Abstracts
New evidence concerning the pathophysiology of migraine has come from the results of therapeutic transcranial magnetic stimulation (tTMS). The instantaneous responses to single pulses applied during the aura or headache phase, together with a number of other observations, make it unlikely that cortical spreading depression is involved in migraine. tTMS is considered to act by abolishing abnormal impulse activity in cortical pyramidal neurons and a suggestion is made as to how this activity could arise.
migraine; spreading depression; transcranial magnetic stimulation
Novas evidências referentes à fisiopatologia da enxaqueca são o resultado de estimulação magnética transcraniana terapêutica (tTMS). As respostas imediatas a pulsos simples aplicados durante as fases de aura ou de cefaleia, em associação a diversas outras observações, tornam improvável a ideia de que a depressão alastrante esteja envolvida na enxaqueca. Considera-se que tTMS tenha sua ação abolindo atividade anormal de impulsos em neurônios corticais piramidais, sugerindo que esta atividade tenha um papel desencadeante.
enxaqueca; depressão alastrante; estimulação magnética transcraniana
Three different mechanisms - vascular, trigeminovascular and cortical hyperexcitability -
have been proposed for the pathogenesis of migraine and have recently been the subject
of extensive reviews11 .Goadsby PJ, Charbit AR, Andreou AP, Akerman S, Holland PR.
Neurobiology of migraine. Neuroscience. 2009;161(2):327-41.
http://dx.doi.org/10.1016/j.neuroscience.2009.03.019
https://doi.org/10.1016/j.neuroscience.2...
,22 .Tietjen GE. Migraine as a systemic vasculopathy. Cephalalgia.
2009;29(9):989-996.
http://dx.doi.org/10.1111/j.1468-2982.2009.01937.x
https://doi.org/10.1111/j.1468-2982.2009...
,33 .Cutrer FM. Pathophysiology of migraine. Semin Neurol.
2010;30(2):120-30. http://dx.doi.org/10.1055/s-0030-1249222
https://doi.org/10.1055/s-0030-1249222...
,44 .Pietrobon D, Moskowitz MA. Pathophysiology of migraine. Ann Rev
Physiol, 2013;75:365-91.
http://dx.doi.org/10.1146/annurev-physiol-030212-183717
https://doi.org/10.1146/annurev-physiol-...
. In our own assessment55 .McComas A, Upton A. Therapeutic transcranial magnetic stimulation
in migraine and its implications for a neuroinflammatory hypothesis.
Inflammopharmacology. 2009;17(2):68-75.
http://dx.doi.org/10.1007/s10787-009-8058-7
https://doi.org/10.1007/s10787-009-8058-...
we presented the results of therapeutic transcranial
magnetic stimulation (tTMS) as evidence that, in at least some patients, virtually all
migraine symptoms during the headache phase are cortical in origin and that a
neuroinflammatory mechanism could not be involved in the initiation and maintenance of
the headache; instead, the evidence clearly pointed to cortical hyperexcitability66 .Aurora SK, Wilkinson F. The brain is hyperexcitable in migraine.
Cephalalgia. 2007;27(12):1442-53.
http://dx.doi.org/10.1111/j.1468-2982.2007.01502.x
https://doi.org/10.1111/j.1468-2982.2007...
. We now use the results of tTMS once
more, this time as part of an argument against one of the central tenets of migraine
pathophysiology, namely, that cortical spreading depression (CSD) is responsible for the
aura.
CORTICAL SPREADING DEPRESSION (CSD)
In 1944 Aristides Leão reported the ability to produce temporary suppression of electrical activity in the rabbit cortex by any one of several manoeuvres77 .Leão AAP. Spreading depression of activity in the cerebral cortex. J Neurophysiol. 1944;7(6):359-90.. The latter included intense electrical stimulation of the brain surface, the application of KCl solution, and mechanical deformation. Once induced, the depression of spontaneous EEG activity traveled slowly into neighbouring regions of cortex (Figure 1). The cortical spreading depression (CSD) could be blocked by an incision in the cortex or by the topical application of a local anaesthetic (cocaine HCl). Leão subsequently reported that CSD was associated with marked dilatation of the pial arteries and increased blood flow; a smaller vasoconstriction sometimes followed88 .Leão AAP. Pial circulation and spreading depression of activity in the cerebral cortex. J Neurophysiol. 1944;7(6):391-6.. Leão extended his observations by demonstrating that CSD took place in the more superficial layers of cortex99 .Leão AAP, Morison RS. Propagation of spreading cortical depression. J Neurophysio.l 1945;8(1):33-45.. In a final paper, published after his return to Brazil from the United States, Leão described a slow, negative-positive potential that accompanied the onset of EEG suppression1010 .Leão AAP. Further observations on the spreading depression of activity in the cerebral cortex. J Neurophysiol. 1947;10(6):409-14..
Spreading depression in rabbit brain. Electrical stimuli were delivered through electrodes (S, inset) over frontal cortex, and recordings made through paired electrodes 1-7 placed progressively more posteriorly. Thirty seconds after stimulation there is depression of cortical activity under the nearest pair of recording electrodes (1-2, inset), and within a further 4 minutes the depression has reached the last electrode (7, inset).
Leão’s observations have been repeatedly confirmed and new observations added. Thus
CSD is associated with the efflux of potassium from neurons and glia into the
extracellular space, and with a cellular influx of sodium, calcium and water; the
concentration of glutamate also rises in the extracellular space1111 .Smith JM, Bradley DP, James MF, Huang CL-H. Physiological studies
of cortical spreading depression. Biol Rev Camb Philos Soc. 2006;81(4):457-81.
http://dx.doi.org/10.1017/S1464793106007081
https://doi.org/10.1017/S146479310600708...
,1212 .Charles A, Brennan KC. Cortical spreading depression—new insights
and persistent questions. Cephalalgia. 2009;29(10):1115-24.
http://dx.doi.org/10.1111/j.1468-2982.2009.01983.x
https://doi.org/10.1111/j.1468-2982.2009...
. The onset of the CSD is associated with
depolarization of both astrocytes and neurons, being larger and more rapid in the
former1313 .Sugaya E, Takato M, Noda Y. Neuronal and glial activity during
spreading depression in cerebral cortex of cat. J Neurophysiol.
1975;38(4):822-41.. In the neurons there
is a transient burst of impulses, which ceases with further depolarization. It is
thought that CSD propagates through the dendritic network and/or through gap
junctions between astrocytes.
HISTORICAL LINKAGE OF CSD TO MIGRAINE
Leão himself speculated that CSD might be linked in some way to migraine, in part
because of its slow passage across the cortex; he was, however, thinking in terms of
the dilatation of the pial arteries rather than the change in electrical activity. A
few years prior to Leão’s first publication, the experimental psychologist, Karl
Lashley, gave detailed and lucid descriptions of his own visual auras; the
scintillations were interpreted as the result of intense excitation in the visual
cortex, while the enlarging scotomas were attributed to inhibition1414 .Lashley KS. Patterns of cerebral integration indicated by the
scotomas of migraine. Arch Neurol Psychiatry. 1941;46(2):331-9.
http://dx.doi.org/10.1001/archneurpsyc.1941.02280200137007
https://doi.org/10.1001/archneurpsyc.194...
. Taking the length of the striate
cortex to be 67 mm, and allowing 20 minutes for the visual phenomena to reach the
margin of the temporal visual field from their central origin, Lashley calculated
that the underlying cortical process traveled rostrally at approximately 3 mm/minute
from the occipital pole. The possible link between CSD and the migraine aura was not
pointed out until 1958, in the form of a brief note by Milner1515 .Milner PM. Note on a possible correspondence between the scotomas
of migraine and spreading depression of Leão. Electroencephalogr Clin
Neurophysiol. 1958;10(4):705.
http://dx.doi.org/10.1016/0013-4694(58)90073-7
https://doi.org/10.1016/0013-4694(58)900...
. In the following year Hubel and Wiesel published
the results of a microelectrode study of the cat visual cortex, reporting the
presence of orientation sensitive cells in the primary visual area1616 .Hubel DH, Wiesel TN. Receptive fields of single neurons in the cat
striate cortex. J Physiol. 1959;148(3)-574-91.
http://dx.doi.org/10.1113/jphysiol.1959.sp006308
https://doi.org/10.1113/jphysiol.1959.sp...
. The likelihood that excitation
of these cells was responsible for the flickering zigzag lines of a visual aura
should have been immediately obvious, but instead had to wait 12 years before
appearing in print1717 .Richards W. The fortification illusions of migraine. Sci Am.
1971;224:88-96..
The proposition that CSD might be responsible, not only for the visual symptoms but
for other types of aura as well, was a logical extension, and one developed by
Vincent and Hadjikhani1818 .Vincent MB, Hadjikhani N. Migraine aura and related phenomena:
beyond scotomata and scintillations. Cephalalgia. 2007:27(12):1368-77.
http://dx.doi.org/10.1111/j.1468-2982.2007.01388.x
https://doi.org/10.1111/j.1468-2982.2007...
. These
authors sent out questionnaires to migraineurs and discovered that many had complex
symptoms before the headache; these included difficulties in recognizing objects and
faces, in recalling names, and in speech. The authors suggested that the nature and
temporal sequence of symptoms could be explained by the course taken by CSD over the
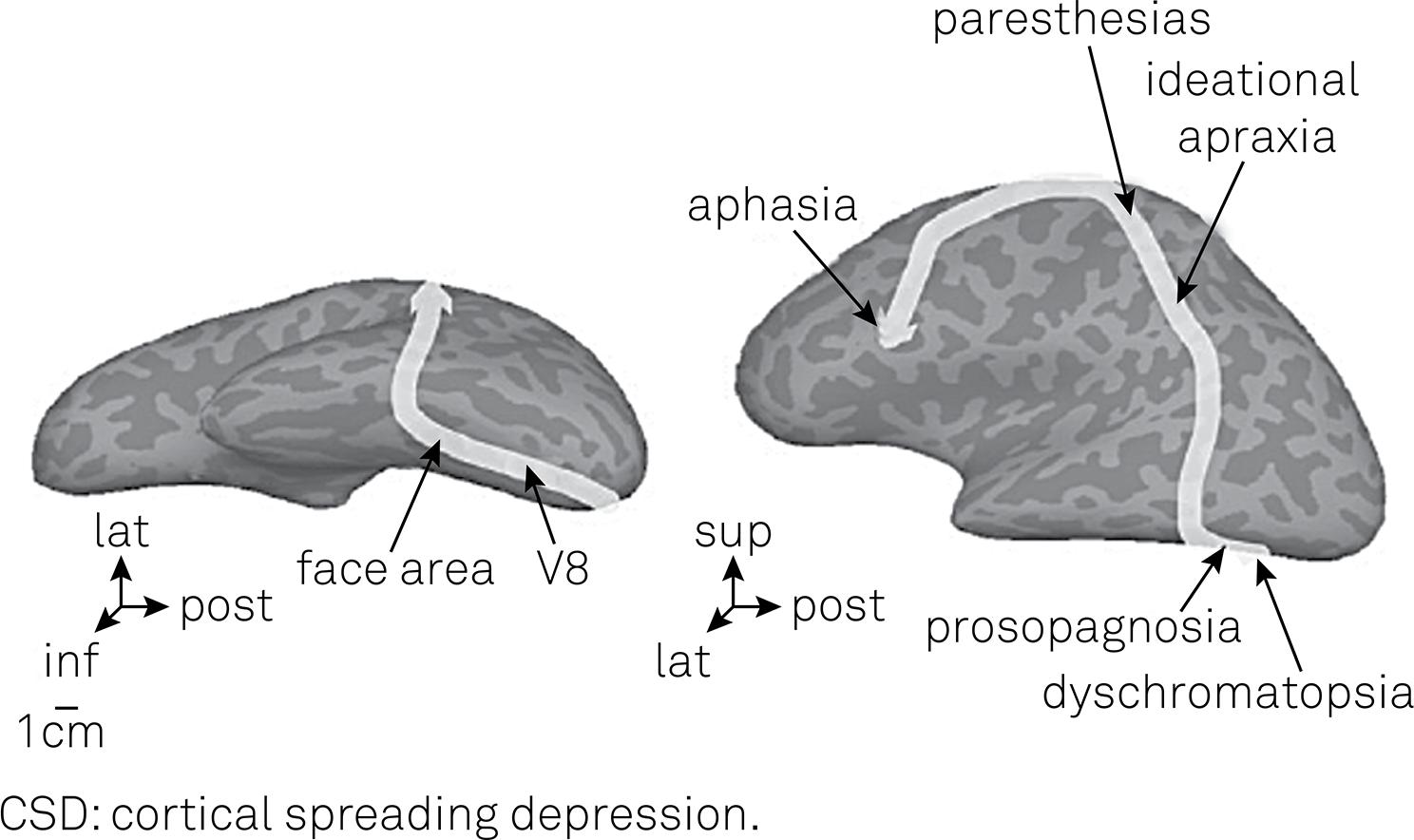
surface of the affected hemisphere (Figure
2).
Postulated track of CSD in a patient, following initial visual symptoms (dyschromatopsia) and culminating in aphasia18. Although CSD is known to have difficulty crossing fissures, in this patient it would have had to travel across 20 or so (dark grey areas in figure). Figure reproduced by permission of Blackwell Publishing.
The possibility that CSD might be directly responsible for the headache as well as
the aura was suggested by Moskowitz1919 .Moskowitz MA. The neurobiology of vascular head pain. Ann Neurol.
1984;16(2):157-68. http://dx.doi.org/10.1002/ana.410160202
https://doi.org/10.1002/ana.410160202...
at the end of a comprehensive review of the innervation of
the intracranial arteries. Moskowitz postulated that the rise in extracellular
potassium during CSD would depolarize and excite the nociceptive fibres in the
ophthalmic division of the trigeminal nerve surrounding the pial arteries; this, by
itself, would cause pain, as would reflex changes in the walls of other vessels. The
finding of increased levels of CGRP (calcitonin gene related peptide) in jugular
venous blood during migraine attacks2020 .Goadsby PJ, Edvinsson L, Ekman R. Vasoactive peptide release in the
extracerebral circulation of humans during migraine headache. Ann Neurol.
1990;28(2):183-7. http://dx.doi.org/10.1002/ana.410280213
https://doi.org/10.1002/ana.410280213...
was consistent with Moskowitz’s scheme, as it suggested
that this neuropeptide, perhaps with others, was being liberated from the
depolarized trigeminal afferent fibres. In an extension of the hypothesis, Burstein
proposed that increased impulse traffic in the same fibres would enhance the
excitability of second-order trigeminal neurones in the brain stem, thereby
interfering with pain-gating mechanisms2121 .Burstein R. Deconstructing migraine headache into peripheral and
central sensitization. Pain. 2001;89(2-3):107-10.
http://dx.doi.org/10.1016/S0304-3959(00)00478-4
https://doi.org/10.1016/S0304-3959(00)00...
,2222 .Noseda R, Burstein R. Migraine pathophysiology: anatomy of the
trigeminovascular pathway and associated neurological symptoms, cortical
spreading depression, sensitization, and modulation of pain. Pain.
2013;154:S44-53. http://dx.doi.org/10.1016/j.pain.2013.07.021
https://doi.org/10.1016/j.pain.2013.07.0...
.
DOES CSD OCCUR IN MIGRAINE?
As already noted, it is generally accepted that CSD occurs in migraine patients and is responsible not only for a visual aura but for other types of aura as well. After weighing the evidence for and against this concept, including new information gained from single-pulse tTMS, we consider the opposite to be true – that it is very unlikely that CSD has any place in the pathophysiology of migraine.
Evidence in favour of CSD in migraine
Blood flow studies
As already noted, Leão reported that the spreading depression in exposed
rabbit brain was associated with marked dilatation of the pial arteries,
with resulting increases in blood flow and in the oxygenation level of the
venous blood (‘in the veins, the rate of flow is strikingly increased, and
these vessels promptly become as scarlet as the arteries88 .Leão AAP. Pial circulation and spreading depression of activity in
the cerebral cortex. J Neurophysiol. 1944;7(6):391-6..) The vasodilatation spread
outwards in the affected hemisphere, following the depression of neural
activity; at a given site it reached its peak in 0.5 – 3 minutes and was
over in another 1.5 – 3 minutes. The vasodilatation was occasionally
followed by a prolonged reduction in arterial caliber. Given Leão’s
description, it was logical to see whether there was altered cortical blood
flow during migraine. The initial studies, undertaken in Copenhagen, were
based on the detection of regional brain radioactivity at different times
following intracarotid injections of radioactive xenon; the testing was
undertaken during spontaneous or induced migraine auras2323 .Olesen J, Larsen B, Lauritzen M. Focal hyperemia followed by
spreading oligemia and impaired activation of rCBF in classic migraine. Ann
Neurol. 1981;9(4):344-52.
http://dx.doi.org/10.1002/ana.410090406
https://doi.org/10.1002/ana.410090406...
. The results were interpreted as evidence
of decreased cortical blood flow (oligaemia) during the aura; the reduction
was first noted in the posterior part of the brain and it then spread into
parietal and temporal areas at a rate of 2-3 mm/min2424 .Lauritzen M. Pathophysiology of the migraine aura. The spreading
depression theory. Brain. 1994;117(1):199-210.
http://dx.doi.org/10.1093/brain/117.1.199
https://doi.org/10.1093/brain/117.1.199...
; rather similar results were subsequently
obtained with PET imaging2525 .Woods RP, Iacoboni M, Mazziotta JC. Brief report: bilateral
spreading cerebral hypoperfusion during spontaneous migraine headache. N Engl J
Med. 1994;331(25):1689-92.
http://dx.doi.org/10.1056/NEJM199412223312505
https://doi.org/10.1056/NEJM199412223312...
.
With the advent of functional MRI, it has been possible to examine cortical
blood flow with greater temporal and spatial resolution than was previously
possible. Perhaps the most impressive results have come from brain oxygen
level dependent (BOLD) measurements2626 .Cao Y, Welch KM, Aurora S, Vikingstad EM. Functional MRI-BOLD of
visually triggered headache in patients with migraine. Arch Neurol.
1999;56(5):548-54. http://dx.doi.org/10.1001/archneur.56.5.548
https://doi.org/10.1001/archneur.56.5.54...
. In one such study, in which 3 subjects were
examined while experiencing a total of 5 visual auras, there was evidence of
vasodilatation which began in the visual cortex (area V3A) and traveled
slowly over adjacent cortical areas; the vasodilatation was followed by a
period of oligaemia2727 .Hadjikhani N, Sanchez del Rio M, Wu O, Schwartz D, Bakker D, Fischl
B et al. Mechanisms of migraine aura revealed by functional MRI in human visual
cortex. Proc Natl Acad Sci USA. 2001;98(8):4687-92.
http://dx.doi.org/10.1073/pnas.071582498
https://doi.org/10.1073/pnas.071582498...
. In
the example shown in Figure 3, taken
from this study, it can be seen that during the vascular changes the BOLD
responses of the visual cortex to checkerboard stimulation were diminished.
All of these events were consistent with Leão’s classical description of
CSD, though the authors were careful to conclude that their results were
attributable to ‘an electrophysiological event such as
CSD’.
(A) Progression of scintillations and scotoma in left visual field of a patient with migraine aura. (B) Simultaneous recording of BOLD signals in the medial surface of the right occipital lobe, showing the rostral spread of a diminished response to checkerboard stimulation. (C) Projection of retina on to occipital cortex, as determined by BOLD method (red, blue and green areas represent foveal, parafoveal and peripheral retinal projections respectively). From Hadjikhani et al.27, reproduced with permission from the National Academy of Sciences of America.
To summarize this part of the evidence, there can no longer be any doubt that some sort of slowly traveling cortical process is responsible for the visual aura, and presumably for other types of aura. However, although CSD resembles this process in a number of ways, the evidence presently available stops short of proving its presence.
Mutant mouse experiments.
The ability to introduce, into mice, mutations of the CACN1A1
gene responsible for familial hemiplegic migraine (FHM) has
provided a valuable new approach to (studying the pathogenesis of migraine,
including the possible role of CSD. The first such mutation to be
investigated was R192Q2828 .Maagdenberg AM, Pietrobon D, Pizzorusso T, Kaja S, Broos LA,
Cesetti T et al. A Cacna1a knockin migraine mouse model with
increased susceptibility to cortical spreading depression. Neuron.
2004;41(5):701-10.
http://dx.doi.org/10.1016/S0896-6273(04)00085-6
https://doi.org/10.1016/S0896-6273(04)00...
,2929 .Cao YQ, Piedras-Rentería ES, Smith GB, Chen G, Harata NC, Tsien RW.
Presynaptic Ca2+ channels compete for channel type-preferring slots
in altered neurotransmission arising from Ca2+ channelopathy. Neuron.
2004;43(3):387-400.
http://dx.doi.org/10.1016/j.neuron.2004.07.014
https://doi.org/10.1016/j.neuron.2004.07...
and this was followed by the S218L mutation
responsible for a more severe form of FHM3030 .Maagenberg AM, Pizzorusso T, Kaja S, Repolilli N, Shapovalova M,
Hoebeek FE et al. High cortical spreading depression susceptibility and
migraine-associated symptoms in Cav2.1 S218L mice. Ann Neurol.
210;67(1):85-98. http://dx.doi.org/10.1002/ana.21815
https://doi.org/10.1002/ana.21815...
. In both types of knockin mouse model,
abnormalities were reported in the CSDs elicited by 100ms current pulses;
the thresholds were found to be reduced and the rate of spread increased.
However, while the results for the homozygous S218 model are strikingly
different from those for wild-type mice, the abnormalities described in the
heterozygous R192Q model are less convincing. Thus the thresholds for mutant
and wild-type mice show 15-fold and 40-fold ranges respectively (Figure 4B), reflecting the difficulty in
obtaining consistent values - much of the stimulating current would have
been shunted through tissue and cerebrospinal fluid overlying the cortex.
Further, although the published figure does indeed show that the passage of
CSD between the two recording sites is faster in a mutant than in a
wild-type mouse, in both types of mouse the velocity appears to be several
times higher between the stimulating electrodes and the nearest recording
electrode (Figure 4A).
(A) Slow waves, associated with spreading depressions, in wild-type (wt) and R192Q K1 mutant mice. Positions of stimulating and recording electrodes shown at top. In the wild type mouse the wave takes approximately 60 s to travel the 2.1 mm separating the recording electrodes, for a velocity of 2.1 mm/min. However, the same wave required only 20 s or so to travel the 4.8 mm separating the stimulating electrodes from the nearest recording electrode, corresponding to a velocity of 14.4 mm/min. The extra distances incurred because of the curvature of the hemispheres have been ignored. (B) Cumulative distributions of thresholds for eliciting CSD in mutant and wild-type mice. From van den Maagdenberg et al.30; reproduced with permission of Elsevier.
Anti-migraine drugs and CSD
Several studies have examined the possibility that anti-migraine drugs might
affect CSD, with the implication that a positive finding would provide
further, albeit circumstantial, evidence for the role of CSD in the
production of aura and headache3131 .Costa C, Tozzi A, Rainero I, Cupini LM, Calabresi P, Ayata C et al.
Cortical spreading depression as a target for anti-migraine agents. J Headache
Pain. 2013;14(1):62. http://dx.doi.org/10.1186/1129-2377-14-62
https://doi.org/10.1186/1129-2377-14-62...
. The results have been variable; for example,
topiramate suppresses CSD effectively inrats3232 .Unekawa M, Tomita Y, Toriumi H, Suzuki N. Suppressive effect of
chronic peroral topiramate on potassium-induced cortical spreading depression in
rats. Cephalalgia. 2012;32(7):518-27.
http://dx.doi.org/10.1177/0333102412444015
https://doi.org/10.1177/0333102412444015...
but not in all cats3333 .Akerman S, Goadsby PJ. Topiramate inhibits cortical spreading
depression in rat and cat: impact in migraine aura. Neuroreport.
2005;16(2):1383-7.
http://dx.doi.org/10.1097/01.wnr.0000175250.33159.a9
https://doi.org/10.1097/01.wnr.000017525...
, and in both species the CSD suppression
is unaccompanied by changes in bloodflow. In contrast, sumatriptan, a very
effective anti-migraine drug, has no effect on CSD3434 .Read SJ, Hirst WD, Upton N, Parsons AA. Cortical spreading
depression produces increased cGMP levels in cortex and brain stem that is
inhibited by tonabersat (SB-220453) but not sumatriptan. Brain Res.
2001;89(1-2):69-77.
http://dx.doi.org/10.1016/S0006-8993(00)03191-7
https://doi.org/10.1016/S0006-8993(00)03...
.
Earlier evidence against CSD in migraine: brain recordings and neural function
(i). Difficulty in inducing CSD in human subjects
In his first paper Leão77 .Leão AAP. Spreading depression of activity in the cerebral cortex.
J Neurophysiol. 1944;7(6):359-90.
reported that it was more difficult to evoke CSD in the cat than in the rabbit,
and this distinction between brains with convoluted and smooth surfaces has been
confirmed3535 .Marshall WH, Essig CF. Relation of air exposure of cortex to
spreading depression of Leão. J Neurophysiol.
1951;14(4):265-73.. In the human
brain, which has many more gyri than the cat, McLachlan and Girvin3636 .McLachlan RS, Girvin JP. Spreading depression of Leão in rodent and
human cortex. Brain Res. 1994;666(1):133-6.
http://dx.doi.org/10.1016/0006-8993(94)90295-X
https://doi.org/10.1016/0006-8993(94)902...
were unable to initiate CSD
in the exposed cortices of 23 patients about to undergo resections for
intractable epilepsy. There are three points that deserve emphasis. First, using
the same techniques (KCl application, electrical and mechanical stimulation),
the authors had no difficulty in inducing CSD in rats. Second, the results could
not be explained by anaesthesia since the patients were only receiving pain
medication with fentanyl and droperidol; though both drugs are known to affect
the EEG3737 .Kushikata T, Araki I, Sato T, Hashimoto Y, Ishihara H, Matsuki A.
[EEG pattern during total intravenous anesthesia with droperidol, fentanyl and
ketamine]. Matsui. 1993;42(8):1194-9. Japanese., neither appears to
interfere with CSD generation3838 .Hunfeld M, Pope KJ, Fitzgibbon SP, Willoughby JO, Broberg M.
Effects of anesthetic agents on seizure-induction with intra-cortical injection
of convulsants. Epilepsy Res. 2013;105(1-2):52-61.
http://dx.doi.org/10.1016/j.eplepsyres.2012.12.009
https://doi.org/10.1016/j.eplepsyres.201...
,3939 .Hertle DN, Dreier JP, Woitzik J, Hartings JA, Bullock R, Okonkwo DO
et al. Effect of analgesics and sedatives on the occurrence of spreading
depolarizations accompanying acute brain injury. Brain. 2012;135(8):2390-8.
http://dx.doi.org/10.1093/brain/aws152
https://doi.org/10.1093/brain/aws152...
. Indeed, the fact that droperidol may terminate
migraine attacks4040 .Nerenberg RH, Friedman BW. Migraine: an evidence-based update.
Emerg Med. 2014;46(7):294-316. and yet not
affect CSD constitutes another argument against a role for the latter in the
pathogenesis of the headache. Third, four of the patients had suffered from
migraine, though without auras. Although there have since been reports of CSD
detection in human subjects, using electrocorticography sometimes combined with
scalp EEG, it is relevant that the CSD was only associated with major brain
lesions, either trauma4141 .Fabricius M, Fuhr S, Bhatia R, Boutelle M, Hashemi P, Strong AJ et
al. Cortical spreading depression and peri-infarct depolarization in acutely
injured human cerebral cortex. Brain. 2006;129(3):778-90.
http://dx.doi.org/10.1093/brain/awh716
https://doi.org/10.1093/brain/awh716...
,
subarachnoid haemorrhage or stroke4242 .Drenckhahn C, Winkler MKL, Major S, Scheel M, Kang EJ, Pinczolits A
et al. Correlates of spreading depolarization in human scalp
electroencephalography. Brain. 2012;135(3):853-68.
http://dx.doi.org/10.1093/brain/aws010
https://doi.org/10.1093/brain/aws010...
.
EEG recordings during migraine aura
Although it is technically possible to detect CSD in human subjects with scalp electrodes (see above), any depression of EEG activity during either the aura or the headache phase of migraine is mild or absent; there may be some increase in slow wave activity but, in a proportion of patients, the EEG may be quite normal4343 .Sand T. EEG in migraine: a review of the literature. Funct Neurol. 1991;6(1):7-22..
Preservation of neural function during the aura
It is well known that sensory function may be lost during an aura, the best example being the scotoma that replaces the scintillations as the latter progress across the visual field. However, the association is not invariable, for in a minority of subjects the fortification spectra may be superimposed on a normal visual image. Similarly, subjects who describe numbness and tingling can still detect touch. Such preservation of function is incompatible with a process, such as CSD, that, at least in animal models, abolishes all neural activity.
New evidence against CSD in migraine: therapeutic transcranial magnetic stimulation
Transcranial magnetic stimulation (TMS) has been employed as a means of exploring CNS
function for almost 30 years, its great advantage over electrical stimulation being
that it is painless and penetrates the skull more readily; nevertheless the magnetic
pulse is converted to an electrical one on intersecting suitably disposed biological
conductors (axons and dendrites). Depending on the choice of target and stimulus
parameters, TMS can be employed to either excite or block function in a neural
circuit4444 .Hallett M. Transcranial magnetic stimulation and the human brain.
Nature. 2000;406(6792):147-50.
http://dx.doi.org/10.1038/35018000
https://doi.org/10.1038/35018000...
. The possibility of
TMS being a useful treatment for migraine was raised in 2004 when it was found that
a course of repetitive TMS, when applied to the left dorsolateral prefrontal cortex
on alternate days, reduced both the frequency and the intensity of migraine attacks;
moreover, the benefit persisted for at least one month4545 .Brighina F, Piazza A, Vitello G, Aloisio A, Palermo A, Daniele O et
al. rTMS of the prefrontal cortex in the treatment of chronic migraine:a pilot
study. J Neurol Sci. 2004;227(1):67-71.
http://dx.doi.org/10.1016/j.jns.2004.08.008
https://doi.org/10.1016/j.jns.2004.08.00...
. Similar, but unexpected, findings were
subsequently reported in two migraineurs treated in a similar way for
depression4646 .O’Reardon JP, Fontecha JF, Cristancho MA, Newman S. Unexpected
reduction in migraine and psychogenic headaches following rTMS treatment for
major depression: a report of two cases. CNS Spectr. 2007;12(12):921-5.
http://dx.doi.org/10.1017/S1092852900015716
https://doi.org/10.1017/S109285290001571...
. Our own interest
in tTMS for migraine arose from observations made on one of the authors, a migraine
sufferer himself, and on patient volunteers4747 .Fisher MJ. Goodbye headaches: a gun that shoots magnetic pulses
puts and end to an age-old scourge. Discover. 2004;25(8):28-9.. Unlike the aforementioned studies, TMS was applied at
the onset of the headache and given as a single pulse, sometimes repeated once, to
the back of the head; the headache was found to diminish and sometimes to disappear
altogether. The working hypothesis - a mistaken one (see below) - was that
stimulation of the cortex in advance of a CSD was halting the progress of the
latter, rather as the creation of a firebreak may stop a forest fire. Any CSD
present during the attack was presumed to have commenced in the occipital cortex,
since visual symptoms constitute the most common aura. Since that time there have
been several controlled trials of single or double pulse TMS administered to the
occipital cortex during the migraine attack, and in all cases there have been
positive outcomes55 .McComas A, Upton A. Therapeutic transcranial magnetic stimulation
in migraine and its implications for a neuroinflammatory hypothesis.
Inflammopharmacology. 2009;17(2):68-75.
http://dx.doi.org/10.1007/s10787-009-8058-7
https://doi.org/10.1007/s10787-009-8058-...
,4848 .Clarke BM, Upton ARKamath MV, Al-Harbi T, Castellanos CM.
Transcranial magnetic stimulation for migraine:clinical effects. J Headache
Pain. 2006;7(5):341-6.
http://dx.doi.org/10.1007/s10194-006-0329-8
https://doi.org/10.1007/s10194-006-0329-...
,4949 .Mohammed YM, Hughes G, Kothari R, Nkrumah M, Fischell S, Robert F
et al. Self-administered transcranial magnetic stimulation (TMS) during the aura
phase improves and aborts headaches. Headache. 2006;46:839.,5050 .Lipton RB, Dodick DW, Silberstein SD, Saper JR, Aurora SK, Pearlman
SH et al. Single-pulse transcranial magnetic stimulation for acute treatment of
migraine with aura: a randomised, double-blind, parallel-group, sham-controlled
trial. Lancet Neurol. 2010;9(4):373-80.
http://dx.doi.org/10.1016/S1474-4422(10)70054-5
https://doi.org/10.1016/S1474-4422(10)70...
,5151 .Lipton RB, Pearlman SH. Transcranial magnetic stimulation in the
treatment of migraine. Neurotherapeutics. 2010;7(2):204-12.
http://dx.doi.org/10.1016/j.nurt.2010.03.002
https://doi.org/10.1016/j.nurt.2010.03.0...
. In the latter two trials, the patients were able to
deliver the magnetic pulses themselves, using a portable hand-held device designed
specifically for that purpose.
While the above studies were conceived on the basis of a pivotal role for CSD in
migraine, additional studies with tTMS made it unlikely that CSD is responsible for
either the aura or the headache. The crucial observation was that, in some subjects,
the effects of tTMS were immediately apparent. No one showed this effect more
convincingly than K.B., a 71-year old woman with a long history of familial
migraine, initially basilar in its symptomatology but later progressing to daily
episodes associated with multiple body pains, tingling, hallucinations,
quadriplegia, coma and respiratory arrest55 .McComas A, Upton A. Therapeutic transcranial magnetic stimulation
in migraine and its implications for a neuroinflammatory hypothesis.
Inflammopharmacology. 2009;17(2):68-75.
http://dx.doi.org/10.1007/s10787-009-8058-7
https://doi.org/10.1007/s10787-009-8058-...
,5252 .Upton A, McComas A. Abolition of migraine by transcranial magnetic
simulation. Abstracts of 40th Meeting of the Canadian Congress of Neurological
Sciences; 2005 June 14-18; Otawwa. CalgarY: Canadian Congress of Neurological
Sciences; 2005. (Can J Neurol Sci. 2005;32(2 suppl 1):S71.,5353 .McComas AJ. The artful chameleon: an exploration of migraine and
medicine. West Flamborough: Alkat Neuroscience; 2006.. It was found that, when directed to the appropriate site
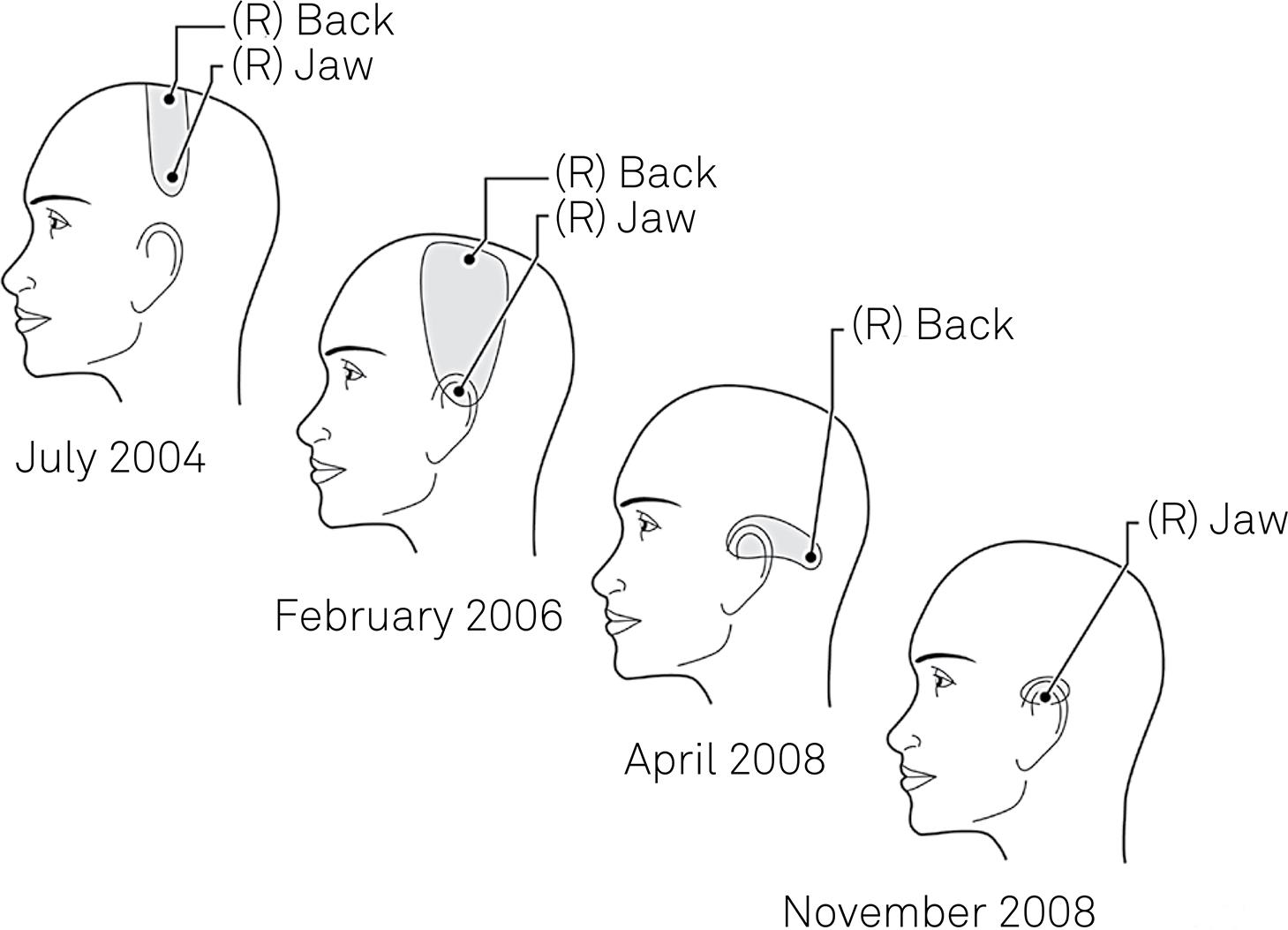
on the hemisphere opposite to the symptoms, tTMS could abolish each symptom
instantaneously (Figure 5). The
significance of the rapidity of action of tTMS is considered below, while Table summarizes the evidence for, and against,
a role for CSD in the genesis of aura and headache in migraine.
Optimal TMS tilted coil placements for abolishing different regions of pain (shaded) in patient KB, at the start of her treatment in July 2004. From McComas 53. The positions roughly correspond to the homoncular map in the postcentral gyrus, as determined by Penfield & Boldrey62.
Mode of action of electrical (and magnetic) stimulation of cortex
The results of tTMS make it clear that CSD could not be involved in either the aura or the headache of migraine. The first reason is that, as Leão described in his first paper, electrical stimulation (and hence TMS) is unable to stop CSD, whether applied before or after the traveling disturbance reaches the stimulus location on the cortex77 .Leão AAP. Spreading depression of activity in the cerebral cortex. J Neurophysiol. 1944;7(6):359-90.. The second reason is that CSD involves such a profound disturbance of water, electrolytes and neurotransmitters, that it is inconceivable that these could be corrected instantly by any means, including TMS. Clearly, then, TMS is doing something else, but what?
The first attempt to understand the local effects of stimulating the cortex appears
to have been made by Adrian5454 .Adrian ED. The spread of activity in the cerebral cortex. J
Physiol. 1937;88(2):127-61.
http://dx.doi.org/10.1113/jphysiol.1936.sp003427
https://doi.org/10.1113/jphysiol.1936.sp...
in
anaesthetized rabbits; he recorded a local negativity generated by superficial
elements in the cortex. With stronger, repeated stimulation the evoked surface
potentials became positive due to depolarization of deeper cortical structures; at
the same time the responses spread further across the cortex. Much later Krnjevic et
al.55 .McComas A, Upton A. Therapeutic transcranial magnetic stimulation
in migraine and its implications for a neuroinflammatory hypothesis.
Inflammopharmacology. 2009;17(2):68-75.
http://dx.doi.org/10.1007/s10787-009-8058-7
https://doi.org/10.1007/s10787-009-8058-...
showed that single
cortical shocks, especially when applied to the deeper layers, produced powerful
inhibition of spontaneous and evoked neural activity in the cortex. In a second
paper the same authors identified basket cells as the inhibitory neurones; these
cells were at the appropriate depth and their axon terminals formed nests around the
bodies of the pyramidal cells5656 .Krnjević K, Randić M, Straughan DW. Nature of a cortical inhibitory
process. J Physiol. 1966;184(1):49-77.
http://dx.doi.org/10.1113/jphysiol.1966.sp007903
https://doi.org/10.1113/jphysiol.1966.sp...
.
While the excitation of the basket cells would account for the ability of a single
TMS pulse to abruptly stop ongoing activity in pyramidal cells producing migraine
symptoms, the cause of the latter activity remains elusive. One explanation is that
the activity is generated by high frequency oscillations in the distal axons of
pyramidal cells, and impulses then travel antidromically and excite the cell bodies
(Figure 6). Normally this back-propagation
would be prevented by intervening axoaxonic inhibitory synapses5757 .Dugladze T, Schmitz D, Whittington MA, Vida I, Gloveli T.
Segregation of axonal and somatic activity during fast network oscillations.
Science. 2012;336(6087):1458-61.
http://dx.doi.org/10.1126/science.1222017
https://doi.org/10.1126/science.1222017...
. Another, not exclusive, possibility is that the
activity in the pyramidal cells is triggered by fast rhythmic bursting neurones in
the cortex5858 .Cunningham MO, Whittington MA, Bibbig A, Roopun A, LeBeau FE, Vogt
A et al. A role for fast rhythmic bursting neurons in cortical gamma
oscillations in vitro. Proc Natl Acad Sci USA.
2004;101(18):7152-7.. In either instance
the spread of the excitatory activity, and hence the spread of migraine symptoms,
would be mediated by gap junctions reported to exist between the axons of pyramidal
cells5959 .Traub R, Bibbig A, LeBeau FE, Cunningham MO, Whittington MA.
Persistent gamma oscillations in superficial layers of rat auditory neocortex:
experiment and model. J Physiol. 2005;562(1):3-8.
http://dx.doi.org/10.1113/jphysiol.2004.074641
https://doi.org/10.1113/jphysiol.2004.07...
.
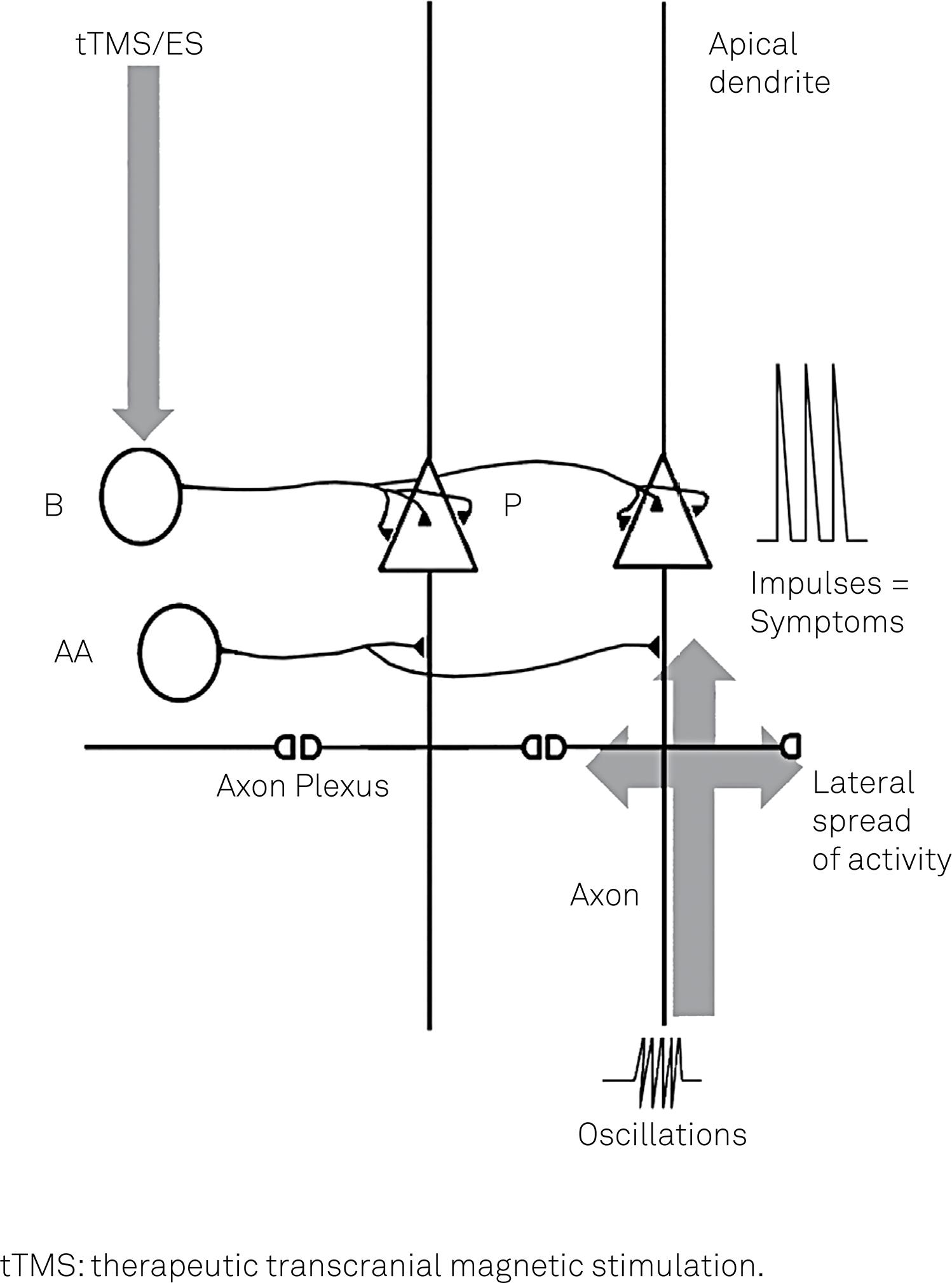
Hypothetical scheme to explain the spreading neural activity responsible for migraine auras and headache. Fast oscillatory activity is present in the distal axons of principal pyramidal neurons (P) in cortical layers 2-4. This activity produces ‘spikelets’ which travel back towards the cell bodies of the P neurons and also across gap junctions formed with neighbouring axons (the axon plexus), causing a slow spread of activity in the cortex. The spikelets do not usually invade the cell bodies because of intervening inhibitory synapses from axoaxonic cells (AA); should the latter not operate, however, the spikelets will set off fully formed impulses in the P cells, causing migraine symptoms. tTMS and electrical stimulation (ES) are effective because they stimulate the basket cells (B), which then powerfully inhibit the P cells. The scheme is based on work by Traub et al.59 and Dugladze et al.57.
The emerging picture
On the basis of the findings and considerations reported above, it is now possible to suggest what may actually be happening in the brain during an attack of migraine:
(i). Almost all the varied symptoms of migraine, including the headache, arise from abnormal impulse activity in the pyramidal cells within the outer layers (II and III) of the cerebral cortex; it is such cells in the primary somatosensory receiving area (S1) that are responsible for the headache. The cause of the neural hyperactivity is not known; back-propagation of impulses from the distal axon is only one of several possibilities. At a molecular level the defect could involve altered calcium fluxes at synapses, in keeping with results in mutant mice expressing the CACN1A gene, but other mechanisms are possible. Diminished serotonergic control of the cortex from the brain stem is a different type of mechanism that may also operate. Whatever the cause of the hyperactivity in pyramidal cells, the process slowly spreads, probably through gap junctions between axons.
(ii). The transiently hyperactive cells in V1 (the primary visual area), responsible for the fortification spectra during a visual aura, are the orientation-sensitive ones. However, V1 also contains colour-sensitive cells and excitation of these accounts for the coloured ‘blobs’ experienced by some patients in their auras.
(iii). The spreading mechanism is not confined to the visual cortex, but may occur in the somatosensory cortex as well. An example is given by Liveing in his classic monograph, in which a patient ‘felt a numbness in her right leg, ascending to the trunk, right arm and face’ 6060 .Liveing E. On megrim, sick-headache and some allied disorders: a contribution to the pathology of nerve-storms. London: Churchill; 1873..
(iv). The presence of multiple symptoms preceding or during the headache is due to
independent islands of cortical hyperactivity, rather than to spread of
hyperactivity over long distances. This conclusion comes from the observation that
different symptoms can occur within seconds of each other, as in patient KB
described earlier55 .McComas A, Upton A. Therapeutic transcranial magnetic stimulation
in migraine and its implications for a neuroinflammatory hypothesis.
Inflammopharmacology. 2009;17(2):68-75.
http://dx.doi.org/10.1007/s10787-009-8058-7
https://doi.org/10.1007/s10787-009-8058-...
,5353 .McComas AJ. The artful chameleon: an exploration of migraine and
medicine. West Flamborough: Alkat Neuroscience; 2006..
(v). During an attack of migraine TMS can be instantaneously effective by exciting the cortical basket cells and thereby inhibiting the firing of the pyramidal cells.
(vi). Finally and importantly, cortical spreading depression is not involved in either the aura or the headache of migraine. It would therefore be appropriate to refer to the process responsible for these phenomena simply as ‘migrainous neural activity’ (MNA).
UNANSWERED QUESTIONS
As already noted, we do not know what sets off the excessive impulse activity in the cortical pyramidal cells, though possible mechanisms have been identified. Similarly, it is not clear why tTMS should work in some patients and not in others, nor is it obvious why the reduction in the intensity of the headache may sometimes take several minutes to appear. Again, why should it be possible to improve the headache by applying tTMS to the back of the head, when the headache is due to neural activity in the primary somatosensory area? We also need to learn why, in a severely affected patient, the optimal site for tTMS may move and the intervention eventually lose its efficacy, as was the case with patient KB6161 .McComas AJ. TMS in a severe case of complex migraine. Lecture Notes of the ICB Seminar. 2009;83:164-9. (see above and Figure 7). Though these questions remain to be answered, it is our submission that the elimination of cortical spreading depression as a migraine mechanism is an important step in resolving the pathophysiology of this puzzling disorder.
References
-
1Goadsby PJ, Charbit AR, Andreou AP, Akerman S, Holland PR. Neurobiology of migraine. Neuroscience. 2009;161(2):327-41. http://dx.doi.org/10.1016/j.neuroscience.2009.03.019
» https://doi.org/10.1016/j.neuroscience.2009.03.019 -
2Tietjen GE. Migraine as a systemic vasculopathy. Cephalalgia. 2009;29(9):989-996. http://dx.doi.org/10.1111/j.1468-2982.2009.01937.x
» https://doi.org/10.1111/j.1468-2982.2009.01937.x -
3Cutrer FM. Pathophysiology of migraine. Semin Neurol. 2010;30(2):120-30. http://dx.doi.org/10.1055/s-0030-1249222
» https://doi.org/10.1055/s-0030-1249222 -
4Pietrobon D, Moskowitz MA. Pathophysiology of migraine. Ann Rev Physiol, 2013;75:365-91. http://dx.doi.org/10.1146/annurev-physiol-030212-183717
» https://doi.org/10.1146/annurev-physiol-030212-183717 -
5McComas A, Upton A. Therapeutic transcranial magnetic stimulation in migraine and its implications for a neuroinflammatory hypothesis. Inflammopharmacology. 2009;17(2):68-75. http://dx.doi.org/10.1007/s10787-009-8058-7
» https://doi.org/10.1007/s10787-009-8058-7 -
6Aurora SK, Wilkinson F. The brain is hyperexcitable in migraine. Cephalalgia. 2007;27(12):1442-53. http://dx.doi.org/10.1111/j.1468-2982.2007.01502.x
» https://doi.org/10.1111/j.1468-2982.2007.01502.x -
7Leão AAP. Spreading depression of activity in the cerebral cortex. J Neurophysiol. 1944;7(6):359-90.
-
8Leão AAP. Pial circulation and spreading depression of activity in the cerebral cortex. J Neurophysiol. 1944;7(6):391-6.
-
9Leão AAP, Morison RS. Propagation of spreading cortical depression. J Neurophysio.l 1945;8(1):33-45.
-
10Leão AAP. Further observations on the spreading depression of activity in the cerebral cortex. J Neurophysiol. 1947;10(6):409-14.
-
11Smith JM, Bradley DP, James MF, Huang CL-H. Physiological studies of cortical spreading depression. Biol Rev Camb Philos Soc. 2006;81(4):457-81. http://dx.doi.org/10.1017/S1464793106007081
» https://doi.org/10.1017/S1464793106007081 -
12Charles A, Brennan KC. Cortical spreading depression—new insights and persistent questions. Cephalalgia. 2009;29(10):1115-24. http://dx.doi.org/10.1111/j.1468-2982.2009.01983.x
» https://doi.org/10.1111/j.1468-2982.2009.01983.x -
13Sugaya E, Takato M, Noda Y. Neuronal and glial activity during spreading depression in cerebral cortex of cat. J Neurophysiol. 1975;38(4):822-41.
-
14Lashley KS. Patterns of cerebral integration indicated by the scotomas of migraine. Arch Neurol Psychiatry. 1941;46(2):331-9. http://dx.doi.org/10.1001/archneurpsyc.1941.02280200137007
» https://doi.org/10.1001/archneurpsyc.1941.02280200137007 -
15Milner PM. Note on a possible correspondence between the scotomas of migraine and spreading depression of Leão. Electroencephalogr Clin Neurophysiol. 1958;10(4):705. http://dx.doi.org/10.1016/0013-4694(58)90073-7
» https://doi.org/10.1016/0013-4694(58)90073-7 -
16Hubel DH, Wiesel TN. Receptive fields of single neurons in the cat striate cortex. J Physiol. 1959;148(3)-574-91. http://dx.doi.org/10.1113/jphysiol.1959.sp006308
» https://doi.org/10.1113/jphysiol.1959.sp006308 -
17Richards W. The fortification illusions of migraine. Sci Am. 1971;224:88-96.
-
18Vincent MB, Hadjikhani N. Migraine aura and related phenomena: beyond scotomata and scintillations. Cephalalgia. 2007:27(12):1368-77. http://dx.doi.org/10.1111/j.1468-2982.2007.01388.x
» https://doi.org/10.1111/j.1468-2982.2007.01388.x -
19Moskowitz MA. The neurobiology of vascular head pain. Ann Neurol. 1984;16(2):157-68. http://dx.doi.org/10.1002/ana.410160202
» https://doi.org/10.1002/ana.410160202 -
20Goadsby PJ, Edvinsson L, Ekman R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol. 1990;28(2):183-7. http://dx.doi.org/10.1002/ana.410280213
» https://doi.org/10.1002/ana.410280213 -
21Burstein R. Deconstructing migraine headache into peripheral and central sensitization. Pain. 2001;89(2-3):107-10. http://dx.doi.org/10.1016/S0304-3959(00)00478-4
» https://doi.org/10.1016/S0304-3959(00)00478-4 -
22Noseda R, Burstein R. Migraine pathophysiology: anatomy of the trigeminovascular pathway and associated neurological symptoms, cortical spreading depression, sensitization, and modulation of pain. Pain. 2013;154:S44-53. http://dx.doi.org/10.1016/j.pain.2013.07.021
» https://doi.org/10.1016/j.pain.2013.07.021 -
23Olesen J, Larsen B, Lauritzen M. Focal hyperemia followed by spreading oligemia and impaired activation of rCBF in classic migraine. Ann Neurol. 1981;9(4):344-52. http://dx.doi.org/10.1002/ana.410090406
» https://doi.org/10.1002/ana.410090406 -
24Lauritzen M. Pathophysiology of the migraine aura. The spreading depression theory. Brain. 1994;117(1):199-210. http://dx.doi.org/10.1093/brain/117.1.199
» https://doi.org/10.1093/brain/117.1.199 -
25Woods RP, Iacoboni M, Mazziotta JC. Brief report: bilateral spreading cerebral hypoperfusion during spontaneous migraine headache. N Engl J Med. 1994;331(25):1689-92. http://dx.doi.org/10.1056/NEJM199412223312505
» https://doi.org/10.1056/NEJM199412223312505 -
26Cao Y, Welch KM, Aurora S, Vikingstad EM. Functional MRI-BOLD of visually triggered headache in patients with migraine. Arch Neurol. 1999;56(5):548-54. http://dx.doi.org/10.1001/archneur.56.5.548
» https://doi.org/10.1001/archneur.56.5.548 -
27Hadjikhani N, Sanchez del Rio M, Wu O, Schwartz D, Bakker D, Fischl B et al. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci USA. 2001;98(8):4687-92. http://dx.doi.org/10.1073/pnas.071582498
» https://doi.org/10.1073/pnas.071582498 -
28Maagdenberg AM, Pietrobon D, Pizzorusso T, Kaja S, Broos LA, Cesetti T et al. A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron. 2004;41(5):701-10. http://dx.doi.org/10.1016/S0896-6273(04)00085-6
» https://doi.org/10.1016/S0896-6273(04)00085-6 -
29Cao YQ, Piedras-Rentería ES, Smith GB, Chen G, Harata NC, Tsien RW. Presynaptic Ca2+ channels compete for channel type-preferring slots in altered neurotransmission arising from Ca2+ channelopathy. Neuron. 2004;43(3):387-400. http://dx.doi.org/10.1016/j.neuron.2004.07.014
» https://doi.org/10.1016/j.neuron.2004.07.014 -
30Maagenberg AM, Pizzorusso T, Kaja S, Repolilli N, Shapovalova M, Hoebeek FE et al. High cortical spreading depression susceptibility and migraine-associated symptoms in Cav2.1 S218L mice. Ann Neurol. 210;67(1):85-98. http://dx.doi.org/10.1002/ana.21815
» https://doi.org/10.1002/ana.21815 -
31Costa C, Tozzi A, Rainero I, Cupini LM, Calabresi P, Ayata C et al. Cortical spreading depression as a target for anti-migraine agents. J Headache Pain. 2013;14(1):62. http://dx.doi.org/10.1186/1129-2377-14-62
» https://doi.org/10.1186/1129-2377-14-62 -
32Unekawa M, Tomita Y, Toriumi H, Suzuki N. Suppressive effect of chronic peroral topiramate on potassium-induced cortical spreading depression in rats. Cephalalgia. 2012;32(7):518-27. http://dx.doi.org/10.1177/0333102412444015
» https://doi.org/10.1177/0333102412444015 -
33Akerman S, Goadsby PJ. Topiramate inhibits cortical spreading depression in rat and cat: impact in migraine aura. Neuroreport. 2005;16(2):1383-7. http://dx.doi.org/10.1097/01.wnr.0000175250.33159.a9
» https://doi.org/10.1097/01.wnr.0000175250.33159.a9 -
34Read SJ, Hirst WD, Upton N, Parsons AA. Cortical spreading depression produces increased cGMP levels in cortex and brain stem that is inhibited by tonabersat (SB-220453) but not sumatriptan. Brain Res. 2001;89(1-2):69-77. http://dx.doi.org/10.1016/S0006-8993(00)03191-7
» https://doi.org/10.1016/S0006-8993(00)03191-7 -
35Marshall WH, Essig CF. Relation of air exposure of cortex to spreading depression of Leão. J Neurophysiol. 1951;14(4):265-73.
-
36McLachlan RS, Girvin JP. Spreading depression of Leão in rodent and human cortex. Brain Res. 1994;666(1):133-6. http://dx.doi.org/10.1016/0006-8993(94)90295-X
» https://doi.org/10.1016/0006-8993(94)90295-X -
37Kushikata T, Araki I, Sato T, Hashimoto Y, Ishihara H, Matsuki A. [EEG pattern during total intravenous anesthesia with droperidol, fentanyl and ketamine]. Matsui. 1993;42(8):1194-9. Japanese.
-
38Hunfeld M, Pope KJ, Fitzgibbon SP, Willoughby JO, Broberg M. Effects of anesthetic agents on seizure-induction with intra-cortical injection of convulsants. Epilepsy Res. 2013;105(1-2):52-61. http://dx.doi.org/10.1016/j.eplepsyres.2012.12.009
» https://doi.org/10.1016/j.eplepsyres.2012.12.009 -
39Hertle DN, Dreier JP, Woitzik J, Hartings JA, Bullock R, Okonkwo DO et al. Effect of analgesics and sedatives on the occurrence of spreading depolarizations accompanying acute brain injury. Brain. 2012;135(8):2390-8. http://dx.doi.org/10.1093/brain/aws152
» https://doi.org/10.1093/brain/aws152 -
40Nerenberg RH, Friedman BW. Migraine: an evidence-based update. Emerg Med. 2014;46(7):294-316.
-
41Fabricius M, Fuhr S, Bhatia R, Boutelle M, Hashemi P, Strong AJ et al. Cortical spreading depression and peri-infarct depolarization in acutely injured human cerebral cortex. Brain. 2006;129(3):778-90. http://dx.doi.org/10.1093/brain/awh716
» https://doi.org/10.1093/brain/awh716 -
42Drenckhahn C, Winkler MKL, Major S, Scheel M, Kang EJ, Pinczolits A et al. Correlates of spreading depolarization in human scalp electroencephalography. Brain. 2012;135(3):853-68. http://dx.doi.org/10.1093/brain/aws010
» https://doi.org/10.1093/brain/aws010 -
43Sand T. EEG in migraine: a review of the literature. Funct Neurol. 1991;6(1):7-22.
-
44Hallett M. Transcranial magnetic stimulation and the human brain. Nature. 2000;406(6792):147-50. http://dx.doi.org/10.1038/35018000
» https://doi.org/10.1038/35018000 -
45Brighina F, Piazza A, Vitello G, Aloisio A, Palermo A, Daniele O et al. rTMS of the prefrontal cortex in the treatment of chronic migraine:a pilot study. J Neurol Sci. 2004;227(1):67-71. http://dx.doi.org/10.1016/j.jns.2004.08.008
» https://doi.org/10.1016/j.jns.2004.08.008 -
46O’Reardon JP, Fontecha JF, Cristancho MA, Newman S. Unexpected reduction in migraine and psychogenic headaches following rTMS treatment for major depression: a report of two cases. CNS Spectr. 2007;12(12):921-5. http://dx.doi.org/10.1017/S1092852900015716
» https://doi.org/10.1017/S1092852900015716 -
47Fisher MJ. Goodbye headaches: a gun that shoots magnetic pulses puts and end to an age-old scourge. Discover. 2004;25(8):28-9.
-
48Clarke BM, Upton ARKamath MV, Al-Harbi T, Castellanos CM. Transcranial magnetic stimulation for migraine:clinical effects. J Headache Pain. 2006;7(5):341-6. http://dx.doi.org/10.1007/s10194-006-0329-8
» https://doi.org/10.1007/s10194-006-0329-8 -
49Mohammed YM, Hughes G, Kothari R, Nkrumah M, Fischell S, Robert F et al. Self-administered transcranial magnetic stimulation (TMS) during the aura phase improves and aborts headaches. Headache. 2006;46:839.
-
50Lipton RB, Dodick DW, Silberstein SD, Saper JR, Aurora SK, Pearlman SH et al. Single-pulse transcranial magnetic stimulation for acute treatment of migraine with aura: a randomised, double-blind, parallel-group, sham-controlled trial. Lancet Neurol. 2010;9(4):373-80. http://dx.doi.org/10.1016/S1474-4422(10)70054-5
» https://doi.org/10.1016/S1474-4422(10)70054-5 -
51Lipton RB, Pearlman SH. Transcranial magnetic stimulation in the treatment of migraine. Neurotherapeutics. 2010;7(2):204-12. http://dx.doi.org/10.1016/j.nurt.2010.03.002
» https://doi.org/10.1016/j.nurt.2010.03.002 -
52Upton A, McComas A. Abolition of migraine by transcranial magnetic simulation. Abstracts of 40th Meeting of the Canadian Congress of Neurological Sciences; 2005 June 14-18; Otawwa. CalgarY: Canadian Congress of Neurological Sciences; 2005. (Can J Neurol Sci. 2005;32(2 suppl 1):S71.
-
53McComas AJ. The artful chameleon: an exploration of migraine and medicine. West Flamborough: Alkat Neuroscience; 2006.
-
54Adrian ED. The spread of activity in the cerebral cortex. J Physiol. 1937;88(2):127-61. http://dx.doi.org/10.1113/jphysiol.1936.sp003427
» https://doi.org/10.1113/jphysiol.1936.sp003427 -
55Krnjević K, Randić M, Straughan DW. An inhibitory process in the cerebral cortex. J Physiol. 1966;184(1):16-48. http://dx.doi.org/10.1113/jphysiol.1966.sp007902
» https://doi.org/10.1113/jphysiol.1966.sp007902 -
56Krnjević K, Randić M, Straughan DW. Nature of a cortical inhibitory process. J Physiol. 1966;184(1):49-77. http://dx.doi.org/10.1113/jphysiol.1966.sp007903
» https://doi.org/10.1113/jphysiol.1966.sp007903 -
57Dugladze T, Schmitz D, Whittington MA, Vida I, Gloveli T. Segregation of axonal and somatic activity during fast network oscillations. Science. 2012;336(6087):1458-61. http://dx.doi.org/10.1126/science.1222017
» https://doi.org/10.1126/science.1222017 -
58Cunningham MO, Whittington MA, Bibbig A, Roopun A, LeBeau FE, Vogt A et al. A role for fast rhythmic bursting neurons in cortical gamma oscillations in vitro. Proc Natl Acad Sci USA. 2004;101(18):7152-7.
-
59Traub R, Bibbig A, LeBeau FE, Cunningham MO, Whittington MA. Persistent gamma oscillations in superficial layers of rat auditory neocortex: experiment and model. J Physiol. 2005;562(1):3-8. http://dx.doi.org/10.1113/jphysiol.2004.074641
» https://doi.org/10.1113/jphysiol.2004.074641 -
60Liveing E. On megrim, sick-headache and some allied disorders: a contribution to the pathology of nerve-storms. London: Churchill; 1873.
-
61McComas AJ. TMS in a severe case of complex migraine. Lecture Notes of the ICB Seminar. 2009;83:164-9.
-
62Penfield W, Boldrey E. Somatic motor and sensory representation in the cerebral cortex of man as studied by electrical stimulation. Brain. 1937;60(4):389-443. http://dx.doi.org/10.1093/brain/60.4.389
» https://doi.org/10.1093/brain/60.4.389
Publication Dates
-
Publication in this collection
Aug 2015
History
-
Received
02 Dec 2014 -
Reviewed
29 Mar 2015 -
Accepted
17 Apr 2015