Abstracts
X-linked adrenoleukodystrophy (X-ALD) is a recessive X-linked disorder associated with marked phenotypic variability. Female carriers are commonly thought to be normal or only mildly affected, but their disease still needs to be better described and systematized. OBJECTIVES: To review and systematize the clinical features of heterozygous women followed in a Neurogenetics Clinic. METHODS: We reviewed the clinical, biochemical, and neuroradiological data of all women known to have X-ADL. RESULTS: The nine women identified were classified into three groups: with severe and aggressive diseases; with slowly progressive, spastic paraplegia; and with mildly decreased vibratory sensation, brisk reflexes, and no complaints. Many of these women did not have a known family history of X-ALD. CONCLUSIONS: Heterozygous women with X-ADL have a wide spectrum of clinical manifestations, ranging from mild to severe phenotypes.
adrenoleukodystrophy; X-linked disorder; heterozygous carriers
A adrenoleucodistrofia ligada ao X (ADL-X) é uma doença recessiva ligada ao X, associada à grande variabilidade clínica. Mulheres heterozigotas portadoras do gene causador da doença são consideradas, tradicionalmente, como clinicamente normais ou com fenótipo clínico muito discreto. No entanto, a apresentação clínica deste grupo necessita ser melhor caracterizada e sistematizada. OBJETIVOS: Revisar e sistematizar as principais características clínicas de mulheres heterozigotas para ADL-X, seguidas em serviço de neurogenética. MÉTODOS: Foram revisados os principais achados clínicos, bioquímicos e neurorradiológicos das mulheres seguidas no serviço com o diagnóstico bioquímico de ADL-X. RESULTADOS: Nove mulheres foram identificadas e classificadas em três grupos: com doença grave e incapacitante; com evolução mais insidiosa e sintomas de paraparesia espástica; e com sintomas discretos apresentando diminuição da sensibilidade vibratória, reflexos vivos, mas sem queixas clínicas. A maioria dessas mulheres não possuía história familiar positiva para ALD-X. CONCLUSÕES: Mulheres heterozigotas para ALD-X apresentam um amplo espectro de manifestações clínicas, variando desde um fenótipo leve, subclínico até apresentações graves e incapacitantes.
adrenoleucodistrofia; doença recessiva ligada ao X; portadoras heterozigotas
ARTICLE
X-linked adrenoleukodystrophy in heterozygous female patients: women are not just carriers
Adrenoleucodistrofia ligada ao X em pacientes femininas heterozigotas: mulheres não são meras portadoras
Charles Marques LourençoI; Gustavo Novelino SimãoII; Antonio Carlos SantosIII; Wilson Marques JrIV
INeurogenetics Division, Clinics Hospital, School of Medicine of Ribeirão Preto, University of São Paulo, Ribeirão Preto SP, Brazil
IIDepartment of Internal Medicine, School of Medicine of Ribeirão Preto, University of São Paulo, Ribeirão Preto SP, Brazil
IIIDepartment of Neurosciences and Behavior Sciences, School of Medicine of Ribeirão Preto, University of São Paulo, Ribeirão Preto SP, Brazil
IVNeurogenetics Division, Clinics Hospital; Department of Neurosciences and Behavior Sciences, School of Medicine of Ribeirão Preto, University of São Paulo; National Science and Technology Institute (INCT) for Translational Medicine, Ribeirão Preto SP, Brazil
Correspondence Correspondence: Wilson Marques Jr. Departamento de Neurociências e Ciências do Comportamento Faculdade de Medicina de Ribeirão Preto - USP Avenida Bandeirantes 3.900 14049-900 Ribeirão Preto SP - Brasil E-mail: wmjunior@fmrp.usp.br
ABSTRACT
X-linked adrenoleukodystrophy (X-ALD) is a recessive X-linked disorder associated with marked phenotypic variability. Female carriers are commonly thought to be normal or only mildly affected, but their disease still needs to be better described and systematized.
OBJECTIVES: To review and systematize the clinical features of heterozygous women followed in a Neurogenetics Clinic.
METHODS: We reviewed the clinical, biochemical, and neuroradiological data of all women known to have X-ADL.
RESULTS: The nine women identified were classified into three groups: with severe and aggressive diseases; with slowly progressive, spastic paraplegia; and with mildly decreased vibratory sensation, brisk reflexes, and no complaints. Many of these women did not have a known family history of X-ALD.
CONCLUSIONS: Heterozygous women with X-ADL have a wide spectrum of clinical manifestations, ranging from mild to severe phenotypes.
Key words: adrenoleukodystrophy, X-linked disorder, heterozygous carriers.
RESUMO
A adrenoleucodistrofia ligada ao X (ADL-X) é uma doença recessiva ligada ao X, associada à grande variabilidade clínica. Mulheres heterozigotas portadoras do gene causador da doença são consideradas, tradicionalmente, como clinicamente normais ou com fenótipo clínico muito discreto. No entanto, a apresentação clínica deste grupo necessita ser melhor caracterizada e sistematizada.
OBJETIVOS: Revisar e sistematizar as principais características clínicas de mulheres heterozigotas para ADL-X, seguidas em serviço de neurogenética.
MÉTODOS: Foram revisados os principais achados clínicos, bioquímicos e neurorradiológicos das mulheres seguidas no serviço com o diagnóstico bioquímico de ADL-X.
RESULTADOS: Nove mulheres foram identificadas e classificadas em três grupos: com doença grave e incapacitante; com evolução mais insidiosa e sintomas de paraparesia espástica; e com sintomas discretos apresentando diminuição da sensibilidade vibratória, reflexos vivos, mas sem queixas clínicas. A maioria dessas mulheres não possuía história familiar positiva para ALD-X.
CONCLUSÕES: Mulheres heterozigotas para ALD-X apresentam um amplo espectro de manifestações clínicas, variando desde um fenótipo leve, subclínico até apresentações graves e incapacitantes.
Palavras-Chave: adrenoleucodistrofia, doença recessiva ligada ao X, portadoras heterozigotas.
X-linked adrenoleukodystrophy (X-ALD) is an X-linked recessive disorder that affects the brain white matter and that is associated with adrenal insufficiency1. The disease most commonly affects boys during early childhood and initially presents with either adrenal insufficiency or a variety of neurological manifestations, such as hyperkinetic behavior, neurological regression, sensorineural deafness and optic atrophy, although most patients eventually develop the whole neurological picture. As white matter demyelination relentlessly progresses, the neurological status steadily becomes worse, resulting in complete disability, vegetative state, and death. Adrenomyeloneuropathy (AMN), a less severe clinical manifestation2, causes progressive myelopathy that predominantly affects the corticospinal tract in the lower limbs of adolescents and young adult males. This condition is frequently mistakenly diagnosed as multiple sclerosis (MS)3. In such patients, the presence of adrenal dysfunction strongly suggests AMN4.
Although women carriers are usually considered normal or only very mildly affected, it has, in fact, been observed that at least half of the heterozygous females present neurological manifestations that vary in severity from mild hyperreflexia and vibratory sense impairment, with little or no functional disability to severe spastic paraparesis that requires a wheelchair5. As in males, females show no correlation between clinical severity and very long chain fatty acids (VLCFA) blood levels6. It seems, however, that there is a correlation between the skewing of the X chromosome and the specific mutation harbored by the patient6. Adrenal insufficiency has also been rarely detected in heterozygotes5-7.
To date, there are no systematic studies that address the prevalence of neurological and adrenal dysfunctions in ALD female heterozygotes. Additionally, their clinical characteristics have not been properly documented. In this report, we review the clinical features of the heterozygous women who were followed in our Neurogenetics Division, Clinics Hospital, School of Medicine of Ribeirão Preto, University of São Paulo, Ribeirão Preto, in São Paulo.
METHODS
In this study, we have included all females with VLCFA profiles showing increased C26:C22 and C24:C22, regardless of their clinical status or family history, followed in our hospital. They were submitted to a detailed clinical examination, nerve conduction studies (NCS), and brain/spinal cord magnetic resonance imaging (MRI). They all provided an informed consent (Clinics Hospital, School of Medicine of Ribeirão Preto, n. 3172/2009).
RESULTS
Nine women showed abnormal VLCFA profiles compatible with a heterozygous state for X-ALD.
Their clinical manifestations suggest the existence of three distinct groups (Table): with a severe and aggressive neurological disease, mimicking the male X-ALD phenotype; with a slowly progressive spastic paraparesis; and with no complaints, having only mild abnormalities detected on neurological examination, including decreased vibratory sensation and brisk reflexes, with no apparent progression. There was no difference between the three groups with respect to the biochemical profile.
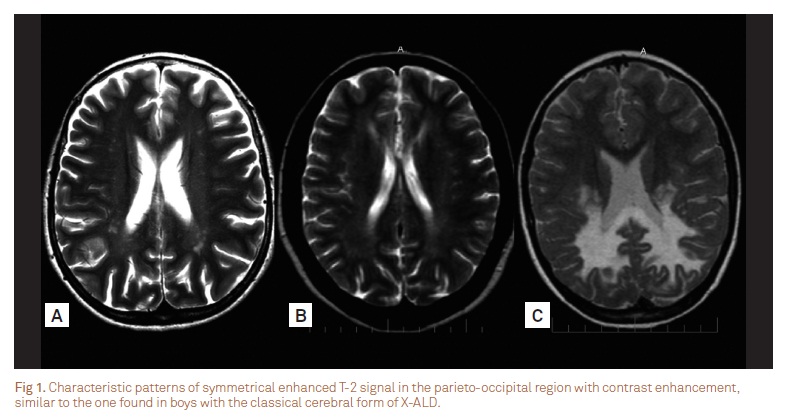
The first group consisted of three patients who presented with neurological regression and white mater abnormalities. They exhibited mental impairment, cerebellar ataxia, and spastic paraplegia. Psychotic behavior was noted in the most severely affected girl, who also presented seizures. Two patients presented learning disabilities before the physical manifestations onset. The brain MRI of patient 1 was virtually identical to the pattern found in males with ALD of childhood onset (Fig 1). Patient 2 was initially diagnosed with childhood onset MS and was treated accordingly (Fig 2).
In the second group, four patients showed spastic paraparesis as the main symptom. Two of them were mother and daughter, who were initially diagnosed as having autosomal dominant hereditary spastic paraplegia. The daughter's manifestations appeared in the second decade of life, whereas the mother developed symptoms in her forties. In this group, the symptoms were slowly progressive, but they were considered debilitating mainly due to the presence of bladder incontinence. Mood disorder was observed in three patients. Interestingly, only one patient had a known family history (son and brother) of ADL. Their brain MRI was initially normal, but mild abnormalities of the subcortical white matter were later developed.
In the third group, two patients were diagnosed exclusively due to their positive family history. They both reported "cramps" and occasional discomfort when walking long distances. On examination, vibration was mildly decreased distally in the lower limbs, and tendon jerks were brisk at knees and ankles.
Karyotype (G-banding) analysis was performed in these patients, but no abnormalities were found in 100 metaphases. Endocrinological, ophthalmological, and audiometric evaluations, NCS and spinal cord MRIs were all normal.
DISCUSSION
The phenotype of X-ALD varies widely among affected patients, even within the same family, but five major clinical subtypes have been identified in affected males8. These include childhood cerebral ALD, the most common phenotype, which affects 48% of male patients under 11 years-old; AMN, the next most common form (26%), which affects men over 21 years-old; the adolescent cerebral form (5%), which is observed between the ages of 11 to 21 years; Addison's disease without neurological involvement (11%); and the adult cerebral form (3%). Additionally, there are reports of a rare olivopontocerebellar atrophy presentation that affects mainly adults with late-onset manifestations of X-ADL9.
Neurological manifestations in ALD heterozygous females were previously thought to be relatively uncommon. However, data from the Kennedy Institute10 showed that 15 to 20% of these patients develop symptomatic spastic paraparesis. In addition, neurological examination of these women showed that almost 55% of them had at least mild neurological abnormalities. This spectrum of manifestations resembled our patients from the first and second groups. Such manifestations were somewhat similar to males with AMN but were milder, slowly progressive, and of late onset. Adrenal dysfunction was extremely rare in female patients.
In our patients, stiffness and clumsiness in the legs were usually the first symptoms to appear (90%). The most common clinical signs were hyperreflexia (100%), spastic paraparesis (76%), and impaired sense of vibration (88%). Neuropathy was not observed in our series of patients, although it was presented in other series11. Two of our patients from the second group developed a late and incapacitating sphincter disturbance. Cerebral involvement is considered uncommon in female carriers12, but three of our patients presented cerebral demyelination, and two developed severe neurological regression. Fluctuating manifestations and adrenal dysfunction were not observed in our cohort5.
The diagnosis of ALD is rarely made in women without an affected male relative. It is possible that many patients are initially misdiagnosed as having MS, until a family history of ALD is detected3. Interestingly, only three women in our cohort had a positive family history, suggesting that women, even those with mild evidence of bilateral corticospinal tract and/or decreased vibration perception in the lower limbs, should have their VLCFA levels tested.
Brain MRI identify characteristic changes in the majority of males with ALD and in about 50% of those with AMN, and may even detect abnormalities in asymptomatic patients13. A total of 16% of symptomatic females may present white matter abnormalities in the internal capsule, cerebral peduncle, and optic radiation10. In general, our patients showed only very mild abnormalities; although those with serious neurological regression showed multiple areas of cerebral demyelination (Fig 1 and 2). In our patients, MRI of the spinal cord was always normal, in opposition to previous reports suggesting that a decreased spinal cord diameter could be a marker of the onset of symptoms in heterozygous females12.
At present, there is no proven prevention or curative therapy for ALD in female carriers, and none of our patients received a specific therapy14. In addition to a supportive follow-up with no unnecessary investigation, a proper genetic counseling may be extremely useful for these patients and their families15. The wide range of clinical variability, which seems to be associated with ADL female carriers, should prompt interested clinical investigators to organize a multicenter systematic study to better define the associated clinical spectrum.
Received 06 January 2012
Received in final form 1 March 2012
Accepted 19 March 2012
Conflict of interest: There is no conflict of interest to declare.
- 1. Moser HW. Adrenoleukodystrophy: phenotype, genetics, pathogenesis and therapy. Brain 1997;120:1485-1508.
- 2. Budka H, Sluga E, Heiss WD. Spastic paraplegia associated with Addison's disease: adult variant of adreno-leukodystrophy. J Neurol 1976;213:237-250.
- 3. Dooley JM, Wright BA. Adrenoleukodystrophy mimicking multiple sclerosis. J Can Sci Neurol 1985;12:73-74.
- 4. Blevins Jr LS, Shankroff J, Moser HW, Ladenson PW. Elevated plasma adrenocorticotropin concentration as evidence of limited adrenocortical reserve in patients with adrenomyeloneuropathy. J Clin Endocrinol Metab 1994;78:261-265.
- 5. Powers JM, Moser HW, Moser AB, Ma CK, Elias SB, Norum RA. Pathologic findings in adrenoleukodystrophy heterozygotes. Arch Pathol Lab Med 1987;111:151-153.
- 6. Maier EM, Kammerer S, Muntau AC, Wichers M, Braun A, Roscher AA. Symptoms in carriers of adrenoleukodystrophy relate to skewed X inactivation. Ann Neurol 2002;52:683-688.
- 7. Moser HW, Moser AB, Naidu S, Bergin A. Clinical aspects of adrenoleukodystrophy and adrenomyeloneuropathy. Dev Neurosci 1991;13:254-261.
- 8. O'Brien TJ, Gates PG, Byrne E. Symptomatic female heterozygotes for adrenoleukodystrophy: a report of two unrelated cases and review of the literature. J Clin Neurosci 1996;3:166-170.
- 9. Marsden CD, Obeso JA, Lang AE. Adrenoleukomyeloneuropathy presenting as spinocerebellar degeneration. Neurology 1982; 32:1031-1032.
- 10. Moser HW, Mahmood A, Raymond GV. X-linked adrenoleukodystrophy. Nat Clin Pract Neurol 2007;3:140-151.
- 11. Jung HH, Wimplinger I, Jung S, Landau K, Gal A, Heppner FL. Phenotypes of female adrenoleukodystrophy. Neurology 2007; 20:960-961.
- 12. Fatemi A, Barker PB, Uluğ AM, Nagae-Poetscher LM, Beauchamp NJ, Moser AB, et al. MRI and proton MRSI in women heterozygous for X-linked adrenoleukodystrophy. Neurology 2003;60:1301-1307.
- 13. Eichler F, Mahmood A, Loes D, Bezman L, Lin D, Moser HW, et al. Magnetic resonance imaging detection of lesion progression in adult patients with X-linked adrenoleukodystrophy. Arch Neurol 2007;64:659-664.
- 14. Mahmood A, Dubey P, Moser HW, Moser A. X-linked adrenoleukodystrophy: therapeutic approaches to distinct phenotypes. Pediatr Transplant 2005;9(Suppl 7):S55-S62.
- 15. Moser HW, Raymond GV, Dubey P. Adrenoleukodystrophy: new approaches to a neurodegenerative disease. JAMA 2005; 294:3131-3134.
Correspondence:
Publication Dates
-
Publication in this collection
23 July 2012 -
Date of issue
July 2012
History
-
Received
06 Jan 2012 -
Accepted
19 Mar 2012 -
Reviewed
01 Mar 2012