Abstracts
Neurological disorders associated with glutamic acid decarboxylase (GAD) antibodies are rare pleomorphic diseases of uncertain cause, of which stiff-person syndrome (SPS) is the best-known. Here, we described nine consecutive cases of neurological disorders associated with anti-GAD, including nine patients with SPS and three cases with cerebellar ataxia. Additionally, four had hypothyroidism, three epilepsy, two diabetes mellitus and two axial myoclonus.
glutamic acid decarboxylase antibodies; anti-GAD; neurological disorders; stiff-person syndrome; cerebellar ataxia
Distúrbios neurológicos associados com anticorpos anti-GAD são doenças pleomórficas, raras, de causa incerta, das quais a rigidez muscular espasmódica (SPR) é a mais conhecida. Neste estudo, descrevemos nove casos consecutivos de distúrbios neurológicos associados com a presença de anticorpos anti-GAD, incluindo nove pacientes com SPR e três casos com ataxia cerebelar. Adicionalmente, foram encontrados quatro casos com hipotireoidismo, três com epilepsia, dois com diabetes mellitus e dois casos com mioclonia axial.
anticorpos anti-descaboxilase do ácido glutâmico; anti-GAD; distúrbios neurológicos; rigidez muscular espasmódica; ataxia cerebelar
ARTICLE
Neurological disorders associated with glutamic acid decarboxylase antibodies: a Brazilian series
Distúrbios neurológicos associadas a anticorpos anti-GAD (descarboxilase do ácido glutâmico): uma série de casos brasileira
Maurício FernandesI; Renato P. MunhozII; Paulo Eduardo Mestrinelli CarrilhoIII; Walter O. ArrudaI; Paulo J. LorenzoniI; Rosana H. ScolaI; Lineu César WerneckI; Hélio A. G. TeiveI
IMovement Disorders Unit, Neurology Service, Internal Medicine Department, Hospital de Clínicas, Universidade Federal do Paraná (UFPR), Curitiba PR, Brazil
IINeurology Service, Pontifícia Universidade Católica (PUC) do Paraná, Curitiba PR, Brazil
IIINeurology Service, Medical School, Universidade Estadual do Oeste do Paraná (UNIOESTE), Cascavel PR, Brazil
Correspondence Correspondence: Hélio A. G. Teive Rua General Carneiro 1103/102 80060-150 Curitiba PR - Brasil E-mail: hagteive@mps.com.br
ABSTRACT
Neurological disorders associated with glutamic acid decarboxylase (GAD) antibodies are rare pleomorphic diseases of uncertain cause, of which stiff-person syndrome (SPS) is the best-known. Here, we described nine consecutive cases of neurological disorders associated with anti-GAD, including nine patients with SPS and three cases with cerebellar ataxia. Additionally, four had hypothyroidism, three epilepsy, two diabetes mellitus and two axial myoclonus.
Key words: glutamic acid decarboxylase antibodies, anti-GAD, neurological disorders, stiff-person syndrome, cerebellar ataxia.
RESUMO
Distúrbios neurológicos associados com anticorpos anti-GAD são doenças pleomórficas, raras, de causa incerta, das quais a rigidez muscular espasmódica (SPR) é a mais conhecida. Neste estudo, descrevemos nove casos consecutivos de distúrbios neurológicos associados com a presença de anticorpos anti-GAD, incluindo nove pacientes com SPR e três casos com ataxia cerebelar. Adicionalmente, foram encontrados quatro casos com hipotireoidismo, três com epilepsia, dois com diabetes mellitus e dois casos com mioclonia axial.
Palavras-Chave: anticorpos anti-descaboxilase do ácido glutâmico, anti-GAD, distúrbios neurológicos, rigidez muscular espasmódica, ataxia cerebelar.
Neurological disorders associated with glutamic acid decarboxylase (GAD) antibodies include stiff-person syndrome (SPS) and its variants - stiff trunk syndrome, stiff limbs syndrome, progressive encephalomyelitis with rigidity, SPS-plus and paraneoplastic SPS - as well as epilepsy and ataxia1-8. These syndromes are often found in association with other autoimmune disorders, such as diabetes mellitus, in more than one thirds of all cases, and, less commonly, those caused by antithyroid, antinuclear and antiparietal cell antibodies (5-10%)1-3,9-13.
GAD is a cytoplasmic enzyme that accelerates the conversion of glutamic acid to gamma-aminobutyric acid (GABA), an inhibitory neurotransmitter present in the brain and spinal cord. GAD is synthesized mainly in presynaptic GABAergic neurons in the central nervous system and in the βcells in the islets of Langerhans in the pancreas. GAD autoantibodies are found in around 60 to 80% of SPS cases. Although there are two GAD isoforms ¾ GAD65 and GAD67 ¾ the main target for GAD autoantibodies in SPS is GAD659-14.
The pathogenetic role of autoantibodies in SPS is unclear. However, all SPS autoantigens identified to date are synaptic proteins involved in inhibitory synaptic transmission, such as GAD and amphiphysin (presynaptic antigens) and GABA(A) receptor-associated protein (GABARAP) and gephyrin (postsynaptic antigens). The autoantibodies anti-amphiphysin and anti-gephyrin have been linked to paraneoplastic SPS9-15.
The objective of this study was to present the clinical and laboratory findings of 12 patients with neurological disorders associated with anti-GAD antibodies.
METHODS
Twelve consecutive patients with neurological disorders associated with anti-GAD antibodies were evaluated over a 17-year period at the Movement Disorders Unit in the Neurology Service, Hospital de Clínicas, Universidade Federal do Paraná, Pontíficia Universidade Católica do Paraná (Curitiba, PR, Brazil) and Medical School of UNIOESTE (Cascavel, Paraná, Brazil). Extensive workups, including neuroimaging, cerebrospinal fluid (CSF) analysis, electroencephalography (EEG) tracing, electroneuromyography (ENMG) and immunological tests (serum anti-GAD, antithyroid, antinuclear and antiparietal cell antibodies), were carried out.
The Dalakas criteria16 for the diagnosis of typical SPS were used. SPS variants were defined in accordance with Baker et al.5 as stiff trunk syndrome, stiff limb syndrome, progressive encephalomyelitis with rigidity and paraneoplastic SPS.
Seven patients were females. Mean age of diagnosis was 47.25 years (11 to 58 years). Two of the patients have been previously described17,18.
RESULTS
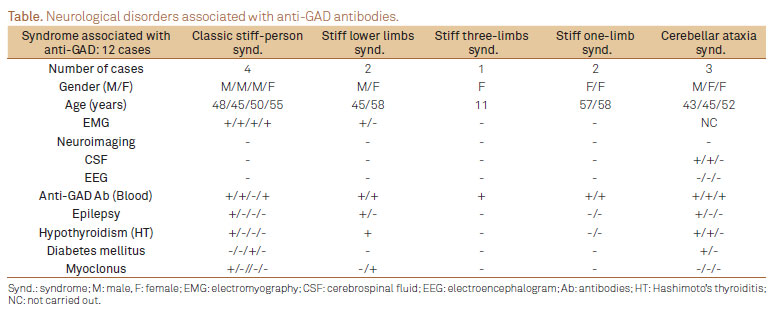
Among a total of nine SPS cases, four presented the classic form and five the variants (two cases of stiff lower limbs, two of stiff one-limb and one of stiff three-limbs). Additionally, we found three cases of cerebellar ataxia. No cases of progressive encephalomyelitis with rigidity or paraneoplastic SPS were found. With one exception, all patients had elevated serum anti-GAD titers, ranging from 20 to 800 U/mL (normal <1).
ENMG results were compatible with a diagnosis of SPS in six cases. Neuroimaging and EEG tracings were normal in all the patients. CSF analysis revealed nonspecific changes (mild pleocytosis) in two cases.
Three patients suffered from epileptic seizures (one with the classic form of SPS, the other with stiff lower limb variant and the last with cerebellar ataxia). Two patients (one with the classic form and the other with stiff lower limb variant) had segmental axial myoclonus.
A total of five patients had clinical and laboratory signs of autoimmune endocrinopathy (four patients with Hashimoto's thyroiditis, two with diabetes mellitus).
The patients with SPS were treated with diazepam (15 to 30 mg/day), baclofen (30 to 60 mg/day) and intravenous immunoglobulin (IVIg, 0.4 g/kg/day for 5 days, followed by single monthly doses). Among the nine SPS patients, seven showed significant clinical improvement and two (with stiff one-limb) were partially responsive. The patients with cerebellar ataxia were treated with IVIg and responded well to this therapy. The Table summarizes the results for all the patients.
Two illustrative cases of neurological disorders associated with anti-GAD antibodies are described below.
ILLUSTRATIVE CASES
CASE 1
A 50-year-old female patient with four years history of progressively worsening signs and symptoms, consisting lumbar muscles contractures, pain and difficulty walking, sought medical attention in her hometown, initially with suspected postural lumbago and later with a possible diagnosis of ankylosing spondylitis. As her condition progressed, increased contracture and rigidity of the paravertebral muscles was observed (mainly in the lumbar region) with hyperlordosis (Fig 1). Her condition fluctuated, alternating between periods when the muscular contracture and rigidity worsened and she experienced pain and difficulty walking, with frequent falls, and periods when there was a partial improvement. She had a positive family history of breast cancer. She had also been treated for breast cancer, and there was no evidence of a relapse. On her latest assessment, in addition to muscular rigidity and bilateral contractures in the paravertebral region, particularly the lumbar region and lower limbs, she sometimes has sudden, jerky, movements and falls to the ground. She reports increasing difficulty walking because of severe muscular rigidity and is taking baclofen (40 mg/day) and diazepam (30 mg/day.)
The patient underwent extensive additional tests, including MR neuroimaging of the skull and cervical/thoracic and lumbosacral spine, which revealed L4-L5 discopathy. The following tests were unremarkable: CSF analysis, complete blood count, fasting blood sugar, lipid profile, creatinine, TSH, T3, T4F, TGO, TGP, amylase, VDRL, electrolytes, mammography, chest X-ray, abdominal echography, whole-body PET-CT, antinuclear antibody (ANA), protein electrophoresis, muscle enzymes, erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP). Bone densitometry showed osteopenia. ENMG showed the presence of continuous muscular activity (Fig 2). The patient is receiving IVIg therapy with a progressive improvement in her clinical picture.
CASE 2
A 52-year-old female patient who presented initially with diplopia had difficulty walking and had progressively worsening imbalance, which deteriorated markedly over 20 days. She had an ophthalmic assessment, which proved inconclusive, and was referred for neurological assessment in her hometown. This revealed gait ataxia, difficulty with tandem gait and the presence of horizontal gaze-evoked nystagmus, as well as a complaint of diplopia. The patient was receiving thyroid hormone therapy (100 µg/day). Further investigation revealed normal routine blood tests, except for ESR (60). Brain neuroimaging (CT and MRI) was normal. CSF analysis revealed the presence of 16 cells (100% lymphomononuclear), but no other abnormalities. Over time the patient complained of nausea and vomiting, and used an antiemetic (metoclopramide), dimenhydrinate and clonazepam. Viral cerebellitis was suspected. She later presented with a clinical picture of urinary infection, which subsequently improved with norfloxacin. The clinical picture of ataxia remained unchanged, and 30 days later the patient had a generalized tonic-clonic seizure while asleep, which was repeated the following day. The patient was hospitalized and phenytoin was administered intravenous (i.v.). Neurological examination revealed cerebellar ataxia with nystagmus and diplopia; a new brain MRI was normal. Phenytoin was replaced by lamotrigine in a daily dose of 200 mg, and methylprednisolone pulse therapy (1 g/day for 5 days) was started. At the end of the five days, the methylprednisolone was substituted by prednisone (40 mg/day). New CSF tests were unremarkable, and a further detailed investigation, including tests for vasculitis and screening for occult neoplasms, infections (serologic tests to detect antibodies against HIV, Epstein-Barr, varicella-zoster and herpes simplex viruses) and Hu, Ri and Yo antibodies, was normal. EEG tracing was also unremarkable.
Because the patient's clinical picture deteriorated, she was referred for specialized assessment. She continued to complain of dizziness, imbalance and diplopia, and neurological examination revealed marked gait ataxia (she could not perform tandem gait unsupported) with horizontal and vertical gaze-evoked nystagmus, as well as slight dysarthria.
As autoimmune cerebellar ataxia was suspected, further investigations were again carried out, including routine blood chemistry, VDRL, complete blood count, coagulogram, ESR, liver function, thyroid tests, anti-DNA tests, a new MRI and a new CSF examination, as well as measurement of antigliadin, antithyroglobulin and antiperoxidase antibodies, all of which were unremarkable, except for the anti-GAD antibody test (39 IU/mL; normal value < 1.0 IU/mL).
As the diagnosis was cerebellar ataxia and epileptic seizures associated with anti-GAD antibodies, the patient underwent IVIg treatment (0.4 g/kg/day) for five days followed by a single monthly i.v. dose (for 12 months) and, subsequently, azathioprine p.o. (2 mg/kg/day). There was a gradual improvement in the clinical picture and, one year after the start of the specific treatment, neurological examination showed only mild gait ataxia (more apparent in tandem gait) and the presence of mild horizontal nystagmus in both extreme gazes. Lamotrigine (200 mg/day) p.o. and clonazepam (1.0 mg/night) were continued; complete blood count and liver function tests were normal and azathioprine was discontinued.
DISCUSSION
Classical SPS is a rare neurological disorder characterized by severe and incapacitating axial and proximal limb rigidity and stiffness due to continuous motor unit activity1-8. This unusual syndrome was first described by Moersch and Woltman in 1956, who defined it as "progressive fluctuating muscular rigidity and spasm" ("Stiff-man" syndrome) after reporting one case and studying 13 others19.
Rigidity is often exacerbated by anxiety, sudden movements or external stimuli, causing intermittent painful spasms and frequently leading to skeletal deformity. Exaggerated lumbar lordosis and pain are almost universal findings, and diagnosis is supported by a finding of anti-GAD antibodies in the blood or CSF2. SPS frequently presents with psychiatric symptoms, such as anxiety, phobias and depression, and psychogenic movement disorder is a common misdiagnosis1.
In our series of patients with neurological disorders associated with anti-GAD antibodies, we found nine patients with SPS and its variants. Four had a classical clinical presentation of SPS, and the other five presented with SPS variants (two with stiff lower limbs, two with stiff one-limb and one with stiff three-limbs). ENMG examination was diagnostic in all the patients with classical SPS and stiff lower limbs syndrome; however, in the patients with stiff three-limbs or stiff one-limb syndrome, it was not diagnostic, and the diagnosis was confirmed after high serum anti-GAD antibodies had been detected. Autoimmune endocrinopathies, particularly diabetes mellitus and hypothyroidism (Hashimoto's thyroiditis), were found in some of the patients. Two patients suffered from epileptic seizures and segmental myoclonus affecting the trunk.
Clinically, stiff-person syndrome presents as neurological paraneoplastic syndromes in about 5% of cases1,20,21. In such cases, paraneoplastic stiff-person syndrome is usually associated with amphiphysin or anti-gephyrin antibodies20. While a few cases of paraneoplastic SPS have been described in anti-GAD-antibody-positive patients with thymoma and other solid tumors, a finding of anti-GAD antibodies in patients with other classical paraneoplastic neurological syndromes is rare10,20.
Progressive encephalomyelitis is a rare form of SPS characterized by brainstem, extrapyramidal and spinal cord dysfunction with acute onset of painful rigidity and muscle spasm in association with nystagmus, opsoclonus, ophthalmoparesis, deafness, dysarthria, dysphagia and autonomic dysfunction21,22.
In our series, there were no cases of paraneoplastic SPS or progressive encephalomyelitis with rigidity.
Ataxia associated with anti-GAD antibodies is a very rare condition and is considered to be a component of a polyglandular autoimmune syndrome in patients with circulating anti-GAD antibodies (10%)23. Most patients with ataxia and anti-GAD antibodies are women and have insulin-dependent diabetes mellitus. In general, there is a slowly progressive pancerebellar syndrome, and cerebellar atrophy is sometimes found in MRI examinations1-3,23,24. Three of our patients had acquired secondary cerebellar ataxia associated with anti-GAD antibodies, and two, whose CSF analysis revealed mild pleocytosis, had Hashimoto's thyroiditis. One patient in this group had diabetes mellitus and epileptic crises without myoclonus. There was no difference in anti-GAD levels between this group of cerebellar ataxia patients and the SPS group.
The prevalence of neurological disorders associated with anti-GAD antibodies in epileptic patients is low (less than 10%)25,26. In this series, we found two patients who suffered from epileptic seizures (one from the SPS group and the other from the cerebellar ataxia group). Some authors recommend actively searching for anti-GAD antibodies in patients with atypical refractory epilepsy either on its own or in association with diabetes mellitus or cerebellar ataxia25,26.
The heterogeneity of the neurological presentation in patients with neurological syndromes associated with anti-GAD antibodies is probably related to the widespread distribution of GABAergic neurons in the central nervous system as these cells have a large amount of GAD22.
In conclusion, in this series of neurological disorders associated with anti-GAD antibodies, we found nine patients with stiff-person syndrome (the classical form and its variants) and three with cerebellar ataxia. Four had hypothyroidism, three epilepsy, two diabetes mellitus and two had segmental myoclonus affecting the trunk.
ACKNOWLEDGMENTS
The authors would like to thank all the neurologists who provided clinical and laboratory data on their patients.
Received 01 April 2012
Received in final form 19 April 2012
Accepted 26 April 2012
Conflict of interest: There is no conflict of interest to declare.
- 1. Hadavi S, Noyce AJ, Leslie RD, Giovannoni G. Stiff person syndrome. Pract Neurol 2011;11:272-282.
- 2. Saiz A, Blanco Y, Sabater L, et al. Spectrum of neurological syndromes associated with glutamic acid decarboxylase antibodies: diagnostic clues for this association. Brain 2008;131:2553-2563.
- 3. Vasconcelos OM, Dalakas MC. Stiff-person syndrome. Curr Treat Options Neurol 2003;5:79-90.
- 4. Lorish TR, Thorsteinsson G, Howard FM Jr. Stiff-Man syndrome updated. Mayo Clin Proc 1989;64:629-636.
- 5. Barker RA, Revesz T, Thom M, Marsden CD, Brown P. Review of 23 patients affected by the stiff man syndrome: clinical subdivision into stiff trunk (man) syndrome, stiff limb syndrome, and progressive encephalomyelitis with rigidity. J Neurol Neurosurg Psychiatry 1998;65:633-640.
- 6. Gershanik OS. Stiff-person syndrome. Parkinsonism Relat Disord 2009;15:S130-S134.
- 7. Meinck HM, Thompson PD. Stiff-Man syndrome and related conditions. Mov Disord 2002;17:853-866.
- 8. Hijazi J, Bedat-Millet AL, Hannequim D. Le syndrome de l'homme raide et autres maladies neurologiques associées aux anticorps anti-GAD. Rev Med Interne 2010;31:23-28.
- 9. Boronat A, Sabater L, Saiz A, Dalmau J, Graus F. GABA B receptor antibodies and anti-GAD-associated neurologic disorders. Neurology 2011;76:795-800.
- 10. Grauss F, Saiz A, Dalmau J. Antibodies and neuronal autoimmune disorders of the CNS. J Neurol 2010;257:509-517.
- 11. Solimena M, Folli F, Aparisi R, Pozza G, De Camilli P. Autoantibodies to GABA-ergic neurons and pancreatic beta cells in stiff-man syndrome. N Engl J Med 1990;322:1550-1560.
- 12. Ali F, Rowley M, Jayakrishnan, Teuber S, Gershwin ME, Mackay IR. Stiff-person syndrome (SPS) and anti-GAD-related CNS degenerations: protean addictions to the autoimmune central neuropathies. J Autoimmun 2011;37:79-87.
- 13. Alexopoulos H, Dalakas MC. A critical update on the immunopathogenesis of Stiff Person Syndrome. Eur J Clin Invest 2010;40:1018-1025.
- 14. Dalakas MC, Fujii M, Li M, McElroy B. The clinical spectrum of anti-GAD antibody-positive patients with Stiff-Person syndrome. Neurology 2000;55:1531-1535.
- 15. Dalakas MC. Advances in the pathogenesis and treatment of patients with Stiff-Person syndrome. Curr Neurol Neurosci Rep 2008;8:48-55.
- 16. Dalakas MC. Stiff-Person syndrome: advances in pathogenesis and therapeutic interventions. Curr Treat Options Neurol 2009;11:102-110.
- 17. Teive HAG, Munhoz RP, Cardoso J, Amaral VCG, Werneck LC. Stiff-three limbs syndrome. Mov Disord 2009;24:311-312.
- 18. Munhoz RP, Fameli H, Teive HAG. Stiff person syndrome as the intial manifestation of systemic lupus erythematosus. Mov Disord 2010;25:516-517.
- 19. Moersch FP, Woltman HW. Progressive fluctuating of muscular rigidity and spasm ("stiff-man" syndrome): report of a case and some observations in 13 other cases. Proc Staff Meet Mayo Clin 1956;31:421-427.
- 20. Grant R, Graus F. Paraneoplastic movement disorders. Mov Disord 2009;24:1715-1724.
- 21. Spay AJ, Chen R. Rigidity and spasm from autoimmune encephalomyelopathies: stiff-person syndrome. Muscle Nerve 2006;34:677-690.
- 22. Pittock SJ, Yoshikawa H, Ahlskog E, et al. Glutamic acid decarboxylase autoimmunity with brainstem, extrapyramidal, and spinal cord dysfunction. Mayo Clin Proc 2006;81:1207-1214.
- 23. Klockgether T. Acquired cerebellar ataxias and differential diagnosis. In: Brice A, Pulst SM (Eds). Spinocerebellar degenerations. The ataxias and spastic paraplegias. Philadelphia, PA, USA: Butterworth Heinemann Elsevier; 2007:61-77.
- 24. Kono S, Miyajima H, Sugimoto M, Suzuki Y, Takahashi Y, Hishida A. Stiff-person syndrome associated with cerebellar ataxia and high glutamic-acid decarboxylase antibody titer. Intern Med 2001;40:968-971.
- 25. Vulliemoz S, Vanini G, Truffert A, Chizzolini C, Seeck M. Epilepsy and cerebellar ataxia associated with anti-glutamic acid decarboxylase antibodies. J Neurol Neurosurg Psychiatry 2007;78:187-189.
- 26. Yoshimoto T, Doi M, Fukai N, et al. Type 1 diabetes mellitus and drug-resistent epilepsy: presence of high titer of anti-glutamic acid decarboxylase autoantibodies in serum and cerebrospinal fluid. Intern Med 2005;44:1174-1177.
Correspondence:
Publication Dates
-
Publication in this collection
12 Sept 2012 -
Date of issue
Sept 2012
History
-
Received
01 Apr 2012 -
Accepted
26 Apr 2012 -
Reviewed
19 Apr 2012