CLINICAL/SCIENTIFIC NOTE
Swallowing dysfunction in hereditary neuropathy with liability to pressure palsies
Distúrbio de deglutição na neuropatia hereditária com suscetibilidade à pressão
Paulo J. LorenzoniI; Rosana H. ScolaI; Juliana CardosoI; Cláudia S.K. KayI; Elmar A. FugmannII; Wilson Marques JrIII; Carlos E. SilvadoI; Lineu C. WerneckI
INeurology Division, Internal Medicine Department, Universidade Federal do Paraná (UFPR), Curitiba PR, Brazil
IIOtorhinolaryngology Department, UFPR
IIINeurology Department, Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo, Ribeirão Preto SP, Brazil
Hereditary neuropathy with liability to pressure palsies (HNPP) is an autosomal dominant peripheral neuropathy characterized by recurrent episodes of paralysis of peripheral nerves, usually after a minor trauma to topographies prone to compression1,2. Involvement of the cranial nerves has rarely been described in these patients3-8. Reports of swallowing dysfunction in patients with HNPP are even rarer.
We report a patient diagnosed with HNPP confirmed by molecular testing who presented with recurrent episodes of swallowing dysfunction (SD).
CASE
A 41-year-old man who presented with recurrent episodes of acute mononeuropathies affecting mainly the ulnar, radial and peroneal nerves followed by spontaneous improvement since the age of 12 years. In the years prior to diagnosis, the patient started experiencing sudden episodes of reversible SD without a specific trigger, but it worsening with progressive increased of volume and solid consistency of bolus food. The SD had a spontaneous improvement after few weeks, but with residual dysfunction when he drinking liquid. Some of his relatives (sisters and nephew) had similar symptoms, but none had SD. He denied episodes of aspiration (airway protection), exposure to drugs or toxic agents.
Cranial nerves showed mild weakness in posterior tongue during the swallow. Gag reflex, taste and sensation of tongue and oropharynx were normal. Mild asymmetrical muscle atrophy was observed in both hands and forearms while weakness was detected in the left first dorsal interosseus dorsal and flexor carpi ulnaris (grades 3 and 4, respectively, according to the Medical Research Council scale). Tendon reflexes were absent in both biceps and left brachioradial and ankle tendon reflexes were also absent. Pain, pinprick and light touch sensory impairment were detected on the territories of the left median, bilateral sural and superficial peroneal nerves.
Nerve conduction studies showed a nonuniform demyelinating neuropathy, mainly the ulnar, radial and peroneal nerves, together with evidence of asymmetrical acute and/or chronic denervation affecting predominantly the first dorsal interosseus, tibialis anterior and gastrocnemius muscles. Nerve conduction studies with similar findings were observed in the patient's sisters and nephew.
Laboratory tests as well as serological tests were normal. Biopsy of the sural nerve showed a reduction in the number of large myelinated fibers and focal thickening of the myelin sheath (formation of tomaculae). Biopsy of the gastrocnemius muscle biopsy showed recent and chronic denervation.
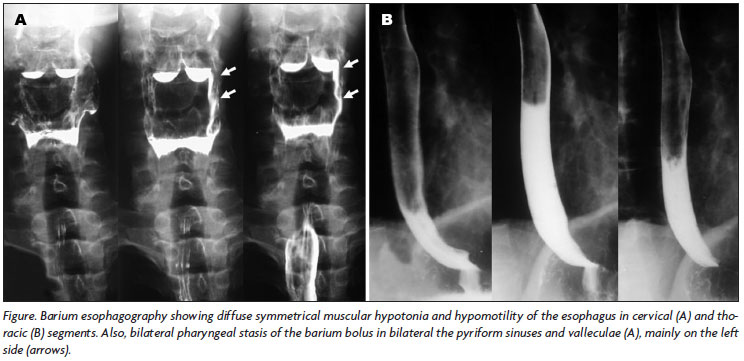
Phonoaudiological evaluation, videoendoscopic and fluoroscopic examination of deglutition revealed normal oral (preparatory and transport) phase; swallowing test (liquid, semisolid and solid consistencies) presented mild nasal escape, normal oral transit time, residual bolus in pyriform sinuses and valleculae cleared by dry swallow and cough, episodes of laryngeal penetration only after great volume of liquid, but with normal coughing reflex, and absence of aspiration or choking; asymmetrical stasis of the bolus (liquid consistency) in the pyriform sinuses and valleculae in the pharyngeal phase of swallowing with a decrease in pharyngeal constriction; penetration in airway remains above vocal folds with normal coughing reflex and no aspiration. Barium esophagography revealed symmetrical muscular hypotonia and hypomotility of the esophagus and bilateral asymmetrical pharyngeal stasis of the barium bolus in the pyriform sinuses and valleculae (Figure).
Cranial magnetic resonance imaging, abdominal ultrasonography and esophagogastroduodenoscopy were normal.
After informed consent was obtained, molecular genetic testing revealed deletion of the 17p11.2 region, which encompasses the PMP22 gene, consistent with the diagnosis of autosomal dominant HNPP.
A diagnosis of HNPP associated with SD (pharyngeal and esophageal) was made on the basis of molecular, endoscopic and radiological findings. During the follow-up period, the patient continued to have acute recurrent episodes of mononeuropathies and SD.
DISCUSSION
HNPP is an uncommon hereditary neuropathy with an estimated prevalence of at least 16/100,000 people9. The disease is usually caused by a heterozygous 1.5-megabase deletion of the PMP22 gene on chromosome 17p11.2, but several point mutations associated with HNPP have been reported10,11. The onset is around the second or third decade of life and 15% of carriers remain asymptomatic2. Carriers are predisposed to focal neuropathies that most frequently involve the brachial plexus and peroneal, ulnar, radial, and median nerves5,12. Several authors have reported involvement of the facial, trigeminal, hypoglossal and recurrent laryngeal nerves3-8. The attacks usually stop within days or weeks and are triggered by compression, slight traction or other minor trauma1,12.
The electrophysiological pattern in patients with HNPP consists of prolonged distal motor latencies in some nerves, multiple conduction slowing at compression sites and only mildly reduced conduction velocities in other segments of the motor nerves1,13. There is also a diffuse reduction in sensory nerve action potential amplitudes1,13. Asymptomatic carriers frequently have prolonged distal motor latencies and abnormal results in sensory conduction studies12.
The mechanism of this susceptibility to compression is unknown. Distal myelinopathy or a simple length-dependent process can not explain the abnormal eletrophysiological findings of HNPP14. The typical pathologic finding in nerve biopsies is a focal sausage-shaped thickening of the myelin sheath in about 25% or more of the internodes, referred to as 'tomaculae'15,16. This finding however is not specific to HNPP and can occur in Charcot-Marie-Tooth disease and acquired neuropathies16.
SD are frequent in neurological diseases and may arise from pharyngeal or esophageal dysphagia17. Deficits of the lower cranial nerves (nerves IX, X, XI and XII) may cause disabling SD3. Pharyngeal dysphagia has previously been reported in HNPP, mainly related to a transient hypoglossal mononeuropathy or associated with recurrent laryngeal nerve palsy3,6,8. Winter and Juel reported an HNPP patient with evident hypoglossal nerve palsy which could partially explain the pharyngeal dysfunction in our patient8. The weakness in the tongue during swallowing, caused by hypoglossal neuropathy could result in pharyngeal dysfunction with stasis in the pyriform sinuses and valleculae3,8.
Esophageal dysphagia may be caused by mechanical lesions or motility disorders17. Our patient presented with an esophageal motility dysfunction, and other causes, such as drugs, achalasia, diffuse esophageal spasm, diabetes mellitus, Chagas' disease and scleroderma were all excluded.
To our knowledge esophageal dysphagia in HNPP has not previously been reported, but in bovine tomaculous neuropathy it has been attributed to bilateral vagus nerve degeneration, with nerve lesions similar to those seen in tomaculous neuropathy in humans18. We suggest that HNPP should be considered in the differential diagnosis of patients with atypical swallowing dysfunction.
ACKNOWLEDGMENTS - We are grateful to all Phonoaudiologists (Per-Oral Endoscopy Service) for review of the videoendoscopic and fluoroscopic examination. The authors would like to thank Prof. Collins by English review of the manuscript.
Received 25 April 2008, received in final form 1 September 2008. Accepted 17 September 2008.
Dra. Rosana Herminia Scola - Serviço de Doenças Neuromusculares / Hospital de Clínicas da UFPR - Rua General Carneiro 181 / 3º andar - 80060-900 Curitiba PR - Brasil. E-mail: scola@hc.ufpr.br
- 1. Dubourg O, Mouton P, Brice A, LeGuern E, Bouche P. Guidelines for diagnosis of hereditary neuropathy with liability to pressure palsies. Neuromuscul Disord 2000;10:206-208.
- 2. Gouider R, LeGuern E, Gugenheim M, et al. Clinical, electrophysiologic, and molecular correlations in 13 families with hereditary neuropathy with liability to pressure palsies and a chromosome 17p11.2 deletion. Neurology 1995;45:2018-2023.
- 3. Corwin HM, Girardet RE. Hereditary neuropathy with liability to pressure palsies mimicking hypoglossal nerve injuries. Neurology 2003;61:1457-1458.
- 4. Iwasaki Y, Iguchi H, Ikeda K, Kano O. CNS involvement in hereditary neuropathy with pressure palsies. Neurology 2007;68:2046.
- 5. Meier C, Moll C. Hereditary neuropathy with liability to pressure palsies: report of two families and review of the literature. J Neurol 1982;228:73-95.
- 6. Ohkoshi N, Kohno Y, Hayashi A, Wada T, Shoji S. Acute vocal cord paralysis in hereditary neuropathy with liability to pressure palsies. Neurology 2001;56:1415.
- 7. Poloni TE, Merlo IM, Alfonsi E, et al. Facial nerve is liable to pressure. Neurology 1998;51:320-322.
- 8. Winter WC, Juel VC. Hypoglossal neuropathy in hereditary neuropathy with liability to pressure palsy. Neurology 2003;61:1154-1155.
- 9. Meretoja P, Silander K, Kalimo H, Aula P, Meretoja A, Savontaus ML. Epidemiology of hereditary neuropathy with liability to pressure palsies (HNPP) in south western Finland. Neuromuscul Disord 1997;7:529-532.
- 10. Chance P, Alderson M, Leppig K, et al. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell 1993;72:143-151.
- 11. Nodera H, Nishimura M, Logigian EL, Herrmann DN, Kaji R. HNPP due to a novel missense mutation of the PMP22 gene. Neurology 2003;60:1863-1864.
- 12. Infante J, Garcia A, Combarros O, et al. Diagnostic strategy for familial and sporadic cases of neuropathy associated with 17p11.2 deletion. Muscle Nerve 2001;24:1149-1155.
- 13. Mouton P, Tardieu S, Gouider R, et al. Spectrum of clinical and electrophysiologic features in HNPP patients with the 17p11.2 deletion. Neurology 1999;52:1440-1446.
- 14. Li J, Krajewski K, Shy ME, Lewis RA. Hereditary neuropathy with liability to pressure palsy: the electrophysiology fits the name. Neurology 2002;58:1769-1773.
- 15. Madrid R, Bradley WG. The pathology of neuropathies with focal thickening of the myelin sheath (tomaculous neuropathy), studies on the formation of the abnormal myelin sheath. J Neurol Sci 1975;25:415-448.
- 16. Sander S, Ouvrier RA, McLeod JG, Nicholson GA, Pollard JD. Clinical syndromes associated with tomacula or myelin swellings in sural nerve biopsies. J Neurol Neurosurg Psychiatry 2000;68:483-488.
- 17. Domenech E, Kelly J. Swallowing disorders. Med Clin N Am 1999;83:97-113.
- 18. Hill BD, Prior H, Blakemore WF, Black PF. A study of pathology of a bovine primary peripheral myelinopathy with features of tomaculous neuropathy. Acta Neuropathol 1996;91:545-548.
Publication Dates
-
Publication in this collection
09 Dec 2008 -
Date of issue
Dec 2008