Abstracts
OBJECTIVE: Juvenile Neuronal Ceroid-Lipofuscinosis (JNCL, CLN 3, Batten Disease) (OMIM #204200) belongs to the most common group of neurodegenerative disorders of childhood. We report the clinical data and molecular analysis of a large Brazilian family. METHOD: Family composed of two consanguineous couples and thirty-two children. Clinical data of ten JNCL patients and molecular analyses on 13 participants were obtained. RESULTS: The large 1.02 kb deletion was detected. The most severe phenotype, with autistic behavior, tics and parkinsonism was seen in a 12-year-old female and a milder phenotype in a 14-year-old male. Nyctalopia was the first symptom in one deceased child. The visual loss of six patients has been first observed in the school and not at home. CONCLUSION: The report highlights the phenotypical intrafamily variation in 10 affected children of this family. The molecular investigation of this large family in our metabolic center turned possible the diagnosis, right approach and genetic counseling.
Batten disease; neuronal ceroid-lipofuscinoses; polymerase chain reaction
OBJETIVO: Lipofuscinose Ceróide Neuronal Juvenil (JNCL, CLN 3, Doença de Batten) (OMIM # 204200) pertence ao grupo mais comum de doenças neurodegenerativas na infância. É causada por mutações no gene CLN3, com padrão de herança recessiva. A deleção de 1,02 kb é a mutação mais comum. Relatamos os dados clínicos e análise molecular de uma família consanguínea numerosa. MÉTODO: Família composta por dois casais consanguíneos e trinta e duas crianças. Foram obtidos dados clínicos de dez pacientes e análises moleculares de 13 participantes. RESULTADOS: Foi detectada deleção de 1,02 kb. O fenótipo mais grave, com comportamento autista, tiques e parkinsonismo foi visto em uma paciente do sexo feminino de 12 anos e o fenótipo mais leve em um paciente do sexo masculino de 14 anos. Nictalopia foi o primeiro sintoma de uma criança falecida. A perda visual de seis pacientes foi observada pela primeira vez na escola e não em casa. CONCLUSÃO: Destaca-se a variação fenotípica intrafamiliar em 10 pacientes. A investigação molecular desta família numerosa tornou possível o diagnóstico, a abordagem correta e aconselhamento genético.
doença de Batten; lipofuscinoses ceróides neuronais; reação em cadeia da polimerase
ARTICLE
Juvenile neuronal ceroid-lipofuscinosis: clinical and molecular investigation in a large family in Brazil
Lipofuscinose ceróide neuronal juvenil: investigação clínica e molecular em uma família grande no Brasil
Eugênia Ribeiro ValadaresI, VI; Mayara Xavier PizarroII; Luiz Roberto OliveiraIII, VI; Regina Helena Caldas de AmorimIII, VI; Tarcísio Márcio Magalhães PinheiroIV; Ulrike GriebenV; Helena Hollanda SantosVI; Rachel Rabelo QueirozII; Guilherme de Castro LopesII; Ana Lúcia Brunialti GodardII
IDepartamento de Propedêutica Complementar, Faculdade de Medicina da UFMG, Belo Horizonte MG, Brazil
IIDepartamento de Biologia Geral, Instituto de Ciências Biológicas da UFMG, Belo Horizonte MG, Brazil
IIIDepartamento de Pediatria, Faculdade de Medicina da UFMG, Belo Horizonte MG, Brazil
IVDepartamento de Medicina Preventiva e Social, Faculdade de Medicina da UFMG, Belo Horizonte MG, Brazil
VOtto Heubner-Zentrum für Kinder und Jugendmedizin, SPZ, Neuropädiatrie, Berlin, Germany
VIHospital das Clínicas, Faculdade de Medicina da UFMG, Belo Horizonte MG, Brazil
Correspondence Correspondence: Eugênia Ribeiro Valadares Av. Alfredo Balena 190 30130-100 Belo Horizonte MG - Brasil E-mail: eugenia@medicina.ufmg.br
ABSTRACT
OBJECTIVE: Juvenile Neuronal Ceroid-Lipofuscinosis (JNCL, CLN 3, Batten Disease) (OMIM #204200) belongs to the most common group of neurodegenerative disorders of childhood. We report the clinical data and molecular analysis of a large Brazilian family.
METHOD: Family composed of two consanguineous couples and thirty-two children. Clinical data of ten JNCL patients and molecular analyses on 13 participants were obtained.
RESULTS: The large 1.02 kb deletion was detected. The most severe phenotype, with autistic behavior, tics and parkinsonism was seen in a 12-year-old female and a milder phenotype in a 14-year-old male. Nyctalopia was the first symptom in one deceased child. The visual loss of six patients has been first observed in the school and not at home.
CONCLUSION: The report highlights the phenotypical intrafamily variation in 10 affected children of this family. The molecular investigation of this large family in our metabolic center turned possible the diagnosis, right approach and genetic counseling.
Key words: Batten disease, neuronal ceroid-lipofuscinoses, polymerase chain reaction.
RESUMO
OBJETIVO: Lipofuscinose Ceróide Neuronal Juvenil (JNCL, CLN 3, Doença de Batten) (OMIM # 204200) pertence ao grupo mais comum de doenças neurodegenerativas na infância. É causada por mutações no gene CLN3, com padrão de herança recessiva. A deleção de 1,02 kb é a mutação mais comum. Relatamos os dados clínicos e análise molecular de uma família consanguínea numerosa.
MÉTODO: Família composta por dois casais consanguíneos e trinta e duas crianças. Foram obtidos dados clínicos de dez pacientes e análises moleculares de 13 participantes.
RESULTADOS: Foi detectada deleção de 1,02 kb. O fenótipo mais grave, com comportamento autista, tiques e parkinsonismo foi visto em uma paciente do sexo feminino de 12 anos e o fenótipo mais leve em um paciente do sexo masculino de 14 anos. Nictalopia foi o primeiro sintoma de uma criança falecida. A perda visual de seis pacientes foi observada pela primeira vez na escola e não em casa.
CONCLUSÃO: Destaca-se a variação fenotípica intrafamiliar em 10 pacientes. A investigação molecular desta família numerosa tornou possível o diagnóstico, a abordagem correta e aconselhamento genético.
Palavras-chave: doença de Batten, lipofuscinoses ceróides neuronais, reação em cadeia da polimerase.
The Neuronal Ceroid-Lipofuscinoses (NCL's) are the most common group of neurodegenerative disorders of childhood, characterized by the lysosomal accumulation of autofluorescent material, which resembles ceroid and lipofuscin, in patient's tissues1. This accumulation on neuronal tissue leads to neocortical neurons death2. The global incidence of NCL's is 1 to 8 in 100,000 births3. In the Portuguese population, which is more similar to Brazilians from Minas Gerais State, the prevalence of overall NCL is estimated in 1.55 per 100,000 live births, distributed in CLN3 (42.3%), CLN 2 (11.5%) and CLN 1 (3.8%)4.
The first report of NCL was in 1826 by Stengal, who investigated four siblings presenting progressive blindness, epilepsy, cognitive decline and motor dysfunction. Later, in 1903, Batten reported the juvenile form of the disease (CLN 3)5. Until recently, there had been four main forms of NCL's classified on age of onset and storage material at the ultrastructural level, i.e., infantile (CLN 1), classical late infantile (CLN 2), juvenile (CLN 3), and adult forms (CLN 4). However, due to recent advances in molecular and biochemical analysis, it is considered that the NCL's are classified into ten genetic forms (CLN1-10), only CLN 4 and CLN 9 not yet molecularly characterized (OMIM). The number of NCL forms can still grow, since syndromes with seizures and a variable degree of psychomotor deterioration will probably get in this group of disorders2. Most variants manifest cell death and dysregulated sphingolipid metabolism, suggesting that the proteins defective in these disorders may interact along a common pathway6.
NCLs are autosomal recessive diseases, except for the adult form (CLN 4) that can present both dominant and recessive autosomal patterns of inheritance2. Juvenile-onset Neuronal Ceroid-Lipofuscinoses (JNCL, CLN 3, Batten Disease) (OMIM #204200) is caused by mutations in CLN3 gene. This gene is located at chromosome region 16p12. The gene is constituted by at least 15 exons7 and CLN 3 can be caused by at least 25 different mutations, specially within exons 6 to 8 and 13. A common mutation achieving the rate of 81% worldwide is a deletion of 1.02kb8 that eliminates exons 7 and 8, resulting in a truncated protein9. The CLN3 mutations cause only partial loss of function of this lysosomal enzyme2.
The symptoms in classic or typical JNCL often begin after 4 years of age with progressive visual loss and other neurological dysfunction (dementia; seizures; pyramidal, extrapyramidal, and cerebellar signs). Before the recent progress in molecular genetic studies, diagnosis of NCLs was based on age at onset, clinical course, and pathological findings of fingerprint profiles and/or vacuoles in the lymphocytes by electron microscopy10.
In 27 homozygous patients for the 1.02-kb deletion in Finland, visual failure was noticed at 4-10 years old (100%), onset of epilepsy at 8-13 years old (92%), parkinsonian signs at 12-15 years of age (30%) and at 17-29 years of age (22%); and death occurred between 10 and 28 year11. In this study great clinical variability between different families and also within the families was noticed, and 22% of patients had good motor performance between 14 and 22 years old. Problems related to behavior, emotions and thought have clinically been some of the main features of JNCL patients. The psychiatric symptoms include anxiety, aggressive behavior, depression, hallucinations and psychotic symptoms. Female patients with JNCL have more difficult psychiatric problems than their male counterparts12.
We report the clinical data and molecular analysis of a large consanguineous Brazilian family from Minas Gerais State, highlighting the intrafamilial phenotypical variation in 10 affected children. We also focus in the importance of the molecular analysis to establish the diagnosis and to turn possible an accurate genetic counseling.
METHOD
Studied family
Patients were identified by medical students in formal activity at a poor village in Minas Gerais State. Their family is composed of two consanguineous couples (brother and sister who got married to their first-cousins, also brothers) and their kindred of thirty-two children. The clinical evaluation of patients was performed by the research group in their hometown, without access to major clinical propedeutics other than those already done. We studied the clinical data of ten JNCL patients of this family.
The project was approved by the Ethics Committee on Research of Universidade Federal de Minas Gerais under the number ETIC 337/06, on November 29th (2006). The study was performed in accordance with the ethical standards laid down in the1964 Declaration of Helsinki. The written informed consent was given by all participants; parents gave it for their children.
Molecular analysis
Molecular analyses were performed on 13 participants including 3 parents, 4 symptomatic and 6 asymptomatic children. Samples of 10 mL of peripheral blood were collected from each member of the family (clinically diagnosed or not) totalizing 13 individuals. Next, genomic DNA was extracted and PCR amplification of the exons 6, 7, 8 and 13 were conducted using previously described primers13. Allele-specific PCR amplification was conducted due to obtain differential amplification of a normal DNA fragment (729pb) and a mutated DNA fragment (426pb). The primers and the method were extracted from the literature14. Sequencing was performed to verify if there was any additional mutation in exons 6 and 13 of all patients. Exons 7 and 8 were sequenced in carriers only, using their normal allele, once we are dealing with a large deletion.
RESULTS
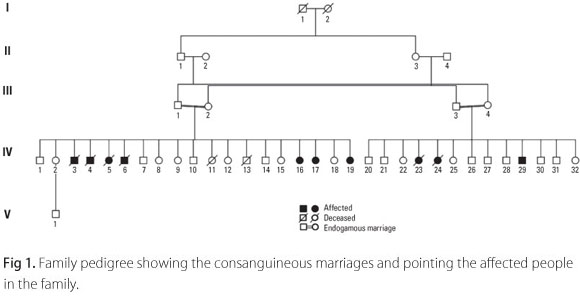
The family is composed by two related couples and their kindred (Fig 1). The first couple is individuals III-1 and III-2, with their kindred of nineteen children. Seven of their sibs were affected by JNCL, four deceased (individuals IV-3, IV-4, IV-5 and IV-6) and three alive (individuals IV-16, IV-17 and IV-19). The second couple is individuals III-3 and III-4 with thirteen children. Three of their sibs were affected by JNCL, only one alive (individual IV-29).
Molecular analyses were performed on 3 parents and 10 children of the family, 4 symptomatic and 6 asymptomatic (III-2, III-3, III-4, IV-12, IV-15, IV-16, IV-17, IV-18, IV-19, IV-29, IV-30, IV-31 e IV-32). The PCR amplification of exons 6, 7, 8 and 13 were obtained from the patients who carried at least one normal allele. The exons 7 and 8 of affected individuals could not be amplified. It suggested a large deletion. Through the allele-specific PCR the molecular diagnostic was obtained (Fig 2). The 1.02kb deletion in both of the mutated alleles was detected in all symptomatic children. All the others analyzed (parents and 6 asymptomatic children) were heterozygote for this mutation. No additional mutation was detected through sequencing.
A total of 10 patients with JNCL were detected in this family, but only the four alive (individuals IV-16, IV-17, IV-19 and IV-29) were seen by the authors and had a molecular diagnosis. Data of the six deceased ones was obtained from parents. The clinical findings from the 10 JNCL patients are shown in Table.
The first symptom in 9 of 10 affected children was visual failure (IV-3, IV-4, IV-5, IV-6, IV-16, IV-19, IV-23, IV-24 e IV-29) and learning disorder in early school years in one (IV-17). Visual loss of 6 patients have been first observed in the school (IV-4, IV-5, IV-16, IV-23, IV-24 e IV-29) and not at home. One child started with nyctalopia (IV-24) as first symptom, different from the others that manifested visual loss both night and day since the beginning. As the disease advanced seven children showed progressive dementia, two temper tantrums and angry outbursts, and autistic behavior emerged in one. Within the group of alive patients, patient IV-17, a 12 year-old female presented the most severe phenotype, with autistic behavior, tics and parkinsonism. Her brain CT was normal. The less severe phenotype was observed in patient IV-19, a 7 year-old female with visual loss since she was six years-old and mild learning disability. The 14 year-old male (patient IV-29) with a milder phenotype, can walk and communicate well.
Eight of nine patients had seizures starting between 8 and 12 years of age; the youngest patient, the 7 year-old female had no seizure until now (IV-19). Death occurred between ages of 11 and 19 years-old (IV-3, IV-4, IV-5, IV-6, IV-23 e IV-24). One blind 12 year-old girl drowned in a river (IV-23).
DISCUSSION
The molecular analysis demonstrated the 1.02kb deletion in the CLN3. The allele-specific PCR gave us a very clear diagnostic. The sequencing was performed only to verify if there were additional mutations and we did not find any of those. As we are dealing with endogamous marriages this result was already expected. The lost part of the gene results in protein truncation. It disables the CLN3 protein capacity of localizing itself in the lysosome9; affecting its correct function and resulting in autofluorescent material storage.
The clinical findings of ten patients, four males and six females, were described. Only four of them are alive and were seen by the authors. Data of the deceased ones were obtained from the parents. The age range of onset of disease was 6 to 9 years-old, in agreement with literature. Death occurred probably related to the natural course of the disease between ages of 11 and 19 years, except for one blind 12-year-old girl who drowned in a river. An intrafamilial phenotypical variation was observed, as expected11.
Nyctalopia was the first symptom in one deceased child (IV-24), different from the others that manifested visual loss both night and day. We have not found in the literature any case of isolated nyctalopia. Vitamin A deficiency as a cause of nyctalopia was not investigated in this patient. Severe visual loss was probably responsible by the chaotic eye movements presented by patient IV-16.
In the group of the four alive patients, the only male patient has a milder phenotype, what might be explained by observations that female patients have more behavior problems than male patients12.
Patient IV-17 presented the most severe phenotype, with autistic behavior, tics and parkinsonism. Learning disorder before visual failure, an unusual finding, can be a predictor of a worsen outcome of the disease, as observed in this patient. It probably indicates a precocious onset of dementia, due to a more rapid degenerating process. In her case a normal brain CT at age of 8 had no correlation with the outcome. On her evaluations equivocal Babinski sign was observed. It was reported that the interobserver reliability and validity of the Babinski sign for identifying upper motor neuron weakness are limited15.
The visual loss of six patients has been first observed in the school and not at home. It can be a sign of negligence of observation by their parents for many reasons: poorness, excessive work, large kindred. The age of onset of visual loss does not seem to be related to the prognosis of the disease in this family.
Brazil is an emergent country, but there are still limitations to realize clinical propedeutics, as brain magnetic resonance imaging (NMR), electroencephalogram (EEG) and electroretinography (ERG), in certain regions. The molecular investigation of this large family in our metabolic center turned possible the diagnosis, right approach and genetic counseling.
Received 22 July 2010
Received in final form 27 July 2010
Accepted 03 August 2010
Support: Research supported by a grant from Fundação de Amparo à Pesquisa do Estado de Minas Gerais - FAPEMIG/SUS 005/2006 - EDT 3261-06
- 1. IEM Digest. The function of CLN3P, the Batten disease protein. Mol Gen Metab 2008;93:269-274.
- 2. Peltonen L, Savukoski M, Vesa J. Genetics of the neuronal ceroid lipofuscinoses. Current Opinion in Gen Dev 2000;10:299-305.
- 3. Kovács AD, Pearce DA. Attenuation of AMPA receptor activity improves motor skills in a mouse model of juvenile Batten disease. Exp Neurol 2008; 209:288-291.
- 4. Teixeira C, Guimarães A, Bessa C, et al. Clinicopathological and molecular characterization of neuronal ceroid-lipofuscinoses in the Portuguese population. J Neurol 2003;250:661-667.
- 5. Wisniewski KE, Kida E, Connell F, Zhong N. Neuronal ceroid lipofuscinoses: research update. Neurol Sci 2000;21(Suppl):S49-S56.
- 6. Persaud-Sawin DA, Mousallem T, Wang C, Zucker A, Kominami E, Boustany RM. Neuronal ceroid-lipofuscinoses: a common pathway? Pediatr Res 2007; 61:146-152.
- 7. Mitchison HM, Munroe PB, O'Rawe AM, et al. Genomic structure and complete nucleotide sequence of the batten disease gene, CLN3. Genomics 1997;40:346-350.
- 8. Järvelä I, Mitchison HM, Munroe PB, O'Rawe AM, Mole SE, Syvanen AC. Rapid diagnostic test for the major mutation underlying Batten disease. J Med Genet 1996;33:1041-1042.
- 9. Kwon JM, Rothberg PG, Leman AR, Weimer JM, Mink JW, Pearce DA. Novel CLN3 mutation predicted to cause complete loss of protein function does not modify the classical JNCL phenotype. Neuroscience Letters 2005; 387:111-114.
- 10. Wisniewski KE, Kaczmarski A, Kida E, et al. Reevaluation of neuronal ceroid lipofuscinoses: atypical juvenile onset may be the result of CLN2 mutations. Mol Gen Metab 1999;66:248-252.
- 11. Järvelä I, Autti T, Lamminranta S, Aberg L, Raininko R, Santavuori P. Clinical and magnetic resonance imaging findings in Batten disease: analysis of the major mutation (1.02-kb deletion). An Neurol 1997;42:799-802.
- 12. Bäckman ML, Santavuori PR, Åberg LE, Aronen ET. Psychiatric symptoms of children and adolescents with juvenile neuronal ceroid-lipofuscinoses. J Intellec Disab Res 2005;49:25-32.
- 13. Munroe B, Mitchison HM, O'Rawe AM, et al. Spectrum of mutations in the Batten disease gene, CLN3. Am J Hum Genet 1997;61:310-316.
- 14. Taschner PEM, Vos N, Breuning MH. Rapid detection of the major deletion in the Batten disease gene CLN3 by allele specific PCR. J Med Genet 1997; 34:955-956.
- 15. Miller TM, Johnston C. Should the Babinski sign be part of the routine neurologic examination? Neurology 2005;65:1165-1168.
Publication Dates
-
Publication in this collection
18 Feb 2011 -
Date of issue
Feb 2011
History
-
Received
22 July 2010 -
Accepted
27 July 2010