Abstracts
Dopa-responsive dystonia (DRD) is an inherited metabolic disorder now classified as DYT5 with two different biochemical defects: autosomal dominant GTP cyclohydrolase 1 (GCH1) deficiency or autosomal recessive tyrosine hydroxylase deficiency. We report the case of a 10-years-old girl with progressive generalized dystonia and gait disorder who presented dramatic response to levodopa. The phenylalanine to tyrosine ratio was significantly higher after phenylalanine loading test. This condition had two different heterozygous mutations in the GCH1 gene: the previously reported P23L mutation and a new Q182E mutation. The characteristics of the DRD and the molecular genetic findings are discussed.
dystonia; levodopa; dopa-responsive dystonia; guanosine triphosphate cyclohydrolase 1; GCH1 gene
Distonia dopa-responsiva (DRD), classificada como DYT5, é um erro inato do metabolismo que pode ser causado por dois diferentes tipos de defeito bioquímico: deficiência de GTP ciclo-hidrolase 1 (GCH1) (autossômica dominante) ou de tirosina hidroxilase (autossômica recessiva). Descrevemos o caso de menina de 10 anos com distonia generalizada progressiva e alteração da marcha com importante melhora após uso de levodopa. A relação fenilalanina/tirosina estava aumentada após teste de sobrecarga com fenilalanina. O estudo molecular mostrou que o paciente apresenta uma combinação hererozigótica de mutação no gene GCH1: a já conhecida mutação P23L e uma nova mutação Q182E. Discutem-se as características da DRD e as alterações genéticas possíveis.
distonia; levodopa; distonia dopa-responsiva; guanosina trifosfato ciclo-hidrolase 1; gene GCH1
SCIENTIFIC NOTES
A novel missense mutation pattern of the GCH1 gene in dopa-responsive dystonia
Novo padrão de mutação missense no gene GCH1 na distonia dopa-responsiva
Rosana H. ScolaI; Carla CarducciII; Vanise G. AmaralI; Paulo J. LorenzoniI; Helio A.G. TeiveI; Teresa GiovannielloII; Lineu C. WerneckI
INeurology Division, Internal Medicine Department, Hospital de Clínicas, Universidade Federal do Paraná, Curitiba PR, Brazil (UFPR)
IIDepartment of Experimental Medicine and Pathology, University of Rome " La Sapienza" , Rome, Italy
ABSTRACT
Dopa-responsive dystonia (DRD) is an inherited metabolic disorder now classified as DYT5 with two different biochemical defects: autosomal dominant GTP cyclohydrolase 1 (GCH1) deficiency or autosomal recessive tyrosine hydroxylase deficiency. We report the case of a 10-years-old girl with progressive generalized dystonia and gait disorder who presented dramatic response to levodopa. The phenylalanine to tyrosine ratio was significantly higher after phenylalanine loading test. This condition had two different heterozygous mutations in the GCH1 gene: the previously reported P23L mutation and a new Q182E mutation. The characteristics of the DRD and the molecular genetic findings are discussed.
Key words: dystonia, levodopa, dopa-responsive dystonia, guanosine triphosphate cyclohydrolase 1, GCH1 gene.
RESUMO
Distonia dopa-responsiva (DRD), classificada como DYT5, é um erro inato do metabolismo que pode ser causado por dois diferentes tipos de defeito bioquímico: deficiência de GTP ciclo-hidrolase 1 (GCH1) (autossômica dominante) ou de tirosina hidroxilase (autossômica recessiva). Descrevemos o caso de menina de 10 anos com distonia generalizada progressiva e alteração da marcha com importante melhora após uso de levodopa. A relação fenilalanina/tirosina estava aumentada após teste de sobrecarga com fenilalanina. O estudo molecular mostrou que o paciente apresenta uma combinação hererozigótica de mutação no gene GCH1: a já conhecida mutação P23L e uma nova mutação Q182E. Discutem-se as características da DRD e as alterações genéticas possíveis.
Palavras-chave: distonia, levodopa, distonia dopa-responsiva, guanosina trifosfato ciclo-hidrolase 1, gene GCH1.
Hereditary progressive dystonia with marked diurnal fluctuation or Segawas disease (OMIM: #128230) is an inherited form of dopa-responsive dystonia (DRD), classified as an autosomal dominant metabolic disorder that results in dopamine deficiency in the basal ganglia1-4. The incidence is estimated to be between 0.5 and 1 per million people and the typical clinical picture includes onset in childhood, early foot dystonia with progression to multifocal or generalized dystonia, diurnal fluctuation and dramatic response to levodopa (L-dopa)1-3. It is caused by mutation in the guanosine triphosphate (GTP) cyclohydrolase 1 (GCH1)1-3, 5. There is also an autosomal recessive form caused by mutation in the tyrosine hydroxylase gene (OMIM: #650407)3.
This disease was first described in 1971, by Segawa et al, as dystonia with diurnal fluctuation affecting the gait with a beneficial therapeutic response to L-dopa1-5. Diagnosis can be based on clinical findings (including dramatic and sustained response to low doses of L-dopa), plasma levels of phenylalanine/tyrosine after an oral phenylalanine load and molecular analysis of GCH1 gene1-6.
In this study, we describe a patient with progressive limb dystonia whose molecular analysis revealed two different heterozygous missense mutations of the GCH1 gene, one previously described and a novel one.
CASE
A 10-years-old girl presented with progressive difficulty in running since the age of three years. Initial observation revealed difficulty in moving the left upper and lower limbs. At eight years of age she had severe gait difficulties due to involuntary sustained muscle contractions in the lower limbs, tiptoe pattern during walking (wheelchair bound) and worsening of the symptoms as the day progressed, with relief after sleep. She was the first child affected in the family and was born at term of non-consanguineous parents.
Due to symptoms fluctuation she was firstly diagnosed as myasthenia gravis and received pyridostigmine (300 mg per day) and prednisone (20 mg per day). After eight months, without improvement, she was referred to our hospital and the drugs were gradually withdrawn.
On neurological examination the following neurological features were observed: normal cranial nerves; mild hypotrophy of the quadriceps muscle; muscle strength with score 3 in the upper limbs and 2 in the lower limbs according to the Medical Research Council scale; bradykinesia; rigidity in all limbs; cervical dystonia (anterocollis); distal dystonia in the upper and lower limbs at rest exacerbated by voluntary physical activity; increased deep tendon reflexes; and normal sensation.
Both parents were asymptomatic and normal on examination by two neurologists.
The administration of a low-dose of L-dopa/carbidopa (50/12.5 mg per day) resulted on prompt improvement of anterocollis, rigidity and strength. The dose of L-dopa/carbidopa was then increased (100/25mg per day), but she developed erratic myoclonic jerks affecting upper muscle limbs and trunk and a better result was obtained by L-dopa/carbidopa (35/8.75mg per day) and clonazepan 4mg/day. After 30 days she was walking with assistance and with in 6 months of follow-up she was walking independently.
The laboratorial tests (blood count, biochemistry, hepatic and renal function, coagulation tests and cerebrospinal fluid) and electroencephalogram were normal.
To determine if phenylalanine metabolism was compromised the plasma levels of phenylalanine and tyrosine were measured at baseline and 1, 2, 4, and 6 hours after an oral phenylalanine load (100 mg/kg). Before loading phenylalanine and tyrosine concentrations were normal, but the phenylalanine levels were higher than control subjects after 1, 2, 4 and 6 hours post-load (Table). No increase in plasma tyrosine was observed after the phenylalanine load (Table). The phenylalanine to tyrosine ratio (Table) was significantly higher after four hours post-load (normal: < 7.5).
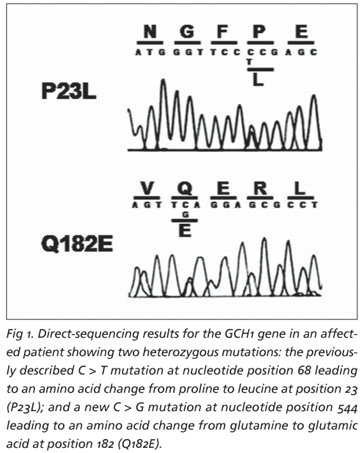
Genomic DNA was extracted from peripheral blood lymphocytes and all six exons, intron-exon boundaries and 5UTR-region of the GCH1 gene were amplified separately using the polymerase chain reaction (PCR), according to standard procedures2, 3. The fragments were amplified and analyzed by direct sequencing using Big Dye Terminator v1.1 (Applied Biosystems) on a 310 ABI PRISM genetic analyzer. The direct sequence analysis showed a heterozygous C > T mutation at position 68 (c.68 C>T) of the GCH1 gene, leading to an amino acid change from nonpolar proline (P) to nonpolar leucine (L) at position 23 (P23L) and a new heterozygous C > G transition at nucleotide position 544 (c.544 C>G) of the GCH1 gene, producing substitution of the non-charged amino acid glutamine (Q) by negatively charged glutamic acid (E) at position 182 (Q182E) (Fig 1). The mendelian inheritance was confirmed in the parents, with the mother being a P23L carrier and the father a Q182E carrier (Fig 2). Our patient is therefore a compound heterozygote for two mutant alleles in the GCH1 gene: the P23L mutation, of maternal origin, and the Q182E mutation, of paternal origin (Fig 2). The new Q182E mutation was not found in 100 normal chromosomes.
After a follow-up of four years, the patient sustained the excellent response to low doses of levodopa.
All studies were done following informed consent of parents.
DISCUSSION
Genetic studies of autosomal dominant DRD firstly revealed linkage to the long arm of chromosome 14 and later showed this to be caused by a heterozygous mutation in the GCH1 gene located on 14q22.1-q22.27,8. This gene has a high degree of variability, and to date more than 100 different mutations have been identified in the GCH1 coding region5,9,10. The disease is usually confirmed by means of biochemical tests, and abnormalities of this gene were only observed in 20 to 87% of patients2,8,9,11,12. GCH1 is responsible for catalyzing the formation of tetrahydrobiopterin (BH4), an essential cofactor of tyrosine hydroxylase, which is the rate-limiting step for dopamine biosynthesis2,13,14. Where as, the decrease of neopterin as well as biopterin in cerebrospinal fluid suggested the decrease of GCH1 enzyme as the cause of DRD2,5,13,14.
The main clinical features of dopa-responsive dystonia are onset of dystonia at 4-8 years of age, usually in the lower limbs, causing an abnormal gait, diurnal fluctuation of signs and symptoms and other associated features such as hyperreflexia and extensor plantar reflexes, parkinsonian signs (such as mask faces), or tremor1-5,15,16. Although diurnal variation is a typical feature of DRD, characterized by worsening of symptoms from day to day and improving after sleep, as in our patient, such marked fluctuation is present only in half patients with DRD1-5,15. DRD patients can have a marked intrafamilial variability of the clinical manifestations between affected members of the same family14,16.
Although individuals with DRD never develop hyperphenylalaninemia, a subclinical defect in phenylalanine metabolism caused by partial BH4 deficiency in the liver can often be detected by the phenylalanine loading test, which analyzes plasma phenylalanine-to-tyrosine ratios following an oral phenylalanine load6.
In our patient, the phenotype included dystonia after a dramatic response to L-dopa trial, and molecular analysis revealed a compound heterozygosity for two mutations in the GCH1 gene: a previously described mutation of maternal origin (P23L) and a novel mutation of paternal origin (Q182E).
The P23L mutation in the GCH1 allele was previously described in four different families17,18. It is particularly interesting the fact that this sequence change was previously detected in a patient with DRD combined with another mutation, suggesting that P23L could represent a rare polymorphism in the population or, alternatively, that this mutation could only be pathogenic in association with another mutation, as in our case18.
The new Q182E mutation that was found in our patient led to the substitution of a neutrally charged amino acid by a negatively charged one, a net charge alteration that can adversely affect the structure and function of the GCH1 gene at critical positions of the amino acid sequence. Another mutation (Q182X) that caused DRD was previously reported in this same amino acid position, highlighting the important functional role of this part of the protein10. The Q182E mutation in the paternal allele might be sufficient to reduce GCH1 activity below a critical threshold, although neither mutation alone was sufficient to cause disease in either of the carrier parents18. These observations agree with the hypothesis that the new mutation, in combination with the other previously described, is responsible for the phenotype in our case.
DRD should be considered in any child who presents with paroxysmal or progressive hypertonia of unknown etiology, and its dramatic response to L-dopa has to be emphasized. Fluctuations in response, increased dose requirements, or long-term adverse effects such as the " on-off" phenomenon have not been described4, 12, 19-21. The detection of the specific mutation is important to confirm the diagnosis, and to provide accurate treatment and genetic counseling to the patients and their family. Treatment of these patients is one of the most satisfying experiences in neurology4, 19.
Acknowledgment The authors are grateful to Drs Carlo Marrone, MD and Ehrenfried Wittig, MD for their collaboration in the case.
Received 29 June 2007, received in final form 31 August 2007. Accepted 2 October 2007.
Dra. Rosana Herminia Scola - Serviço de Doenças Neuromusculares / Hospital de Clínicas da UFPR - Rua General Carneiro 181 / 3º andar - 80060-900 Curitiba PR - Brasil. E-mail: scola@hc.ufpr.br
- 1. Segawa M. Hereditary progressive dystonia with marked diurnal fluctutation. Brain Dev 2000; 22 (Suppl 1): S65-S80.
- 2. Segawa M, Nomura Y, Nishiyama N. Autosomal dominant guanosine triphosphate cyclohydrolase I deficiency (Segawa disease). Ann Neurol 2003;54:S32-S45.
- 3. Tarsy D, Simon D. Current concepts: dystonia. N Engl J Med 2006;355: 818-829.
- 4. Araujo AQC, Miranda SBM. Doença de Segawa: distonia progressive sensível à L-dopa. Arq Neuropsiquiatr 1993; 51: 532-536.
- 5. Venna N, Sims KB, Grant PE. Case 26-2006: a 19-year-old woman with difficulty walking. N Engl J Med 2006; 355: 831-839.
- 6. Hyland K, Fryburg JS, Wilson WG, et al. Oral phenylalanine loading in dopa-responsive dystonia: a possible diagnostic test. Neurology 1997; 48: 1290-1297.
- 7. Nygaard TG, Wilhelmsen KC, Risch NJ, et al. Linkage mapping of dopa-responsive dystonia (DRD) to chromosome 14q. Nat Genet 1993;5:386-391.
- 8. Ichinose H, Ohye T, Takahashi E, et al. Hereditary progressive dystonia with marked diurnal fuctuation caused by mutations in the GTP cyclohydrolase I gene. Nat Genet 1994; 8: 236-242.
- 9. Furukawa Y. Genetics and biochemistry of dopa-responsive dystonia: significance of striatal tyrosine hydroxylase protein loss. Adv Neurol 2003; 91: 401-410.
- 10. BIOMD database. Available at: http://www.bh4.org/BH4_databases_biomdb2.asp (accessed August 25, 2007).
- 11. Furukawa Y, Kish SJ. Dopa-responsive dystonia: recent advances and remaining issues to be addressed. Mov Disord 1999;14:709-715.
- 12. Steinberger D, Korinthenberg R, Topka H, Berghauser M, Wedde R, Muller U. Dopa-responsive dystonia: mutation analysis of GCH1 and analysis of therapeutic doses of L-dopa. Neurology 2000;55:1735-1737.
- 13. Cheng WW, Kong CK. Girl with dopa-responsive dystonia. J Pediatric Child Health 2001;37:300-305.
- 14. Hahn H, Trant MR, Brownstein MJ, Harper RA, Milstien S, Butler IJ. Neurologic and psychiatric manifestations in a family with a mutation in exon 2 of the guanosine triphosphate-cyclohydrolase gene. Arch Neurol 2001;58:749-755.
- 15. Segawa M, Hosana A, Miyagawa F, Nomura Y, Imai H. Hereditary progressive dystonia with marked diurnal fluctuation. Adv Neurol 1976; 14:215-233.
- 16. Lopez-Laso E, Camino R, Mateos ME, et al. Dopa-responsive infantile hypokinetic rigid syndrome due to dominant guanosine triphosphate cyclohydrolase 1 deficiency. J Neuro Sci 2007;256:90-93.
- 17. Leuzzi V, Carducci C, Carducci C, Cardona F, Artiola C, Antonozzi I. Autosomal dominant GTP-CH deficiency presenting as a dopa-responsive myoclonus-dystonia syndrome. Neurology 2002;59:1241-1243
- 18. Jarman PR, Bandmann O, Marsden CD, Wood NW. GTP cyclohydrolase I mutations in patients with dystonia responsive to anticholinergic drugs. J Neurol Neurosurg Psychiatry 1997; 63: 304-308.
- 19. Jan MMS. Misdiagnoses in children with dopa-responsive dystonia. Pediatr Neurol 2004;31:298-303.
- 20. Nygaard TG, Waran SP, Levine RA, Naini AB, Chutorian AM. Dopa-responsive dystonia simulating cerebral palsy. Pediatr Neurol 1994;11: 236-240.
- 21. Van Hove JL, Steyaert J, Matthijs G, et al. Expanded motor and psychiatric phenotype in autosomal dominant Segawa syndrome due to GTP cyclohydrolase deficiency. J Neurol Neurosurg Psychiatry 2006;77:18-23.
Publication Dates
-
Publication in this collection
07 Mar 2008 -
Date of issue
Dec 2007
History
-
Accepted
02 Oct 2007 -
Received
31 Aug 2007