Abstracts
Cerebrotendinous xanthomatosis is a treatable rare autossomal recessive disease characterized by lipid storage secondary to a sterol 27-hydroxylase deficiency in the formation of cholic and chenodeoxycholic acids. We describe two Brazilian brothers with cognitive impairement and chronic diarrhea. One of them also presents bilateral cataracts. Neurological findings were progressive walking deficit, limb ataxia and pyramidal signs. Both patients had bilateral Achilles tendon xanthomata. Magnetic resonance image showed signal alterations in cerebellar hemispheres. We describe these cases with molecular genetic analysis confirming diagnosis and comparing with previous literature. The CYP27A1 gene study showed a C1187T mutation on exon 6.
cerebrotendinous xanthomatosis; sterol 27-hydroxylase; CYP27A1
Xantomatose cerebrotendínea é doença autossômica recessiva tratável causada pelo acúmulo de lipídeos por deficiência da enzima 27-esterol hidroxilase na produção de ácido cólico e deoxicólico. Descrevemos dois irmãos brasileiros com dificuldade cognitiva e diarréia crônica. Um deles apresentava catarata bilateral. Os achados neurológicos foram dificuldade progressiva para deambular, ataxia de membros e sinais piramidais. Ambos tinham xantomas de tendão aquileu bilateralmente. O exame de ressonância magnética revelou áreas de sinal hiperintenso em ambos os hemisférios cerebelares. Descrevemos os casos com diagnóstico genético comparando-os com a literatura. O estudo do gene CYP27A1 demonstrou a mutação C1183T no exon 6.
xantomatose cerebrotendínea; 27-esterol hidroxilase; CYP27A1
Cerebrotendinous xanthomatosis: report of two Brazilian brothers
Xantomatose cerebrotendínea: relato de dois irmãos brasileiros
Marcos Christiano LangeI; Viviane Flumignan ZétolaII; Helio A.G. TeiveIII; Rosana H. ScolaIV; Ana Paula TrentinII; Jorge A. ZavalaI; Eduardo R. PereiraI; Salmo RaskinVI; Lineu C. WerneckV; Erik A. SistermansVII
Serviço de Neurologia do Hospital de Clínicas da Universidade Federal do Paraná, Curitiba, PR, Brazil (UFPR)
IResidente em Neurologia
IIMédico Neurologista
IIIProfessor Assistente
IVProfessor Adjunto
VProfessor Titular
VIMédico Geneticista, Laboratório Genetika, Curitiba - PR Brazil
VIIKlinisch-Genetisch Centrum Nijmegen, Nijmegen - Netherlands
Correspondence Correspondence to Dra. Viviane F. Zétola Serviço de Neurologia, Hospital de Clínicas Rua General Carneiro 181 / sala 1236 80060-900 Curitiba PR - Brasil E-mail: viviane@flumignano.com
ABSTRACT
Cerebrotendinous xanthomatosis is a treatable rare autossomal recessive disease characterized by lipid storage secondary to a sterol 27-hydroxylase deficiency in the formation of cholic and chenodeoxycholic acids. We describe two Brazilian brothers with cognitive impairement and chronic diarrhea. One of them also presents bilateral cataracts. Neurological findings were progressive walking deficit, limb ataxia and pyramidal signs. Both patients had bilateral Achilles tendon xanthomata. Magnetic resonance image showed signal alterations in cerebellar hemispheres. We describe these cases with molecular genetic analysis confirming diagnosis and comparing with previous literature. The CYP27A1 gene study showed a C1187T mutation on exon 6.
Key words: cerebrotendinous xanthomatosis, sterol 27-hydroxylase, CYP27A1.
RESUMO
Xantomatose cerebrotendínea é doença autossômica recessiva tratável causada pelo acúmulo de lipídeos por deficiência da enzima 27-esterol hidroxilase na produção de ácido cólico e deoxicólico. Descrevemos dois irmãos brasileiros com dificuldade cognitiva e diarréia crônica. Um deles apresentava catarata bilateral. Os achados neurológicos foram dificuldade progressiva para deambular, ataxia de membros e sinais piramidais. Ambos tinham xantomas de tendão aquileu bilateralmente. O exame de ressonância magnética revelou áreas de sinal hiperintenso em ambos os hemisférios cerebelares. Descrevemos os casos com diagnóstico genético comparando-os com a literatura. O estudo do gene CYP27A1 demonstrou a mutação C1183T no exon 6.
Palavras-chave: xantomatose cerebrotendínea, 27-esterol hidroxilase, CYP27A1.
Cerebrotendinous xanthomatosis (CTX) is a rare autosomal recessive disease characterized by lipid accumulation in several tissues, mainly in the eye lenses, central nervous system (CNS) and muscle tendons. Mutations in the gene that codes for the hepatic enzyme sterol 27-hydroxylase (CYP27A1) have been found in these patients. We present the first description of a Brazilian family with suggestive clinical presentation and diagnosis confirmation through genetic test of CYP27A1 gene.
CASE
Case 1 A 47 years old white man which symptoms began at 20 years old (according to the mother's information). He presented progressive walking difficulty, becoming unable to walk fast at 40 years old. Since the beginning he presented cognitive difficulties and bilateral swelling of Achilles tendons. He presented delay of the psychomotor development and started walking by one year and a half and he acquired the capacity to run and to go up stairways just at five years of age. The mother referred that he had a history of chronic diarrhea during the childhood. She denied other family members with similar symptoms, except for the patient's younger brother.
Physical examination disclosed no abnormalities except for bilateral swelling of Achilles tendons (Fig 1a). In the neurological examination, he presented mild mental retardation, incomprehensive disarthria, mild bilateral facial palsy without other findings in cranial nerves. His muscular strength was IV - in superior limbs (SL) and IV- in inferior limbs (IL) according to Medical Research Council (MRC) rating. We noted moderate spasticity in IL, preserved trophism and increased deep tendon reflexes with bilateral Babinski sign. The sensibility was preserved and there was retraction in bilateral Aquilles tendon. The patient presented symmetrical dismetry and disdiadococinesy in SL. The cerebellar clinical evaluation of IL was disabled by the limitation of the movements. He had also gait ataxia and walked with support.
Laboratorial exams (blood count, biochemistry, hepatic and renal function, lipid analysis and coagulation tests) were normal. The biochemical analysis for serum cholestanol level, urinary bile alcohol excretion and chenodeoxycholic acid in bile were not done. Cerebrospinal fluid (CSF) was normal. Electrocardiogram (ECG) presented overload of left ventricle. Electroencephalogram (EEG) showed mild diffuse cerebral suffering, without irritation activity. Electrophisiologic studies showed sensorimotor demyelinating polineuropathy with mild axonal characteristics (Table 1). Ophthalmologic exam evidenced atrophy of retinal pigmented epithelium and peripapillar atrophy. Magnetic resonance imaging (MRI) showed hyperintense signal in the postero-inferior segments of both cerebellar hemispheres on T2 WI and 'FLAIR' sequences (Fig 2a). CYP27A1 gene was sequenced according to standard and adequate techniques1. It showed homozygous mutation in exon 6 (C1183T) of the CYP27A1 gene.
Case 2 A, 40 years old white man, case one's brother, which according to the mother's information, presented learning difficulty since childhood, unable to read or to write. He did not finish the basic school. At 35 years of age he began with progressive difficulty to walk and two years after he began to present bilateral swellings of Achilles tendon. He also presented chronic diarrhea and was submitted to a bilateral cataracts surgery in the childhood.
Physical examination disclosed no abnormalities except for bilateral swellings of Achilles tendons (Fig 1b). In the neurological exam he presented mild mental retardation, moderate disarthria, visual handicap and facial palsy without other abnormalities in cranial nerves. The muscle strength was V in SL and IV+ in IL (MRC rating). Other findings were spasticity in IL with preserved trophism, increased deep tendon reflexes with bilateral Babinski and Hoffman signs. Sensibility was preserved. He had walking disturbance with ataxia and spasticity in IL.
Laboratorial exams (blood count, biochemistry, lipid analysis, hepatic and renal function) were normal. The biochemical analysis for serum cholestanol level, urinary bile alcohol excretion and chenodeoxycholic acid in bile were not done. CSF presented mild elevation of proteins (75mg/dl, reference: 35 to 45 mg/dl) with other values in normal limits. ECG presented overload of left cameras. EEG presented with mild diffuse cerebral suffering, without irritation activity. Electrophisiologic studies showed sensorimotor axonal polineuropathy with mild demyelination (Table 1). He was submitted to a muscle biopsy with normal fresh frozen section (HE-trichrome, oil red O, PAS, cresyl violet and siryus red) and histochemical reactions (ATPase, NADH, esterase, myophosphorylase, acid phosphatase, alkaline phosphatase, succinate dehydrogenase, cytochrome-C oxidase and adenylate deaminase). Ophthalmologic exam evidenced incipient cataracts without surgical indication. MRI with hyperintense signal in inferior segments of both cerebellar hemispheres on T2WI and 'FLAIR' (Fig 2b). Genetic analysis showed homozygous mutation in exon 6 (C1183T) of the CYP27A1 gene.
Mother's blood was sequenced and she was a carrier of the same exon 6 CYP27A1 gene mutation.
The DNA extraction was made in Curitiba by one author (SR) and the CYP27A1 analysis was made in Nijmegen, The Netherlands (EAS). In both cases we did not start any treatment due to acquisition difficulty of medication and loss of follow-up.
DISCUSSION
CTX was first described in two cousins, with the presence of cerebellar and pyramidal signs, palate myoclonus, cognitive disability, cataracts and tendon xanthomas, apud Bertini2. The main clinical characteristics are cataracts and premature atherosclerosis, tendon xanthomas with preferential location for the Achilles tendon and neurological manifestations as pyramidal signs, cerebellar ataxia, seizures, cognitive disability, dementia and peripheral neuropathy1,3,4. The findings observed in our patients were cataracts, tendon xanthomas and neurological manifestations, such as cerebellar ataxia, pyramidal signs, cognitive disability and peripheral neuropathy suggesting CTX diagnosis. The combination of chronic diarrhea and bilateral cataracts in the childhood, described in the case 2, was pathognomonic for the disease5. Besides cataracts, other retinal abnormalities have been described in these patients as paleness of the optical disk and senile retinal degeneration, as we could see in the case 16.
Behavioral disturbance as depressed or distimic humor, irritability, appetite reduction, insomnia and fatigue can be present1,3,4. Heart complications, as lipomatosis hipertrophy with atrial sept thickness and lung alterations and osteoporosis were described, none was found in our patients7-9.
Other manifestations have been described, such as chronic myelopathy and spastic paraplegy in association with frontal lobe dementia. The differential diagnosis must be done in patients with early-onset parkinsonism-plus, that usually is associated with walking disturbances, pyramidal signs, cognitive abnormalities and partial L-dopa response10-12.
The diagnosis can be confirmed by biochemical exams, which demonstrate increase of serum cholestanol level and urinary bile alcohol excretion associated with reduction of the chenodeoxycholic acid in bile13. Because technical difficulties these tests were not done in our patients.
Molecular genetic analysis, accomplished in our patients, is obtained by the screening of CYP27A1 gene located in chromosome 2q 23. The genomic structure of CYP27A1 contains nine exons and eight introns with 18,6 kb of DNA. The mature enzyme consists of 498 amino acids and it is expressed in CNS, liver, lungs, duodenum and endothelial cells. Until now, more than 45 mutations were described in the CYP27A1 gene, usually affecting heme or adrenodoxin related domains between exons 6 and 91,13-15. The patients described here presented a homozygous CYP27A1 gene mutation in exon 6 (1183 C>T) that was already described in other populations1,16.
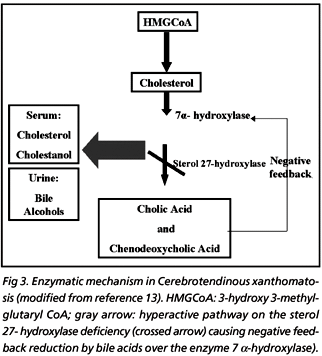
CYP27A1 enzyme (sterol 27-hydroxylase) with two protein cofactors, adrenodoxin and adrenodoxin reductase, hydroxylates a variety of sterols at the C-27 position producing the primary bile acids (cholic and chenodeoxycholic acid)14. In normal individuals, the primary bile acid inhibits the rate-limiting step in their production, the enzyme 7 m-hydroxylase (negative feedback mechanism). The gene mutation causes deficiency of the enzyme sterol 27-hydroxylase, leading to reduced synthesis of cholic acid and almost no chenodeoxycholic acid. Due to the absence of the negative feedback mechanism, the 7 ?-hydroxylase activity is increased, producing excessive cholesterol and cholestanol and accumulating them in many tissues with increased urine excretion of the bile alcohols2,17. The metabolic pathway can be seen in the Figure 3.
The electrophisiologic findings from CTX patients usually show an axonal degeneration, but they commonly are described as a mixed neuropathy, as observed in our cases1. Muscle biopsy is uncharacteristic4.Xanthomatous lesions, lipid crystal clefts and macrophage clusters that are usually seen in the CNS, were not found in the muscle biopsies4. MRI usually demonstrate hyperintense signal in T2 and FLAIR located preferentially in periventricular area, basal ganglia and dentate nuclei of the cerebellum, besides variable degrees of cerebral and cerebellar atrophy (Fig 2A)18-21. Brain spectroscopy demonstrates significant decreases of NAA (N-acetylaspartate) and increases of lactate MRI signals. These results suggests a brain accumulation of metabolic neurotoxins and deficiency of the mitochondria metabolism secondary to the high levels of cholestanol and bile alcohols21,22.
The mechanism for neurological manifestations is not known. A hypothesis suggests that this happens from an increase neuronal apoptosis caused by the cholestanol overload. The presence of apolipoprotein B in CSF indicates penetration of low-density lipoprotein particles from plasma through the blood-brain barrier. These lipoprotein particles may carry cholestanol as well as cholesterol13. Chenodeoxycholic acid replacement therapy is usually associated with improvement in the clinical symptoms, normalization cholesterol synthesis and reestablishment of selective permeability of blood-brain barrier with normalized CSF apolipoprotein and sterol concentrations23. The absence of neurological dysfunction in other lipid disorders (familial hypercholesterolemia or sitosterolemia) further support the hypothesis that cholestanol itself impairs brain function13.
It is important to remember that precocious diagnosis is fundamental due to the good response with chenodeoxycholic acid treatment reducing cholestanol accumulation and promoting lesion regression. The effectiveness of statins is controversial with the possibility of worsening the condition owning to increased low-density lipoprotein uptake as the result of augmented low-density lipoprotein receptor activity13. The detection of the specific mutation in each family is important to definitely confirm the diagnosis, to start treatment as earlier as possible, and to provide accurate genetic counseling to the patients and their family.
Acknowledgements
The authors wish to thanks Dr. Ana C. Bacelar Limeira from CETAC - Curitiba for neuroimage evaluation.
Received 18 February 2004, received in final form 22 June 2004. Accepted 3 August 2004.
- 1. Verrips A, Hoesfloot LH, Steenbergen GCH, et al. Clinical and molecular genetic characteristics of patients with cerebrotendinous xanthomatosis. Brain 2000;123:908-919.
- 2. Bertini E, Dionisi-Vici C, Zeviani M. Metabolic and mitochondrial ataxias. In Pulst SM (ed). Genetics of movement disorders. San Diego: Elsevier,2003:238.
- 3. Lee Y, Lin PY, Chiu NM, Chang WN, Wen JK. Cerebrotendinous xanthomatosis with psychiatric disorders: report of three siblings and literature review. Chang Gung Med J 2002;25:334-340.
- 4. Verrips A, van Engelen BGM, ter Laak H, et al. Cerebrotendinous xanthomatosis: controversies about nerve and muscle: observations in ten patients. Neuromusc Disord 2000;10:407-414.
- 5. Cruysberg JRM, Wevers RA, Tolboom JJM. Juvenile cataract associated with chronic diarrhea in pediatric cerebrotendinous xanthomatosis. Am J Ophthalm 1991;112:606-607.
- 6. Dotti MT, Rufa A, Federico A. Cerebrotendinous xanthomatosis: heterogeneity of clinical phenotype with evidence of previously undescribed ophthalmological findings. J Inherit Metab Dis 2001;24:696-706.
- 7. Kawabata M, Kuriyama M, Mori S, Sakashita I, Osame M. Pulmonary manifestations in cerebrotendinous xanthomatosis. Intern Med 1998;37:922-926.
- 8. Dotti MT, Mondillo S, Plewnia K, Agricola E, Federico A. Cerebrotendinous xanthomatosis: evidence of lipomatous hypertrophy of the atrial septum. J Neurol 1998;245:723-726.
- 9. Berginer VM, Shany S, Alkalay D, et al. Osteoporosis and increased bone fractures in cerebrotendinous xanthomatosis. Metabolism 1993;42:69-74.
- 10. Verrips A, Nijeholt GJ, Barkhof F, et al. Spinal xanthomatosis: a variant of cerebrotendinous xanthomatosis. Brain 1999;122:1589-1595.
- 11. Sugama S, Kimura A, Chen W, et al. Frontal lobe dementia with abnormal cholesterol metabolism and heterozygous mutation in sterol 27-hydroxylase gene (CYP27). J Inherit Metab Dis 2001;24:379-392.
- 12. Grandas F, Anaya F. Early-onset parkinsonism in cerebrotendineous xanthomatosis. Mov Disord 2002;17:1396-1397
- 13. Moghadasian MH, Salen G, Frohlich JJ, Scudamore CH. Cerebrotendinous xanthomatosis: a rare disease with diverse manifestations: arch Neurol 2002;59:527-529.
- 14. Cali JJ, Russell DW. Characterization of the human sterol 27-hydroxylase: a mitochondrial P-450 that catalyzes multiple oxidation reactions in bile acid biosynthesis. J Biol Chem 1991;266:7774-7778.
- 15. Lamon-Fava S, Schaefer EJ, Garuti R, Salen G, Calandra S. Two novel mutations in the sterol 27-hydroxylase gene causing cerebrotendinous xanthomatosis. Clin Genet 2002;61:185-191.
- 16. Cali JJ, Hsieh CL, Francke U, Russell DW. Mutations in the bile acid biosynthetic enzyme sterol 27-hydroxylase underlie cerebrotendinous xanthomatosis. J Biol Chem 1991;266:7779-7783.
- 17. Oftebro H, Björkhem I, Stormer FC, Pedersen JL. Cerebrotendinous xanthomatosis: defective liver mitochondrial hydroxylation of chemodeoxycholic acid precursors. J Lipid Res 1981;22:632-640.
- 18. Dotti MT, Federico A, Signorini E, et al. Cerebrotendinous xanthomatosis (van Bogaert-Scherer-Epstein disease): CT and MR findings. AJNR 1994;15:1721-1726.
- 19. Hokezu Y, Kuriyama M, Kubota R, Nakagawa M, Fujiyama J, Osame M. Cerebrotendinous xanthomatosis: cranial CT and MRI studies in eight patients. Neuroradiology 1992;34:308-312.
- 20. Vanrietvelde F, Lemmerling M, Mespreuve M, Crevits L, De Reuck J, Kunnen M. MRI of the brain in cerebrotendinous xanthomatosis (van Bogaert-Scherer-Epstein disease). Eur Radiol 2000;10:576-578.
- 21. De Stefano N, Dotti M, Mortilla M, Frederico A. Magnetic resonance imaging and spectroscopic changes in brains of patients with cerebrotendinous xanthomatosis. Brain 2000;124:121-131.
- 22. Soffer D, Benharroch D, Berginer V. The neuropathology of cerebrotendinous xanthomatosis revisited: a case report and review of the literature. Acta Neuropathol (Berl) 1995;90:213-220.
- 23. Salen G, Berginer V, Shore V, et al. Increased concentrations of cholestanol and apolipoprotein B in cerebrospinal fluid of patients with cerebrotendinous xanthomatosis: effect of chenodeoxycholic acid. N Engl J Med 1987;316:1233-1238.
Correspondence to
Publication Dates
-
Publication in this collection
25 Apr 2006 -
Date of issue
Dec 2004
History
-
Reviewed
22 June 2004 -
Received
18 Feb 2004 -
Accepted
03 Aug 2004