Abstracts
Acquired hepatocerebral degeneration (AHD) and hepatolenticular degeneration can have similar clinical presentations, but when a chronic liver disease and atypical motor findings coexist, the distinction between AHD and hepatic encephalopathy (HE) can be even more complicated. We describe three cases of AHD (two having HE) with different neuroimaging findings, distinct hepatic diseases and similar motor presentations, all presenting chronic arterial hypertension and weight loss before the disease manifestations. The diagnosis and physiopathology are commented upon and compared with previous reports. In conclusion, there are many correlations among HE, hepatolenticular degeneration and AHD, but the overlapping of AHD and HE could be more common depending on the clinical knowledge and diagnostic criteria adopted for each condition. Since AHD is not considered a priority that affects the liver transplant list, the prognosis in AHD patients remains poor, and flow interruption in portosystemic shunts must always be taken into account.
athetosis; chorea; dyskinesias; dystonia; hepatic encephalopathy; hepatolenticular degeneration; hyperkinesis; movement disorders
A degeneração hepatocerebral adquirida (AHD) e a degeneração hepatolenticular podem ter apresentações clínicas semelhantes, mas quando uma doença hepática crônica e achados motores atípicos coexistem, a distinção entre AHD e encefalopatia hepática (HE) pode ser ainda mais complicada. Descrevemos três casos de AHD (dois tendo HE) com diferentes achados em neuroimagem, doenças hepáticas distintas e apresentações motoras semelhantes, todos com hipertensão arterial e perda de peso antes das manifestações motoras. O diagnóstico e a fisiopatologia são comentados e comparados com relatos prévios. Concluímos que existem muitas correlações entre HE, degeneração hepatolenticular e AHD, mas a sobreposição de HE e AHD pode ser mais comum dependendo do conhecimento clínico e da acurácia dos critérios diagnósticos adotados para cada enfermidade. Como a AHD não é considerada prioridade na lista de transplante hepático, o prognóstico dos pacientes com AHD permanece ruim, e a interrupção do fluxo nos shunts portossistêmicos deve ser sempre considerada.
atetose; coréia; degeneração hepatolenticular; discinesias; distonia; distúrbios do movimento; encefalopatia hepática; hipercinesia
ARTICLE
Acquired hepatocerebral degeneration and hepatic encephalopathy: correlations and variety of clinical presentations in overt and subclinical liver disease
Degeneração hepatocerebral adquirida e encefalopatia hepática: correlações e variedade de apresentações clínicas na doença hepática evidente e subclínica
Fernando G. RomeiroI; Madileine F. AméricoII; Fábio S. YamashiroI; Carlos A. CaramoriI; Arthur O. SchelpIII; Antonio C. SantosIV; Giovanni F. SilvaI
IDepartamento de Clínica Médica, Faculdade de Medicina de Botucatu, Universidade Estadual Paulista (UNESP), Botucatu SP, Brazil
IIInstituto de Ciências Biológicas e da Saúde, Campus Médio Araguaia, Universidade Federal do Mato Grosso (UFMT), Cuiabá MT, Brazil
IIIDepartamento de Neurologia, Psicologia e Psiquiatria, Faculdade de Medicina de Botucatu, Universidade Estadual Paulista (UNESP), Botucatu SP, Brazil
IVDepartamento de Clínica Médica, Divisão de Radiologia, Faculdade de Ribeirão Preto, USP, Ribeirão Preto SP, Brazil
Correspondence Correspondence: Fernando Gomes Romeiro Universidade Estadual Paulista (UNESP) 18618-970 Botucatu SP - Brasil E-mail: fgromeiro@fmb.unesp.br
ABSTRACT
Acquired hepatocerebral degeneration (AHD) and hepatolenticular degeneration can have similar clinical presentations, but when a chronic liver disease and atypical motor findings coexist, the distinction between AHD and hepatic encephalopathy (HE) can be even more complicated. We describe three cases of AHD (two having HE) with different neuroimaging findings, distinct hepatic diseases and similar motor presentations, all presenting chronic arterial hypertension and weight loss before the disease manifestations. The diagnosis and physiopathology are commented upon and compared with previous reports. In conclusion, there are many correlations among HE, hepatolenticular degeneration and AHD, but the overlapping of AHD and HE could be more common depending on the clinical knowledge and diagnostic criteria adopted for each condition. Since AHD is not considered a priority that affects the liver transplant list, the prognosis in AHD patients remains poor, and flow interruption in portosystemic shunts must always be taken into account.
Key words: athetosis, chorea, dyskinesias, dystonia, hepatic encephalopathy, hepatolenticular degeneration, hyperkinesis, movement disorders.
RESUMO
A degeneração hepatocerebral adquirida (AHD) e a degeneração hepatolenticular podem ter apresentações clínicas semelhantes, mas quando uma doença hepática crônica e achados motores atípicos coexistem, a distinção entre AHD e encefalopatia hepática (HE) pode ser ainda mais complicada. Descrevemos três casos de AHD (dois tendo HE) com diferentes achados em neuroimagem, doenças hepáticas distintas e apresentações motoras semelhantes, todos com hipertensão arterial e perda de peso antes das manifestações motoras. O diagnóstico e a fisiopatologia são comentados e comparados com relatos prévios. Concluímos que existem muitas correlações entre HE, degeneração hepatolenticular e AHD, mas a sobreposição de HE e AHD pode ser mais comum dependendo do conhecimento clínico e da acurácia dos critérios diagnósticos adotados para cada enfermidade. Como a AHD não é considerada prioridade na lista de transplante hepático, o prognóstico dos pacientes com AHD permanece ruim, e a interrupção do fluxo nos shunts portossistêmicos deve ser sempre considerada.
Palavras-chave: atetose, coréia, degeneração hepatolenticular, discinesias, distonia, distúrbios do movimento, encefalopatia hepática, hipercinesia.
The neuroimaging and pathological distinctions between chronic acquired hepatocerebral degeneration (AHD) and genetic hepatolenticular degeneration (Wilson's disease) are well elucidated and applied worldwide1-5. In the inherited form there is a metabolic dysfunction which leads to copper deposition in basal ganglia. Consequently, the disease causes parkinsonism, dystonia, and abnormal movements that include athetosis and chorea2,4. In the acquired form, akinetic-rigid symptoms were prevalent6,7. Even so, the disease displays a wide spectrum of symptoms, and it is not rare that patients present other signs and symptoms such as choreoathetosis and dystonia8,9. In AHD physiopathology, manganese deposition in basal nuclei appears to be a key factor10-12.
Hepatic encephalopathy (HE) is a more comprehensive term and is characterized by neuropsychiatric symptoms as a complication of a liver disease. The term is usually used in the presence of hepatic insufficiency or portosystemic shunts, when the central nervous system shows no signs of disease in current neuroimaging exams. Nevertheless, encephalic abnormalities may already have been found, including primary astrocytopathy with type 2 Alzheimer cells in a wide range of brain structures in patients who died from HE13,14. The same cells are also encountered in the brain of patients diagnosed with AHD15,16, showing a common neurological finding in these two situations.
Thus, the question of whether of HE is completely reversible is controversial, and some neurological deficits may persist17,18. However, until now there is no consensus about whether repeated HE episodes constitute a key factor in AHD development. Some reports did not find any relationship between HE and AHD6,8. However, other studies showed a direct relationship between the number of episodes and the probability of AHD occurrence19,20, and some authors prefer the diagnosis of chronic hepatic encephalopathy for patients with a chronic liver disease and long-term neurological signs and symptoms and without apparent signs of manganese deposition in the brain21.
In this paper we describe three AHD cases in which chorea was the prominent sign. All three manifested severe weight loss prior to chorea, and two had persistent HE. Correlations between clinical findings, neuroimaging studies and liver disease severity were discussed. The prognosis and treatment options are briefly described and analyzed.
METHOD
Written informed consent to participate in the study was obtained according to the Declaration of Helsinki. Obtained neuroimages and all other determinations were part of routine examination of patients with neurological symptoms associated with suspected hepatic disease.
Case 1
A 49-year-old man came to the hospital in June 2004 with cirrhosis and chronic arterial hypertension, and reported his consumption of at least 200 ml of brandy (64 g of alcohol) per day for 26 years. He had tense ascites, pronounced edema of both legs and inconstant flapping tremor. Clinical and laboratory tests were compatible with the cirrhosis Child-Pugh score of 12 (Class C). He underwent three additional episodes of overt HE in 2005, when abdominal ultrasonography showed cirrhotic liver, splenomegaly, recanalization of the umbilical vein and collateral veins in the abdomen. Until this time he had shown clear recovery from hepatic encephalopathy and ceased taking lactulose and diuretics, but continued to present chronic somnolence, an attention deficit, impaired concentration, and postural hand tremor. He stopped consuming alcohol and reported substantial weight loss within the year (10 Kg).
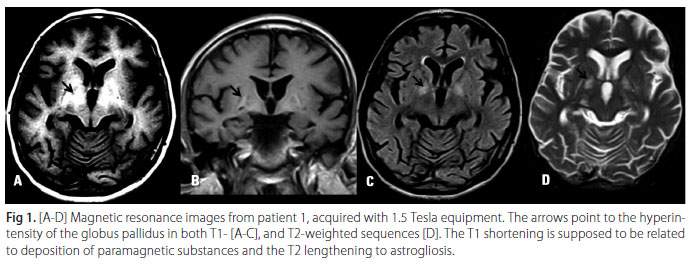
He was readmitted in July 2006 due to abruptly impaired cognitive function associated with involuntary movements. His liver cirrhosis received a Child-Pugh score of 9 (Class B) but he did not present ascites. He had mild consciousness impairment associated with symmetrical ballistic and choreoathetoid movements, prominent orobuccolingual (OBL) dyskinesia and dysarthric speech. Urinary copper and serum ceruloplasmin were normal and slit lamp examination did not show Kayser-Fleischer rings. Cranial Magnetic Resonance Imaging (MRI) showed T1 weighted symmetrical hyperintense globus pallidus and putamen (Fig 1). Haloperidol 2 mg/day and lactulose 20 ml q.i.d. produced a good initial recovery, but did not avoid symptom recurrences. Hepatic transplantation was indicated but his model for end-stage liver disease (MELD) score was still very low. Tremors and dysarthria remained and orobuccolingual dyskinesia became progressively unmanageable. At that time he had clear difficulty swallowing. Since the patient had manifested previous esophageal variceal bleeding, collateral vein embolization was contraindicated.
The patient's motor disturbances were seen only during the progressively rarer moments he was awake. He developed pneumonia, renal insufficiency and pressure ulcers and died after a septic shock episode in December 2007.
Case 2
The second patient is a 56-year-old man who consumes on average 2 liters of brandy (about 320 g of alcohol) daily and tells of alcohol abuse since the age of eight. In March 2009 he underwent neurological treatment for a 4-year history of chorea that initially affected the left hand and more recently the arms, face and trunk, causing a walking disability. He had chronic osteomyelitis and during the treatment suffered a chronic weight loss during the last years (6 Kg). He exhibited vascular spiders, a palpable spleen and an increased liver consistency that led to a diagnostic hypothesis of portal hypertension and cirrhosis. He showed bilateral choreoathetosis including abnormal movements in the face. Endoscopy was used to look for other manifestations of portal hypertension, and portal hypertensive gastropathy was encountered. Abdominal ultrasonography did not reveal portosystemic shunts. Liver biopsy showed no signs of cirrhosis. Cranial MRI was performed using standard axial, sagittal, and coronal spin-echo. T1 and T2 sequence images showed no signs of metal deposits, but did reveal abnormalities in the mesencephalic tegumentum with hypersignal on T2-weighted images (Fig 2). The patient improved after the introduction of haloperidol 2 mg/day and cessation of alcohol consumption.
Case 3
The third patient is a 63-year-old woman who had arterial hypertension, obesity, and diabetes, but never consumed alcohol. She was hospitalized in August 2009 because of a severe HE episode. According to her daughter, she had weakness in both legs five years ago, and involuntary movements of the tongue, neck, arms, and legs in the last eighteen months. After many falling episodes in this period, she has not been able to walk since February 2009. In the same year she began to have gagging episodes associated with feeding, without complaints of dysphagia, and had marked weight loss, which was not quantified because of her difficulty in standing.
Abdominal ultrasonography showed many dilated and tortuous vessels around the left gastric vein, signs of chronic hepatic disease and splenomegaly, but no ascites. Slit lamp examination did not show Kayser-Fleischer rings. Liver biopsy confirmed the diagnosis of cirrhosis and autoimmune hepatitis. Clinical and laboratory data revealed a Child-Pugh score of 8 (Class B). Serum and urinary copper were normal, but serum ammonia was very high (193 µmol/ml). MRI showed no signs of metal deposits or any other specific abnormalities. The patient improved with haloperidol 1 mg/day and lactulose 20 ml q.i.d, but symptoms later reappeared despite the medications.
DISCUSSION
In 1965, Victor et al.20 described the clinical and pathological aspects of acquired hepatocerebral degeneration (AHD) and proposed its universally adopted name. Like other previous works, the study did not find any changes in copper metabolism and linked the symptoms to the blood ammonia levels. Today there appears to be no question that manganese accumulation, among other less evident disturbances, plays a central role in the physiopathology of AHD10-12,19.
Analysis of 1.5 T and 3.0 T nuclear magnetic resonance images (MRI) of our patients showed three distinct patterns. In patient 1, T1-weighted images showed
increased signal in the globus pallidus and putamen, and T2 images reveal increased signal in the globus pallidus. This finding is considered a marker for AHD with manganese deposition in basal nuclei10. The hyperintensity in basal ganglia T1 images is evidence of paramagnetic substance deposit associated with astrocyte organelle proliferation22, associated with lengthening in T2 depending on the increase of fluid linked to gliosis. Complementary studies have shown that despite elevated copper in different areas of the brain, it is manganese that is deposited in the globus pallidus and putamen at levels up to seven times higher than controls in hepatic-insufficiency patients without neurological signs23, and the same finding was observed in those presenting neurological risk10. Other authors showed that although symmetric hyperintense globus pallidus in a T1- weighted cranial MRI in patients with cirrhosis precedes hepatocerebral disease, there is no correlation with clinical symptoms19, and that shortening the T1 relaxation time in MRI (caused by the paramagnetic properties of deposited substances) does not reflect the amount of accumulated cerebral manganese24.
The second case did not show the characteristic radiological T1 hyperintensity that suggests manganese deposition, but clinical manifestations were very similar to cases 1 and 3. Although MRI images showed no abnormalities in the globus pallidus neuroimaging, there were no other reasons for the involuntary movements and the patient had clear signs of portal hypertension. There were reports of AHD without MRI signs of manganese deposition in patients with chronic hepatic disease, moderate portal fibrosis, and canalicular proliferation25,26. Similarly to our case in which MRI shows T1 hypointensity and T2 hyperintensity signal in mesencephalus but also in subcortical and deep white matter, these authors found symmetrical hypointense T1 and hyperintense T2 images in cerebelar peduncle, subcortical and deep white matter25. The pathological correlations in these previous reports demonstrate the presence of microscopic spongy degeneration with numerous Alzheimer type II astrocytes in basal ganglia. The same findings were seen in patients suffering from cirrhosis with MRI exams showing hyperintense globus pallidus in T1 MRI21. Of interest, our non-cirrhotic patient has an apparently normal globus pallidus in MRI. Another finding of Saporta et al.25 that was highlighted in our patients is marked weight loss, which could be the source of toxins release from adipose tissue. Similarly, our patients still had chronic arterial hypertension, but this disease has no clear correlation with the AHD physiopathology. These findings should be described in future reports on AHD to allow a more detailed discussion about the disease's pathogenesis.
Our two cirrhotic patients had marked portal hypertension and clear portosystemic shunts in Doppler ultrasound exam, while the non-cirrhotic patient had only signs of portal hypertension and no evidence of cirrhosis in an adequate liver biopsy. According to this finding and the previous studies, we infer that neural lesions caused by AHD and HE have an almost independent course, and the absence of cirrhosis could explain the lack of HE in the presentation of our second patient. If he returns to alcohol consumption and/or develops liver cirrhosis, probably some signs of HE would appear. Additionally, if every patient with HE could be monitored by high-quality MRI exams, any neurological effect of a hypothetical trigger such as weight loss, arterial hypertension or new portosystemic shunts could be adequately documented.
Both HE and AHD share a common etiopathogenic finding, namely the presence of portosystemic shunts27, but in our second patient we still did not find any signal of portal shunting, even in the presence of portal hypertension. Given that the portosystemic shunts appear to be the first step in the development of type II Alzheimer's cells in both HE and AHD13-15,27, we could hypothesize that this patient had pathologic shunts still not visualized by the Doppler exam at that time.
The third patient shows the classical clinical picture of AHD, with portosystemic shunts and cirrhosis. Her MRI exams did not show T1 hyperintensity, but revealed some findings compatible with spongy degeneration associated with hyperintensity in T2 and Flair. As in the first patient, her clinical presentation was punctuated by severe episodes of HE, which underwent incomplete remission after ammonia reduction treatment.
Previous studies registered the predominance of hypokinetic forms with bradykinesia and rigidity in AHD6,7. Even so, there are many other reports in which chorea emerged as the prominent manifestation. Choreoathetosis is almost invariably present in the face, affecting the tongue, eyes and lips, with the appearance of so called grimacing3,16,20. Nonetheless, even patients with different clinical presentations have an invariable association with direct or indirect signs of portocaval shunts9,11,28,29.
The spectrum of symptoms can be similar in some patients when AHD and HE coexist, but the pathogenesis of both conditions are complex and not fully understood6,7,8,15,30. Given that the AHD physiopathology is not only associated with manganese deposition31, complementary studies are needed to identify other metabolic abnormalities which could elucidate the causes of clinical and neuroimaging variations. According to the previous studies and a detailed analysis of our patients, we recognize a multifactorial physiopathology with a wide range of clinical presentations not always correlated with HE but certainly associated with portal hypertension and different degrees of portosystemic shunts, not always observed in Doppler ultrasonography.
Clinical diagnosis in patients with chronic hepatocerebral degeneration is often difficult to achieve due to the simultaneous symptoms related to HE and findings linked to the chronic form, with more extensive cerebral lesions. The three cases presented herein have extrapyramidal symptoms affecting the face and tongue and were well-controlled initially by means of neuroleptics. We seek to emphasize the multiple pathogenic factors associated with hepatic impairment and the different imaging findings obtained during the etiological investigation.
Both cirrhotic patients presented in the current report had lower MELD scores and visible portosystemic shunts in Doppler exams. This morbid association is a paradox in the prognostic factors associated with cirrhosis, in which a serious complication of hepatic disease has no priority in the liver transplant list because AHD is not considered a special situation. A recent report showed a patient with Parkinsonism and dementia associated with MRI hypersignal in T1, which was interpreted as evidence of encephalic manganese deposition. The authors considered this finding as the determinant factor for indicating a liver transplant32. The indication in this setting is reasonable, since about 20% of patients selected for liver transplant have extrapyramidal signs.
Despite the severity of any neurological impairment caused by liver diseases, a difficult task in these patients is to distinguish HE from other forms of hepatocerebral disease when neuroimaging findings are not typical of AHD or hepatolenticular degeneration. In a recent review, Ferrara and Jankovic3 assumed that movement and cognitive disorders have distinct causes in cirrhosis. Thus, in AHD there is no consciousness impairment and poor response to ammonia reduction, in contrast to what occurs in HE. Therefore, in addition to a careful radiological exam, repeated evaluations of the consciousness level are still necessary to make this differentiation. However, the overlapping of AHD and HE is possible, and despite the fact that each condition requires customized management, interrupting the flow in the portosystemic shunts must always be considered when a short-term liver transplant is not feasible. With this strategy we probably could reduce the neurological deficits seen in the post-transplant patien
Received 2 October 2010
Received in final form 31 January 2011
Accepted 7 February 2011
- 1. Durand F. Wilson's disease. Eur J Gastroenterol Hepatol 2007;19:97-99.
- 2. Brewer GJ. Neurological presenting Wilson's disease. CNS Drugs 2005;19: 185-192.
- 3. Ferrara J, Jankovic J. Acquired hepatocerebral degeneration. J Neurol 2009; 256:320-332.
- 4. Taly AB, Meenakshi-Sundaram S, Sinha S, Swamy HS, Arunodaya GR. Wilson disease description of 282 patients evaluated over 3 decades. Medicine 2007;82:112-121.
- 5. Machado A, Chien HF, Deguti MM, et al. Neurological manifestations in Wilson's disease: report of 119 cases. Mov Disord 2006;21:2192-2196 .
- 6. Burkhard PR, Delavelle J, Du Pasquier R, Spahr L. Chronic parkinsonism associated with cirrhosis: a distinct subset of acquired hepatocerebral degeneration. Arch Neurol 2003;60:521-528.
- 7. Klos KJ, Ahlskog JE, Josephs KA, Fealey RD, Cowl CT, Kumar N. Neurologic spectrum of chronic liver failure and basal ganglia T1 hyperintensity on magnetic resonance imaging. Arch Neurology 2005;62:1385-1390.
- 8. Toghill PJ, Johnston AW, Smith JF. Choreoathetosis in Porto-systemic encephalopathy. J Neurol Neurosurg Psychiatry 1967;30:358-363.
- 9. Jog MS, Lang AE. Chronic acquired hepatocerebral degeneration: case reports and new insights. Mov Disord 1995;6:714-722.
- 10. Klos KJ, Ahlskog J E, Kumar N, et al. Brain metal concentrations in chronic liver failure patients with pallidal T1 MRI hyperintensity. Neurology 2006; 67:1984-1989.
- 11. Condat B, Dusoleil A, Bernardeau M, Roche A, Pelletier G, Buffet C. Chronic acquired hepatocerebral degeneration: the role of manganese and treatment by endovascular occlusion of a porto-cava shunt. Gastroenterol Clin Biol 1999;23:268-270.
- 12. Butterworth RF. Metal toxity, liver disease and neurodegeneration. Neurotox Res 2010;18:100-105.
- 13. Butterworth RF. Neuronal death in hepatic encephalopathy. Metab Brain Dis 2007;22:309-320.
- 14. Thornberry DS, Itabashi HH. Pontine spongy degeneration of white matter associated with hepatic encephalopathy. Arch Pathol Lab Med 1984; 108:564-566.
- 15. Finlayson MH, Superville B. Distribution of cerebral lesions in acquired hepatocerebral degeneration. Brain 1981;104:79-95.
- 16. Lee J, Lacomis D, Comu S, Jacobsohn J, Kanal E. Acquired hepatocerebral degeneration: MR and pathological findings. AJNR Am J Neuroradiol 1998; 19:485-487.
- 17. Lewis MB, Howdle PD. Neurologic complications of liver transplantation in adults. Neurology 2003;61:1174-1178.
- 18. Wijdicks EF, Wiesner RH. Acquired (non-Wilsonian) hepatocerebral degeneration: complex management decisions. Liver Transpl 2003;9:993-994.
- 19. Krieger S, Jauss M, Jansen O, et al. MRI findings in chronic encephalophaty depend on portosystemic shunt: results of a controlled prospective clinical Investigation 1997;27:121-126.
- 20. Victor M, Adams D, Cole M. The acquired (non-Wilsonian) type of chronic hepatocerebral degeneration. Medicine 1965;44:345-396.
- 21. Matsusue E, Kinoshita T, Ohama E, Ogawa T. Cerebral cortical and white matter lesions in chronic hepatic encephalopathy: MR-pathologic correlations. AJNR Am J Neuroradiol 2005;26:347-351.
- 22. Brunberg JA, Kanal E, Hirsch W, VanThiel DH. Chronic acquired hepatic failure: MR imaging of the brain at 1,5 T. AJNR Am J Neuroradiol 1991; 12:909-914.
- 23. Awada A, Sullivan S, Palkar V, Sbeih F, Naufal R, Al Rajeh S. Brain ressonance imaging in non-alcoholic cirrhosis. Eur J Radiol 1995;21:84-88.
- 24. Newland MC, Ceckler TL, Kordower JH, Weiss B. Visualizing manganese in the primate basal ganglia with magnetic ressonance imaging. Exp Neurol 1989;106:251-258.
- 25. Saporta MAC, André C, Bahia PRV, et al. Acquired hepatocerebral degeneration without liver disease. Neurology 2004;63:1981-1982.
- 26. Lee J, Lacomis D, Comu S, et al. Acquired hepatocerebral degeneration: MR and pathologic findings. Am J Neuroradiol 1998;19:485-487.
- 27. Ropper AH, Samuels MA. Chronic acquired (nonWilsonian) hepatocerebral degeneration. In: Adams, Victor's (Eds). Principles of Neurology. Ninth Edition. New York: McGraw Hill; 2009:1099-1100.
- 28. Gerard JM, Vanderhaeghen JJ, Telerman-Toppet N, Cöers C. Choreoathetosis with hepato-cerebral degeneration in a patient with porto-caval shunt. Acta Neurol Belg 1973;73:100-109.
- 29. Spitareli DL, Vitolo S, Fasanaro AM, Valiani R. Choreoathetosis. Uncommon manifestation during chronic liver disease with porto-caval shunt. Rev Neurol 1983;53:293-299.
- 30. Khokhar N, Ahmad A, Batt M M. Acquired hepatocerebral degeneration in hepatitis C infection. J Coll Physicians Surg Pak 2005;15:110-111.
- 31. Krieger D, Krieger S, Jansen O, Gass P, Theilman L, Lichtnecker H. Manganese Chron Encephalophaty 1995;346:270-274.
- 32. Fabiani G, Rogacheski E, Wiederkehr JC, Khouri J, Cianfarano A. Liver transplantation in a patient with rapid onset parkinsonism-Dementia complex induced by manganism secondary to liver failure. Arq Neuroqsiquiatr 2007; 65: 685-688.
Publication Dates
-
Publication in this collection
19 July 2012 -
Date of issue
June 2011
History
-
Reviewed
31 Jan 2011 -
Received
02 Oct 2010 -
Accepted
07 Feb 2011