LETTER
MMMM syndrome (macrocephaly, megalocornea, motor and mental retardation) and refractory epilepsy
Síndrome MMMM (macrocefalia, megalocórnea, retardo mental e motor) e epilepsia refratária
Adriana Marques de MattosI; Aluisio Dias MarquesI; Carla Patrícia Oliveira ParrelaI; Josephine Marie da Cunha FishI; Osvaldo Alfonso Pinto ArtigalasII; Josiane RanzanIII; Maria Isabel Bragatti WinkclerIII; Lygia OhlweilerIII; Rudimar dos Santos RiesgoIV
IMD, Child Neurologist. Hospital de Clínicas de Porto Alegre, Universidade Federal do Rio Grande do Sul, Porto Alegre RS, Brazil
IIMD, Geneticist. Hospital de Clínicas de Porto Alegre, Universidade Federal do Rio Grande do Sul, Porto Alegre RS, Brazil
IIIPh.D. Child Neurologist, Pediatric Neurology Residency Program Preceptor. Hospital de Clínicas de Porto Alegre, Universidade Federal do Rio Grande do Sul, Porto Alegre RS, Brazil
IVPh.D. Child Neurologist, Adjunct Professor of Pediatrics, Head of Pediatric Neurology Unit. Hospital de Clínicas de Porto Alegre, Universidade Federal do Rio Grande do Sul, Porto Alegre RS, Brazil
Correspondence Correspondence Rudimar dos Santos Riesgo Av. Juca Batista 8000 / 415 90780-070 Porto Alegre RS - Brasil E-mail: rriesgo@hcpa.ufrgs.br
The Neuhauser syndrome was first described in 1975. Three siblings in the same family and four other sporadic patients were found to have severe mental retardation, hypotonia, seizures, megalocornea (cornea diameter >13 mm) and hypoplasic irises, associated with minor dysmorphic findings, such as epicanthal folds, frontal bossing and depressed nasal bridge. Neuhauser suggested a possible recessive autosomal inheritance and called the condition MMR syndrome (megalocornea and mental retardation)1.
Since then, several other cases with similar signs and symptoms were reported in the literature, although some also had different and additional clinical findings2-10. For example, Tominaga et al.11 described hearing impairment; Balci et al.12 reported two cases of Neuhauser syndrome with hypoplasia of corpus callosum; Yarar et al.13 found associated Peter's anomaly; and Margari et al.14 cited a transient hypothyroidism during a five year follow up of one case of Neuhauser syndrome.
In 1990, Frydman et al. described two patients with the MMR syndrome who had macrocephaly, and called this variation of the Neuhauser syndrome as the MMMM (macrocephaly, megalocornea, motor and mental retardation) syndrome5.
In 1991, Kimura et al. described a patient with the MMR syndrome associated with hypothyroidism and myelination delay confirmed by cranial magnetic resonance studies6. Other findings described in the literature are bifid uvula, diffuse cortical atrophy, micrognathia, scoliosis, short stature, microcrania, hypertelorism, and hypotonia2,4,6,7,10.
The heterogeneity of this syndrome led Verloes to suggest 5 subtypes:15
Subtype 1: a recessive form, as described by Neuhauser, with iris hypoplasia and minor anomalies; Subtype 2: a recessive form, as described by Franky-Temtay, with camptodactyly, scoliosis and growth retardation; Subtype 3: a recessive form, with normal irises, severe hypotonia, relative or absolute macrocephaly and other minor anomalies; Subtype 4: a possible Frydman type, with normal irises, macrocephaly and obesity; Subtype 5: provisionally unclassifiable cases.
The objective of this study is to describe a case of Neuhauser-type dysmorphism with refractory epileptic seizures.
CASE
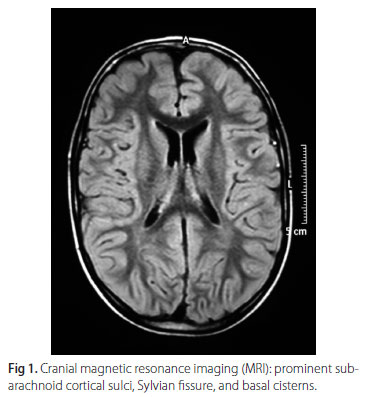
A 5-year-old boy born from non-consanguineous parents had a history of motor and speech development delay, severe mental retardation and marked psychomotor agitation. His motor skills problems were detected since he was eight months old. The clinical picture was compatible with spastic type of cerebral palsy. His electroencephalogram (EEG) showed marked independent bilateral and focal epileptiform discharges in both temporoparietal regions but predominantly in the right hemisphere. Cranial magnetic resonance imaging (MRI) showed prominent subarachnoid cortical sulci, Sylvian fissure, and basal cisterns (Figs 1 and 2).
The patient was taking phenobarbital and had no history of seizures. When discontinuation of phenobarbital was gradually initiated, the patient developed seizures, initially of the tonic type. Carbamazepine was initiated, and seizure control was satisfactory.
Two months later, seizures started again with left sided tonic-clonic limb movements without changes in the level of consciousness. Seizures were controlled only after diazepam and phenytoin were initiated, and patient had no seizures for 24 hours.
Clinical examination revealed severe mental retardation, psychomotor agitation, aggression and self-aggression, little visual contact with examiner, macrocephaly (above 95th percentile), megalocornea, thin and everted lower lip, decreased muscle bulk and tone, symmetrical and adequate muscle strength, and slightly wide-based gait. Preserved symmetrical myotatic reflexes, as well as abdominal and bilateral extensor plantar reflexes, were present. Patient did not react to sound stimuli; pupil diameters were equal, but pupils were poorly reactive to light; eye movements were normal.
Another EEG showed disorganization of base activity with rare sleep spindles, but no asymmetries. Severe multifocal epileptiform discharges were observed over the parietal region of the left hemisphere.
Generalized seizures affecting one side of the body started again; between these seizures, the patient had absence seizures and atonia of the left hemibody. Valproic acid was initiated and carbamazepine was gradually discontinued.
The intensity of seizures was reduced, but they still occurred daily. During gradual increase of valproic acid, the patient had vomits, inappetence, and altered state of consciousness (obnubilation, irritability and aggressiveness).
EEG was repeated and showed a decrease in epileptiform discharges, but was suggestive of diffuse toxic-metabolic encephalopathy. Valproic acid was suspected to be the cause of encephalopathy; therefore, it was discontinued and topiramate was initiated.
After this change in medication, the patient's state of consciousness progressed to normality. Seizure control was achieved with 10 mg/kg/day topiramate.
During hospitalization, several diagnostic exams were performed. Auditory evoked potential revealed bilateral neurosensorial hypoacusis. Ophthalmologic examination confirmed megalocornea; electroretinogram was normal, but visual evoked potential showed reduced visual acuity. Laboratory exams ruled out endocrine abnormalities. An informed consent was obtained with the parents for this publication.
DISCUSSION
The association of mental retardation and megalocornea, first described in 1975, characterizes a syndrome with marked phenotypic heterogeneity. About 36 cases have been reported in the literature, which suggests that it is a rare, non-progressive congenital anomaly associated with dysmorphic traits1,5,9,15.
More recently, other new cases of Neuhauser syndrome have been described (Balci et al., Margari et al., Yarar et al.)12-14, but instead it continues to be a rare clinical situation.
According to Verloes classification, our patient was subtype 4 (Frydman) or MMMM (megalocornea, macrocephaly, mental and motor retardation)5,15. Exams confirmed that he had a megalocornea, severe mental and motor retardation associated with pyramidal-extrapyramidal dysfunction, hypoacusis and macrocephaly, but no endocrine abnormalities. Although epilepsy was also found, it is not one of the diagnostic criteria for this syndrome1,4,15.
In the case reported here, the child had intractable epilepsy, which demonstrates that the syndrome, although not progressive, may be associated with clinical problems that contribute to the clinical deterioration of the patient's health. As far as we know, this is the first report of a brazilian case of Neuhauser syndrome.
Received 30 May 2009
Received in final form 27 August 2009
Accepted 8 September 2009
- 1. Neuhauser G, Kaveggia EG, France TD, Opitz JM. Syndrome of mental retardation, seizures, hypotonic cerebral palsy and megalocorneae, recessively inherited. Z Kinderheilkd 1975;120:1-18.
- 2. Del Giudice E, Sartorio R, Romano A, Carrozzo R, Andria G. Megalocornea and mental retardation syndrome: two new cases. Am J Med Genet 1987;26:417-420.
- 3. Raas-Rothschild A, Berkenstadt M, Goodman RM. Megalocornea and mental retardation syndrome. Am J Med Genet 1988;29:221-223.
- 4. Gronbech-Jensen M. Megalocornea and mental retardation syndrome: a new case. Am J Med Genet 1989;32:468-469.
- 5. Frydman CP, Bleiweiss IJ, Yoo OH, Jagirdar J. Diagnosis of cytomegalovirus infection of the lung in the acquired immune deficiency syndrome (AIDS): by in situ DNA hybridization. Prog AIDS Pathol 1989;1:91-100.
- 6. Kimura M, Kato M, Yoshino K, Ohtani K, Takeshita K. Megolocornea: mental retardation syndrome with delayed myelination. Am J Med Genet 1991;38:132-133.
- 7. Santolaya JM, Grijalbo A, Delgado A, Erdozain G. Additional case of Neuhauser megalocornea and mental retardation syndrome with congenital hypotonia. Am J Med Genet 1992;43:609-611.
- 8. Antinolo G, Rufo M, Borrego S, Morales C. Megalocornea-mental retardation syndrome: an additional case. Am J Med Genet 1994;52:196-197.
- 9. Sarkozy A, Mingarelli R, Brancati F, Dallapiccola B. Primary hypothyroidism and osteopenia associated with Neuhauser syndrome. Am J Med Genet 2002;111:412-414.
- 10. Derbent M, Oto S, Alehan F, et al. Megalocornea-mental retardation (MMR or Neuhauser) syndrome: another case associated with cerebral cortical atrophy and Bifid uvula. Genet Couns 2004;15:477-480.
- 11. Tominaga N, Kondoh T, Kamimura N, et al. A case of megalocornea-mental retardation syndrome complicated with bilateral sensorineural hearing impairment. Pediatr Int 1999;41:392-394.
- 12. Balci S, Teksam O, Gedik S. Megalocornea, macrocephaly, mental and motor retardation: MMMM syndrome (Neuhauser syndrome) in two sisters with hypoplastic corpus callosum. Turk J Pediatr 2002;44:274-277.
- 13. Yarar C, Yakut A, Yildirim N, Yildiz B, Basmak H. Neuhauser syndrome and Peters' anomaly. Clin Dysmorphol 2006;15:249-251.
- 14. Margari L, Presicci A, Ventura P, et al. Megalocornea and mental retardation syndrome: clinical and instrumental follow-up of a case. J Child Neurol 2006;21:893-896.
- 15. Verloes A, Journel H, Elmer C, et al. Heterogeneity versus variability in megalocornea-mental retardation (MMR) syndromes: report of new cases and delineation of 4 probable types. Am J Med Genet 1993;46:132-137.
Publication Dates
-
Publication in this collection
11 Aug 2010 -
Date of issue
Aug 2010