiol 1986;3:158-165.
- 16. Devous MD. Imaging brain function by single-photon emission computer tomography. In Andreasen NC (ed). Brain imaging: application in Psychiatry. Washington: American Psychiatry Press, 1989.
- 17. Montenegro MA, Guerreiro MM, Lopes-Cendes I, Cendes F. Bilateral posterior parietal polymicrogyria: a mild form of congenital bilateral perisylvian syndrome? Epilepsia 2001;42:845-849.
- 18. Marques-de-Faria AP. Estudo genético-clínico de deficientes mentais sem síndrome de Down. Campinas, 1994. Universidade Estadual de Campinas. Tese de doutorado
- 19. Gillberg C. Autism and related behaviours. J Intel Disabil Res 1993;37:343-372.
- 20. Orstavick KH, Stromme P, Torvik A, Skjeldal OH. Macrocephaly, epilepsy, autism, dysmorphic features, and mental retardation in two sisters: a new autosomal recessive syndrome? J Med Genet 1997;34:849-851.
- 21. Howlin P, Wing L, Gould J. The recognition of autism in children with Down syndrome - implications for intervention and some speculations about pathology. Dev Med Child Neurol 1995;37:398-414.
- 22. Gillberg C, Wahlström J. Chromosome abnormalities in infantile autism and other childhood psychosis: a population study of 66 cases. Dev Med Child Neurol 1985;27:293-304.
- 23. Kunugi H, Lee KB, Nanko S. Cytogenetic findings in 250 schizophrenics: evidence confirming an excess of the X chromosome aneuploidies and pericentric inversion of chromosome 9. Schizophr Res 1999;40:43-47.
- 24. Bundey S, Hardy C, Vickers S, Kilpatrick MW, Corbett JA. Duplication of the 15q11-13 region in a patient with autism, epilepsy and ataxia. Dev Med Child Neurol 1994;36:736-742.
- 25. Schroer RJ, Phelan MC, Michaelis RM, et al. Autism and maternally derived aberrations of chromosome 15q. Am J Med Genet 1998;76:327-336.
- 26. Patsalis PC, Sismani C, Hettinger JA, et al. Molecular screening of fragile X (FRAXA) and FRAXE mental retardation syndromes in the Hellenic population of Greece and Cyprus: incidence, genetic variation, and stability. Am J Med Genet 1999;84:184-190.
- 27. Dworniczak B, Aulehla-Scholz C, Kalaydjieva L, et al. Aberrant splicing of phenylalanine hydroxilase mRNA: the major cause of phenylketonuria in parts of Southern Europe. Genomics 1991;11:242-246.
- 28. Minshew NJ. Indices of neuronal function in autism: clinical and biologic implications. Pediatrics 1991;87(Suppl):774-780.
- 29. Courchesne E. New evidence of cerebellar and brainstem hypoplasia in autistic infants, children and adolescent: the MR imaging study by Hashimoto and colleagues. J Autism Dev Disord 1995;25:19-22.
- 30. O'Tuama LA, Treves ST. Brain single-photon emission computed tomography for behavior disorders in children. Sem Nuclear Med 1993;23:255-264.
Publication Dates
-
Publication in this collection
22 Sept 2008 -
Date of issue
June 2003
History
-
Accepted
26 Oct 2002 -
Received
12 June 2002 -
Reviewed
04 Oct 2002