Abstracts
A 5-year-old girl is described as pervasive developmental disorder associated to chromosome 1-4 translocation, being such association not described till this moment in specialized literature.
autistic disorder; child developmental disorders; pervasive; translocation; genetic
Descrevemos uma menina de 5 anos de idade portadora de transtorno invasivo do desenvolvimento associado a translocação dos cromossomos 1-4, associação essa não descrita na literatura especializada até o presente momento.
transtorno autístico; transtornos globais do desenvolvimento infantil; translocação genética
A case of pervasive developmental disorder with chromosomal translocation 1-4
Um caso de transtorno invasivo do desenvolvimento com translocação cromossômica 1-4
Luciana G. TarelhoI; Francisco B. Assumpção Jr.II
Projeto Distúrbios do Desenvolvimento, Departamento de Psicologia Clínica do Instituto de Psicologia (IP) da Universidade de São Paulo, São Paulo SP, Brasil (USP)
IPsiquiatra da Infância e da Adolescência, Mestre em Psiquiatria pelo Departamento de Psiquiatria da Faculdade de Medicina (FM) da USP, Membro do Projeto Distúrbios do Desenvolvimento (PDD) do Instituto de Psicologia da Universidade do IP-USP
IIPsiquiatra da Infância e da Adolescência, Livre Docente em Psiquiatria pela FMUSP, Professor Associado do Departamento de Psicologia Clínica do IP-USP, Coordenador do PDD do IP-USP
ABSTRACT
A 5-year-old girl is described as pervasive developmental disorder associated to chromosome 1-4 translocation, being such association not described till this moment in specialized literature.
Key words: autistic disorder/genetics, child developmental disorders, pervasive, translocation, genetic.
RESUMO
Descrevemos uma menina de 5 anos de idade portadora de transtorno invasivo do desenvolvimento associado a translocação dos cromossomos 1-4, associação essa não descrita na literatura especializada até o presente momento.
Palavras-chave: transtorno autístico/genética, transtornos globais do desenvolvimento infantil, translocação genética.
Pervasive developmental disorders (PDD) are characterized by severe and pervasive impairment in several areas of development: reciprocal social interation skills, communication skills, or the presence of stereotyped behavior, interests, and activities. Such manifestations, indeed, do vary to a very great extent, depending on the individuals level of development and chronological age1. These disorders are usually evident in first years of life and are often associated with some degree of mental retardation, which can range from mild to profound. Autism is the best-known example of a PDD subtype, but, through the years, a number of other subtypes have been proposed as well. The prevalence of this disorder was first estimated to be of about 5:10,000, with a 3:12 man/woman ratio. Recent surveys suggest that the rate of all PDDs is about 60 per 10,000. The prevalence of autism today is estimated at 13 per 10,0003,4. Increased recognition, the broadening of the diagnostic concept and methodological differences across studies may account for most or all of the apparent increase in prevalence, although this cannot be quantified It is described in association with innumerable genetic syndromes5-8 as, for instance, Downs, Prader-Willi or Angelman syndromes and Aarskogs syndromes, among others. The condition is sometimes observed in association with different neurobiological alterations, such as phenylketonuria, congenital rubella, as well as increased serotonin levels in the blood9-11. Various papers12-14 and different authors9, 15-23 also describe chromosomal variations that it is associated with.
The simultaneous occurrence of autism and chromosomal variations can be checked by deletions, translocations or inversions, at a frequency between 5 and 12%20. The chromosomal translocations are the result of the rupturing of two chromosomes and subsequent joining of the segment of one to the fragment of the other, exhibiting clinical manifestations when the transfer is observed of one chromatid to another homologous one; a non-balanced translocation21 thus occurs. The establishment of an association between both occurrences is important due to the possibility provided of pursuing eventual genetic aspects involved in autism, for which reason this case history is presented.
CASE
A five-year-old girl, caucasian, born in and from São Paulo, Brazil, only daughter, was routed to the Development Disturbances Project (DDP), of São Paulo Universitys Institute of Psychology (IP-USP), for diagnostic investigation on September 15, 2005 (Parents gave their informed consent to publish this case). This child was born from a scheduled term cesarean section, with 2.43 kg in weight and 44 cm in stature (regarded as small for her gestational age). She displayed a hypoglycemia episode that was not persistent as well as physiological jaundice, a transitional event that was resolved without treatment not requiring phototherapy and/or exchange transfusion of donor blood with no further intercurrent events. As of one month of age, the child displayed repetitious myoclonic twitching, having no relationship with the period of the day, whether awakening or just being sleepy. At five months, faulty vision was suspected and, following an evoked potential examination, the blame was put on subsequently not confirmed cortical visual impairment.
Being hypotonic, the child steadied her head at one year, began crawling at two, and walked at three. According to the mother, the child spoke a few words when she was one year, but ceased speaking at age two and a half. At present, all she can do is to issue guttural sounds, with no attempted alternate modes of communication, such as by gestures or mimicking. Does not react to playfulness or interact with games of social imitation. As a baby, did not exhibit gestures of anticipation, rarely made visual contact and now, when she does, fails to maintain it. She does not point to objects of interest, but uses the hand of others as a means of obtaining what she desires. No interaction with other children and displays stereotypies (twisting of the hands), biting of anything within her reach, having periods of restlessness, when she cries and screams continuously. Insomnia is likewise described. Mother aged 34 years, father 35 years, healthy, both in administrative jobs. Cousin, on fathers side, suffers from Downs syndrome.
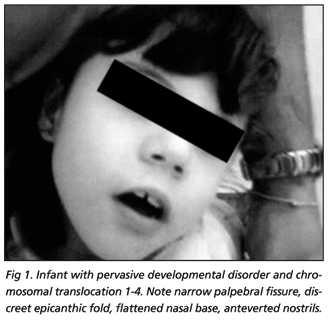
Physical examination exhibited good overall condition, hair with low frontal implantation, narrow palpebral fissure, discreet epicanthic fold, flattened nasal base, anteverted nostrils, flat feet, syndactylism in first third of the second and third right toes. Right-hand divergent strabismus. Cephalic perimeter 44.5 cm, ear 3 cm, biauricular perimeter 26 cm, internal intercanthus distance 3 cm, stature 1.30 m ( Z-score > than +3SD for age or percentile > than 97th according to WHO24) and finger span 1.30 m, weight 18 kg (Z score between median and +2SD for age or percentile between 50th and 85th according to WHO24) (Figs 1 and 2).
Psychic examination, clean, well cared for, sialorrhoea bearer. She exhibits restlessness, with spastic and paretic gait, inattentive and precarious contact with the examiner. Bites objects within reach, with stereotypies of the hands and presence of myoclonic manifestations. She acts with stereotypy and repetition when played with, no imaginative attitudes, displaying a severe cognitive deficit and absent voiced language, except for guttural utterances. Absence of emotional reciprocity, very reduced pragmatism and full dependence on lifes daily activities.
For diagnostic purposes, she underwent evaluation by different scales, which provided ratings compatible with diagnosis of autism (Autism Rating Scale, with 40.5 points, and Evaluation of Autistic Traces, with 33), associated with severe mental retardation (Vineland Adaptive Behavior Scale, with overall score of 29).
On her first visit, brought a G-band karyotype, evidencing 46XX t(1;4)(q42), as well as a cranial magnetic resonance image (MRI) and normal electroencephalogram (EEG). On account of the myoclonic manifestations, a new EEG and cranial MRI were requested, with the patient being routed to São Paulo Universitys Medical Schools Clinics Hospitals Epilepsy Study Center, where she underwent six hours of EEG monitoring. On being examined, electric brain activity was observed with base obscured by beta-range, high-frequency electric brain activity and shortage of elements characteristic of normality. In association, presence of segmentary myoclonic manifestations (over 40), both axial and appendicular, predominantly in the upper limbs, with occurrence during sleep, sleepiness or wakefulness.
Upon repetition of the MRI, no abnormalities were found. She was then sent to the São Paulo Universitys Genetics Center, where a high-resolution karyotype was made, confirming and detailing the previously diagnosed translocation (46, XX, t (1;4) (q42.2 or 42.3;q21.1). Parental karyotype have been investigated and no abnormalities were found. Radiographies made of long bones, knees, hands, elbows, cranium and thorax. Spinal radiography disclosed a deviation from the longitudinal axis, with double convexity in the exam position, osseous reduction and non fusion of the posterior arch of the sacra alta vertebra. In addition TORCHES screen have been made and biochemical investigations included measurement of thyroxine (T4), thyroid-stimulating hormone (TSH), sodium, potassium, calcium, phosphate and plasma amino acid chromatography. No evidence of metabolic disorders were found.
Ultrasonography of the abdomen disclosed small and sparse echogenic images in both renal sinuses, non producers of acoustic shadow suggestive of microlithiasis.
At the time of the first call, she used haloperidol (2 drops per day), with significant improvement in agitation and sleep; however, due to the worsening of myoclonic manifestations, clonazepam was associated, prescribed at a dose of 27 drops per day, with haloperidol being replaced by rispiridona up to a dose of 6 drops per day, with a very important improvement in behavior and satisfactory control of the myoclonic manifestations.
On the basis of the result, characterized by loss in socialization (absence of social smile and visual contact, social isolation), language (echolalia), in addition to stereotypy in behavior and echopraxia, the diagnosis was given of pervasive developmental disorder and severe mental deficiency13, associated with translocation of chromosomes 1-4.
DISCUSSION
Autistic characteristics have been described in a variety of genetic arrangements, but we have not yet been able to establish a causal relationship of the linear type. We have found descriptions of similar situations in Jouberts syndrome, of a recessive autosomal disturbance characterized by total or partial agenisis of the cerebellar vermis25, in which changes in the cerebellum have been reported as associated with autism, with a reduction in the hemisphere parenchyma and neocerebellar vermix, loss of Purkinjes cells and granulomata of the cerebellar neocortex26,27.
We submit a case, in which the child presents, upon physical examination, somatic alterations that, although being unable to characterize any specific syndrome, are linked to the observed chromosomal abnormality. Further displayed, simultaneously, is significant communication impairment, absence of social and emotional reciprocity and stereotypy in motion, thereby meeting the diagnostic criteria for PDD, as recommended by DSM IV-TR1, as well as referenced as of the use of specific scales (Childhood Autism Rating Scale and Evaluation of Autistic Traces), with the severe mental retardation being based on application of the Vineyard Adaptive Behavior Scale, since patient is unwilling to cooperate satisfactorily enough for parametric tests to be applied.
This presentation is not thought to be conclusive, but it is our intention to provide the difficulties inherent to the diagnosis of an autism case associated with severe mental retardation, which difficulties we endeavor to get around by the use of structured scales. Another point to be emphasized is the association between genetic arrangements and autism, since the latter is subject to diverse and heterogeneous causes. It is because different studies of this nature need to be undertaken for the purpose of documenting possible etiological factors involved in cases of autism, seeing that such descriptions may possibly suggest different loci involved in the etiology of PDD28. It is thus important for clinical research to be continued, involving the factors that may clarify the association between genetic and behavioral dispositions.
Since this is the first case to be described in literature correlating such characteristics, that is, coexistence of PDD and translocation of the 1-4 group, the presentation thereof for documentary purposes is deemed important, with other more extensive and detailed studies still being required in this connection.
Acknowledgements The authors would like to acknowledge São Paulo Universitys Genetics Center who gently assigned results of high-resolution karyotype.
Received 17 May 2006, received in final form 18 September 2006. Accepted 6 November 2006.
Dr. Francisco B. Assumpção Jr. - Rua dos Otonis 697 - 04025-002 São Paulo SP - Brasil. E-mail: cassiterides@bol.com.br
-
1American Psychiatric Association (APA). Diagnostic and statistical manual of mental disorders. 4th Edition. (Text Revision). Washington, DC: APA, 2000.
- 2. Gilberg C, Wing L. Autism: not an extremely rare disorder. Acta Psychiatr Scand 1999;99:399-406.
- 3. Charman T. Epidemiology and early identification of autism: research challenges and opportunities. Novartis Found Symp 2003;251 discussion 19-25, 109-11, 281-297.
- 4. Fombonne E. Epidemiology of autistic disorder and other pervasive developmental disorders. J Clin Psychiatry 2005;66(Suppl 10):3-8.
- 5. Assumpção FB Jr. Diagnóstico diferencial. In Schwartzman JS, Assumpção FB Jr (Eds). Autismo infantil. São Paulo: Memnon Editora, 1995:125-146.
- 6. Kuczynski E. Anormalidades cromossômicas esporádicas associadas à síndrome autística. Infanto. Rev Neuropsiq Inf Adol 1996;4:26-36.
- 7. Assumpção FB Jr. Brief report: a case of chromosome 22 alteration associated with syndrome. J Austism Dev Disord 1998;28:253-256.
- 8. Dykens EM, Sutcliffe JS, Levitt P. Autism and 15q11-q13 disorders: behavioral, genetic, and pathophysiological issues. Ment Retard Dev Disabil Res Rev 2004;10:284-291.
- 9. Coleman M. The autistic syndromes. Amsterdan: North-Holand, 1976: 183-208.
- 10. Finnegan J, Quarrington B. Pre, peri and neonatal factors and infantile autism. J Child Psychol Psychiatry 1979;20:119-128.
- 11. Assumpção FB Jr, Kuczynski E. Autismo infantil, transtorno bipolar e retardo mental em portador de síndrome de rubéola congênita. Arq Neuropsiquiatr 2002;60:324-327.
- 12. Assumpção FB Jr. Síndrome XXX (Metafemina) in soggetto portatore di autismo infantile. Phoenix 1995;II:305-309.
- 13. Assumpção FB Jr, Kuczynski E. Autismo e síndrome de Turner. Pediatria Moderna 2000;XXXVI:325-329.
- 14. Ramos ACP, Assumpção FB Jr, Stocco, MR. Fehlbildugen dês Mundes bei Aarskogsyndrom. Sozialpadiatrie 1998;20:96-98.
- 15. Gillberg C. The neurobiology of infantile autism. J Child Psychol Psychiatry 1989;29:257-266.
- 16. Gillberg C. Autism and pervasive development disorders. J Child Psychol Psychiatry 1990;31:99-119.
- 17. Blommquist HK, Bohman M, Edvinsson SO, et al. Frequency of the fragile X syndrome in infantile autism. Clin Genetics 1985;27:113-117.
- 18. Rutter M, Schopler E. Autism and pervasive developmental disorders: concepts and diagnostic issues. J Aut Dev Disorders 1987;17:159-186.
- 19. Bolton P, Rutter M. Genetic influences in autism. Int Rev Psychiat 1990; 2:67-80.
- 20. Gillberg C, Wahlström J. Chromosome abnormalities in infantile autism and other childhood psychoses: a population study of 66 cases. Dev Med Child Neurol 1975;27:293-304.
- 21. Gillberg C. Chromosomal disorders in autism. J Autism Dev Disord 1998;28:415-425.
- 22. Spence SJ. The genetics of autism. Semin Pediatr Neurol 2004;11: 196-204.
- 23. Beiguelman B. Citogenética humana. Rio de Janeiro: Guanabara Koogan, 1982.
-
24WHO ANTHRO 2005, Beta version Feb 17th, 2006. Software for assessing growth and development of the world's children. Geneva: WHO, 2006 (http://www.who.int/childgrowth/software/en/).
- 25. Holroyd S, Reiss AL, Bryan RN. Autistic features in Joubert syndrome: Genetic disorder with agenesis of the cerebelar vermis. Biol Psychiatry 1991;29:287-294.
- 26. Piven J, Arndt S, Bailey JBS, Havercamp SBS, Andreasen NC, Palmer P. An MRI study of brain size in autism. Am J Psychiatry 1995;152:1145-1149.
- 27. Pierce K, Courchesne E. Evidence for a cerebelar role in reduced exploration and stereotyped behavior in autism. Biol Psychiatry 2001;49:655-664.
- 28. Lauritsen MB, Ewald H. The genetics of autism. Acta Psychiatr Scand 2001;103:411-427.
Publication Dates
-
Publication in this collection
23 Mar 2007 -
Date of issue
Mar 2007
History
-
Accepted
06 Nov 2006 -
Reviewed
18 Sept 2006 -
Received
17 May 2006