ABSTRACT
Mitochondrial myopathy, Encephalopathy, Lactic Acidosis, and Stroke-like episodes (MELAS) is a rare mitochondrial disorder. Diagnostic criteria for MELAS include typical manifestations of the disease: stroke-like episodes, encephalopathy, evidence of mitochondrial dysfunction (laboratorial or histological) and known mitochondrial DNA gene mutations. Clinical features of MELAS are not necessarily uniform in the early stages of the disease, and correlations between clinical manifestations and physiopathology have not been fully elucidated. It is estimated that point mutations in the tRNALeu(UUR) gene of the DNAmt, mainly A3243G, are responsible for more of 80% of MELAS cases. Morphological changes seen upon muscle biopsy in MELAS include a substantive proportion of ragged red fibers (RRF) and the presence of vessels with a strong reaction for succinate dehydrogenase. In this review, we discuss mainly diagnostic criterion, clinical and laboratory manifestations, brain images, histology and molecular findings as well as some differential diagnoses and current treatments.
MELAS; mitochondria; myopathy; stroke; encephalopathy; genetics
RESUMO
Miopatia mitocondrial, encefalopatia, acidose lática, e episódios semelhantes a acidente vascular cerebral (MELAS) é uma rara doença mitocondrial. Os critérios diagnósticos para MELAS incluem as manifestações típicas da doença: episódios semelhantes a acidente vascular cerebral, encefalopatia, evidência de disfunção mitocondrial (laboratorial ou histológica) e mutação conhecida em genes do DNA mitocondrial. Na fase inicial da doença, as manifestações clínicas podem não ser uniformes, e sua correlação com a fisiopatologia não está completamente elucidada. Estima-se que as mutações de ponto no gene tRNALeu(UUR) do DNAmt, principalmente a A3243G, sejam responsáveis por cerca de 80% dos casos de MELAS. As alterações morfológicas na biópsia muscular incluem uma grande proporção de fibras vermelhas rasgadas (RRF) e presença de vasos com forte reação para succinato desidrogenase. Nesta revisão, são discutidos os principais critérios diagnósticos, manifestações clínicas e laboratoriais, imagens cerebrais, padrões eletrofisiológicos, histológicos e alterações moleculares, bem como alguns dos diagnósticos diferenciais e tratamentos atuais.
MELAS; mitocôndria; miopatia; acidente vascular cerebral; encefalopatia; genética
The first description of cases with clinical features suggested MELAS was in 197511 Koenigsberger MR, Pellock JM, DiMauro S, Eastwood A.. Juvenile mitochondrial myopathy, short stature and lactic acidosis: a clinical, biochemical, and ultrastructural study (Abstract #80). In: Fifth Annual Meeting of the Child Neurological Society; 1976 October 28-30; Monterey, CA.,22 Shapira Y, Cederbaum SD, Cancilla PA, Nielsen D, Lippe BM. Familial poliodystrophy, mitochondrial myopathy, and lactate acidemia. Neurology. 1975;25(7):614-21. doi:10.1212/WNL.25.7.614
https://doi.org/10.1212/WNL.25.7.614...
. The patients common point was the presence of mitochondrial myopathy associated with brain changes, such as mental retardation, seizures, myoclonus, ophthalmoplegia, retinitis pigmentosa, blindness, calcification in basal ganglia and sudden hemiplegia suggestive of stroke11 Koenigsberger MR, Pellock JM, DiMauro S, Eastwood A.. Juvenile mitochondrial myopathy, short stature and lactic acidosis: a clinical, biochemical, and ultrastructural study (Abstract #80). In: Fifth Annual Meeting of the Child Neurological Society; 1976 October 28-30; Monterey, CA.,22 Shapira Y, Cederbaum SD, Cancilla PA, Nielsen D, Lippe BM. Familial poliodystrophy, mitochondrial myopathy, and lactate acidemia. Neurology. 1975;25(7):614-21. doi:10.1212/WNL.25.7.614
https://doi.org/10.1212/WNL.25.7.614...
. In the following years other cases with similar findings were added to the literature, and in 1984, Pavlakis et al., better characterize the patients who had normal early development, short stature, seizures, alternating hemiparesis, hemianopsia and cortical blindness, through the framework that called MELAS (Mitochondrial myopathy, Encephalopathy, Lactic Acidosis, and Stroke-like episodes)33 Pavlakis SG, Phillips PC, DiMauro S, De Vivo DC, Rowland LP. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: a distinctive clinical syndrome. Ann Neurol.1984;16(4):481-8. doi:10.1002/ana.410160409
https://doi.org/10.1002/ana.410160409...
.
In Brazil, the first report of MELAS was made by Werneck et al. in 1987 who describe a boy who had recurrent episodes of seizures, headache and vomiting associated with focal neurological signs44 Werneck LC, Abdalla H, Lohr A. [MELAS (mitochondrial encephalopathy, lactic acidosis and stroke like episodes): case report]. Arq Neuropsiquiatr. 1987;45(3) 288-94. Portuguese. doi:10.1590/S0004-282X1987000300009
https://doi.org/10.1590/S0004-282X198700...
. The brain computed tomography showed lesions similar to ischemic stroke and calcification of the basal ganglia, ragged-red fiber (RRF) in muscle biopsy, elevation of lactic acid and determination of enzyme activity consistent with the respiratory chain complex IV deficiency also were found44 Werneck LC, Abdalla H, Lohr A. [MELAS (mitochondrial encephalopathy, lactic acidosis and stroke like episodes): case report]. Arq Neuropsiquiatr. 1987;45(3) 288-94. Portuguese. doi:10.1590/S0004-282X1987000300009
https://doi.org/10.1590/S0004-282X198700...
.
WHAT IS THE DIAGNOSTIC CRITERION?
The cases previously published with a diagnosis suggestive of MELAS have been reviewed by Hirano et al. in 1992 with the purpose of obtaining diagnostic criteria for this group of patients55 Hirano M, Ricci E, Koenigsberger MR, Defendini R, Pavlakis SG, DeVivo DC et al.. Melas: an original case and clinical criteria for diagnosis. Neuromuscul Disord.1992;2(2):125-35. doi:10.1016/0960-8966(92)90045-8
https://doi.org/10.1016/0960-8966(92)900...
. After this literature review, the diagnostic criteria for MELAS must include the following events: (1) signs of encephalopathy, often with dementia and seizures, (2) episodes similar to stroke (stroke-like episodes) in young age, and (3) biochemical evidence of mitochondrial dysfunction such as lactic acidosis or RRF in muscle biopsy (Table 1)55 Hirano M, Ricci E, Koenigsberger MR, Defendini R, Pavlakis SG, DeVivo DC et al.. Melas: an original case and clinical criteria for diagnosis. Neuromuscul Disord.1992;2(2):125-35. doi:10.1016/0960-8966(92)90045-8
https://doi.org/10.1016/0960-8966(92)900...
. The diagnosis may also be supported if at least two of the following was present: normal development, recurrent headache and vomiting (Table 1)55 Hirano M, Ricci E, Koenigsberger MR, Defendini R, Pavlakis SG, DeVivo DC et al.. Melas: an original case and clinical criteria for diagnosis. Neuromuscul Disord.1992;2(2):125-35. doi:10.1016/0960-8966(92)90045-8
https://doi.org/10.1016/0960-8966(92)900...
.
Recently, the MELAS Study Group in Japan included obligatory presentation of stroke-like episodes associated to evidence of mitochondrial dysfunction and known mitochondrial gene mutations as diagnostic criterion. In Japan, the disease is also defined using the Japanese diagnostic criteria for MELAS (Table 2)66 Yatsuga S, Povalko N, Nishioka J, Katayama K, Kakimoto N, Matsuishi T et al.. MELAS: a nationwide prospective cohort study of 96 patients in Japan. Biochim Biophys Acta. 2012;1820(5):619-24. doi:10.1016/j.bbagen.2011.03.015
https://doi.org/10.1016/j.bbagen.2011.03...
. Different of the Hirano et al. diagnostic criteria, Japanese’s diagnostic criteria did not consider signs of encephalopathy and includes mitochondrial gene mutations55 Hirano M, Ricci E, Koenigsberger MR, Defendini R, Pavlakis SG, DeVivo DC et al.. Melas: an original case and clinical criteria for diagnosis. Neuromuscul Disord.1992;2(2):125-35. doi:10.1016/0960-8966(92)90045-8
https://doi.org/10.1016/0960-8966(92)900...
,66 Yatsuga S, Povalko N, Nishioka J, Katayama K, Kakimoto N, Matsuishi T et al.. MELAS: a nationwide prospective cohort study of 96 patients in Japan. Biochim Biophys Acta. 2012;1820(5):619-24. doi:10.1016/j.bbagen.2011.03.015
https://doi.org/10.1016/j.bbagen.2011.03...
.
WHAT IS THE PATHOGENESIS?
On clinical evaluation, stroke-like episodes of patients with MELAS are indistinguishable from those reported by patients with acute ischemic stroke, but the pathogenesis of these lesions in patients with MELAS is different and not fully elucidated. The two main hypotheses being considered are: (1) ischemic, with suggestion of a “mitochondrial angiopathy” caused by mitochondrial dysfunction in smooth muscle cells of the small cerebral vessels leading to vascular occlusion with neuronal loss77 Goto Y. Clinical features of MELAS and mitochondrial DNA mutations. Muscle Nerve. 1995;18(Suppl 3):S107-12. doi:10.1002/mus.880181422
https://doi.org/10.1002/mus.880181422...
,88 Mizukami K, Sasaki M, Suzuki T, Shiraishi H, Koizumi J, Ohkoshi N et al.. Central nervous system changes in mitochondrial encephalomyopathy: light and electron microscopic study. Acta Neuropathol. 1992;83(4):449-52. doi:10.1007/BF00713541
https://doi.org/10.1007/BF00713541...
,99 Ohama E, Ohara S, Ikuta F, Tanaka K, Nishizawa M, Miyatake T. Mitochondrial angiopathy in cerebral blood vessels of mitochondrial encephalomyopathy. Acta Neuropathol. 1987;74(3):226-33. doi:10.1007/BF00688185
https://doi.org/10.1007/BF00688185...
,1010 Sakuta R, Nonaka I. Vascular involvement in mitochondrial myopathy. Ann Neurol. 1989;25(6):594-601. doi:10.1002/ana.410250611
https://doi.org/10.1002/ana.410250611...
; and (2) metabolic, due to a “mitochondrial cytopathy”, that trigger energy failure of brain tissue causing neuronal damage1111 Gilchrist JM, Sikirica M, Stopa E, Shanske S. Adult-onset MELAS. Evidence for involvement of neurons as well as cerebral vasculature in strokelike episodes. Stroke. 1996;27(8):1420-3. doi:10.1161/01.STR.27.8.1420
https://doi.org/10.1161/01.STR.27.8.1420...
,1212 Iizuka T, Sakai F, Suzuki N, Hata T, Tsukahara S, Fukuda M et al.. Neuronal hyperexcitability in stroke-like episodes of MELAS syndrome. Neurology. 2002;59(6):816-24. doi:10.1212/WNL.59.6.816
https://doi.org/10.1212/WNL.59.6.816...
,1313 Sparaco M, Bonilla E, DiMauro S, Powers JM. Neuropathology of mitochondrial encephalomyopathies due to mitochondrial DNA defects. J Neuropathol Exp Neurol. 1993;52(1):1-10. doi:10.1097/00005072-199301000-00001
https://doi.org/10.1097/00005072-1993010...
.
Currently, the mechanisms that trigger these episodes are correlated to a combination of these two hypotheses, in which both (neuronal and vascular dysfunction) are responsible for the pathogenesis of stroke-like episodes1414 Iizuka T, Sakai F. Pathogenesis of stroke-like episodes in MELAS: analysis neurovascular cellular mechanisms. Curr Neurovasc Res. 2005;2(1):29-45. doi:10.2174/1567202052773544
https://doi.org/10.2174/1567202052773544...
. Iizuko and Sakai in 2005 proposed a possible mechanism for the formation of stroke-like episodes, based on the main pathological findings related to these episodes (Figure 1): headache, seizures, focal hyperemia, vasogenic edema, lesion progression after the stroke-like episode and neuronal loss1414 Iizuka T, Sakai F. Pathogenesis of stroke-like episodes in MELAS: analysis neurovascular cellular mechanisms. Curr Neurovasc Res. 2005;2(1):29-45. doi:10.2174/1567202052773544
https://doi.org/10.2174/1567202052773544...
.
Iizuka and Sakai’s proposed mechanism of stroke-like episodes (with permission from Curr Neurovasc Res 2005;2:29-45) 14.
WHAT ARE THE GENETIC ABNORMALITIES?
In 1990, Goto et al. and Kobayashi et al., described a point mutation of mtDNA affecting the gene encoding the leucine tRNA (UUR) (tRNALeuUUR) by the exchange at position 3243 of the nucleotide A by G (A3243G) in muscle of MELAS patients1515 Goto YI, Nonaka I, Horai S. A mutation in the tRNA(Leu)(UUR gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990;348(6302):651-3. doi:10.1038/348651a0
https://doi.org/10.1038/348651a0...
,1616 Kobayashi Y, Momoi MY, Tominaga K, Momoi T, Nihei K, Yanagisawa M et al.. A point mutation in the mitochondrial tRNA(Leu)(UUR) gene in MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes). Biochem Biophys Res Comm. 1990;173(3):816-22. doi:10.1016/S0006-291X(05)80860-5
https://doi.org/10.1016/S0006-291X(05)80...
. The tRNALeuUUR, also known asMT-TL1, is located between nucleotides 3230 and 3304 and is responsible for decoding of UUR codons (R = A or G)1717 Finsterer J. Genetic, pathogenetic, and phenotypic implications of the mitochondrial A3243G tRNALeu(UUR) mutation. Acta Neurol Scand. 2007;116(1):1-14. doi:10.1111/j.1600-0404.2007.00836.x
https://doi.org/10.1111/j.1600-0404.2007...
. The A3243G mutation affects the stability of the structure, methylation, aminoacylation and codon recognition of the tRNALeuUUR, more sharply than other mutations in this gene1414 Iizuka T, Sakai F. Pathogenesis of stroke-like episodes in MELAS: analysis neurovascular cellular mechanisms. Curr Neurovasc Res. 2005;2(1):29-45. doi:10.2174/1567202052773544
https://doi.org/10.2174/1567202052773544...
,1717 Finsterer J. Genetic, pathogenetic, and phenotypic implications of the mitochondrial A3243G tRNALeu(UUR) mutation. Acta Neurol Scand. 2007;116(1):1-14. doi:10.1111/j.1600-0404.2007.00836.x
https://doi.org/10.1111/j.1600-0404.2007...
,1818 Hao R, Yao YN, Zheng YG, Xu MG, Wang ED. Reduction of mitochondrial tRNALeu(UUR) aminoacylation by some MELAS-associated mutations. FEBS Lett. 2004;578(1-2):135-9. doi:10.1016/j.febslet.2004.11.004
https://doi.org/10.1016/j.febslet.2004.1...
. This could reduce the functional level of tRNALeuUUR that participates in the process of mitochondrial protein synthesis1414 Iizuka T, Sakai F. Pathogenesis of stroke-like episodes in MELAS: analysis neurovascular cellular mechanisms. Curr Neurovasc Res. 2005;2(1):29-45. doi:10.2174/1567202052773544
https://doi.org/10.2174/1567202052773544...
. After this first description other studies have shown that the A3243G mutation was responsible for most cases of MELAS1919 Ciafaloni E, Ricci E, Shanske S, Moraes CT, Silvestri G, Hirano M et al.. MELAS: clinical features, biochemistry and molecular genetics. Ann Neurol. 1992;31(4):391-8. doi:10.1002/ana.410310408
https://doi.org/10.1002/ana.410310408...
,2020 Goto Y, Horai S, Matsuoka T, Koga Y, Nihei K, Kobayashi M et al.. Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS): a correlative study of the clinical features and mitochondrial DNA mutation. Neurology. 1992;42(3):545-50. doi:10.1212/WNL.42.3.545
https://doi.org/10.1212/WNL.42.3.545...
,2121 Hammans SR, Sweeney MG, Brockington M, Morgan-Hughes JA, Harding AE. Mitochondrial encephalopathies: molecular genetic diagnosis from blood samples. Lancet. 1991;337(8753):1311-3. doi:10.1016/0140-6736(91)92981-7
https://doi.org/10.1016/0140-6736(91)929...
.
In the years following the discovery of mutation A3243G, a second point mutation by substitution of T for C nucleotide at position 3271 (T3271C) in the tRNALeuUURof the mtDNA was also found in patients with MELAS2222 Goto Y, Nonaka I, Horai S. A new mtDNA mutation associated with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS). Biochim Biophys Acta. 1991;1097(3): 238-40. doi:10.1016/0925-4439(91)90042-8
https://doi.org/10.1016/0925-4439(91)900...
. Over the years, other point mutations in mtDNA were found in patients with MELAS, revealing their genetic heterogeneity. However, the A3243G mutation still is responsible for remained about 80% of patients with MELAS and T3271C by approximately 7.5%, but up to 10% of patients with MELAS the mutations of mtDNA remain unknown (Table 3)77 Goto Y. Clinical features of MELAS and mitochondrial DNA mutations. Muscle Nerve. 1995;18(Suppl 3):S107-12. doi:10.1002/mus.880181422
https://doi.org/10.1002/mus.880181422...
,1616 Kobayashi Y, Momoi MY, Tominaga K, Momoi T, Nihei K, Yanagisawa M et al.. A point mutation in the mitochondrial tRNA(Leu)(UUR) gene in MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes). Biochem Biophys Res Comm. 1990;173(3):816-22. doi:10.1016/S0006-291X(05)80860-5
https://doi.org/10.1016/S0006-291X(05)80...
. The mutations in DNAmt genes coding to mitochondrial complex I, as ND5 gene, have been appointed as the second more frequent (Table 3)2323 Zhao D, Hong D, Zhang W, et al.. Mutations in mitochondrially encoded complex I enzyme as the second common cause in a cohort of Chinese patients with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes. J Hum Genet. 2011;56(11):759-64. doi:10.1038/jhg.2011.96
https://doi.org/10.1038/jhg.2011.96...
. The presence of mutation by rearrangement of the deletion type, large or small, has rarely been described in mtDNA associated to MELAS phenotype. The presence of nuclear DNA gene mutations has been also rarely associated to MELAS.
Summary of mutations in mitochondrial DNA in the two most frequent genes associated with MELAS.
Although most MELAS patients have the A3243G point mutation, this mutation is not specific for this group of patients, also found in patients with progressive external ophtalmoplegia (PEO), cardiomyopathy, sensorineural deafness and diabetes mellitus with maternal inheritance2424 Zeviani M, Di Donato S. Mitochondrial disorders. Brain. 2004;127(10):2153-72. doi:10.1093/brain/awh259
https://doi.org/10.1093/brain/awh259...
. Moreover, the clinical and laboratory evaluation of other family members have found relatives with incomplete features for MELAS, usually with maternal inheritance pattern. However, genetic studies have showed that maternal relatives clinically asymptomatic but with the A3243G mutation, might have mitochondrial dysfunction with RRF in muscle biopsy55 Hirano M, Ricci E, Koenigsberger MR, Defendini R, Pavlakis SG, DeVivo DC et al.. Melas: an original case and clinical criteria for diagnosis. Neuromuscul Disord.1992;2(2):125-35. doi:10.1016/0960-8966(92)90045-8
https://doi.org/10.1016/0960-8966(92)900...
,1919 Ciafaloni E, Ricci E, Shanske S, Moraes CT, Silvestri G, Hirano M et al.. MELAS: clinical features, biochemistry and molecular genetics. Ann Neurol. 1992;31(4):391-8. doi:10.1002/ana.410310408
https://doi.org/10.1002/ana.410310408...
. In addition, patients with MELAS concomitant with other mitochondrial diseases such as myoclonic epilepsy associated with ragged red fibers (MERRF), Leigh syndrome and Leber’s hereditary optic neuropathy have been reported with associated mutation of the mtDNA. These data contribute to state that mitochondrial diseases caused by mtDNA point mutations have heterogeneous phenotype77 Goto Y. Clinical features of MELAS and mitochondrial DNA mutations. Muscle Nerve. 1995;18(Suppl 3):S107-12. doi:10.1002/mus.880181422
https://doi.org/10.1002/mus.880181422...
.
The proportion of mutant mtDNA usually is different in tissues of MELAS patients associated to mutations of tRNALeuUUR55 Hirano M, Ricci E, Koenigsberger MR, Defendini R, Pavlakis SG, DeVivo DC et al.. Melas: an original case and clinical criteria for diagnosis. Neuromuscul Disord.1992;2(2):125-35. doi:10.1016/0960-8966(92)90045-8
https://doi.org/10.1016/0960-8966(92)900...
,77 Goto Y. Clinical features of MELAS and mitochondrial DNA mutations. Muscle Nerve. 1995;18(Suppl 3):S107-12. doi:10.1002/mus.880181422
https://doi.org/10.1002/mus.880181422...
,1919 Ciafaloni E, Ricci E, Shanske S, Moraes CT, Silvestri G, Hirano M et al.. MELAS: clinical features, biochemistry and molecular genetics. Ann Neurol. 1992;31(4):391-8. doi:10.1002/ana.410310408
https://doi.org/10.1002/ana.410310408...
,2020 Goto Y, Horai S, Matsuoka T, Koga Y, Nihei K, Kobayashi M et al.. Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS): a correlative study of the clinical features and mitochondrial DNA mutation. Neurology. 1992;42(3):545-50. doi:10.1212/WNL.42.3.545
https://doi.org/10.1212/WNL.42.3.545...
. For these reasons the use of clinically affected tissues, such as muscle, to perform the extraction of mtDNA for the molecular study could be recommended due to better results55 Hirano M, Ricci E, Koenigsberger MR, Defendini R, Pavlakis SG, DeVivo DC et al.. Melas: an original case and clinical criteria for diagnosis. Neuromuscul Disord.1992;2(2):125-35. doi:10.1016/0960-8966(92)90045-8
https://doi.org/10.1016/0960-8966(92)900...
,77 Goto Y. Clinical features of MELAS and mitochondrial DNA mutations. Muscle Nerve. 1995;18(Suppl 3):S107-12. doi:10.1002/mus.880181422
https://doi.org/10.1002/mus.880181422...
,1919 Ciafaloni E, Ricci E, Shanske S, Moraes CT, Silvestri G, Hirano M et al.. MELAS: clinical features, biochemistry and molecular genetics. Ann Neurol. 1992;31(4):391-8. doi:10.1002/ana.410310408
https://doi.org/10.1002/ana.410310408...
,2020 Goto Y, Horai S, Matsuoka T, Koga Y, Nihei K, Kobayashi M et al.. Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS): a correlative study of the clinical features and mitochondrial DNA mutation. Neurology. 1992;42(3):545-50. doi:10.1212/WNL.42.3.545
https://doi.org/10.1212/WNL.42.3.545...
,2525 DiMauro S, Hirano M. MELAS. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH et al., editors. GeneReviews®[Internet]. Seattle (WA): University of Washington, Seattle; 2013 [citado 21 nov 2013]. Disponível em: http://www.ncbi.nlm.nih.gov/books/NBK1233/
http://www.ncbi.nlm.nih.gov/books/NBK123...
. If clinically unaffected tissues were used, such as peripheral blood leukocytes, the pathogenic mutation may be undetectable55 Hirano M, Ricci E, Koenigsberger MR, Defendini R, Pavlakis SG, DeVivo DC et al.. Melas: an original case and clinical criteria for diagnosis. Neuromuscul Disord.1992;2(2):125-35. doi:10.1016/0960-8966(92)90045-8
https://doi.org/10.1016/0960-8966(92)900...
,77 Goto Y. Clinical features of MELAS and mitochondrial DNA mutations. Muscle Nerve. 1995;18(Suppl 3):S107-12. doi:10.1002/mus.880181422
https://doi.org/10.1002/mus.880181422...
,1919 Ciafaloni E, Ricci E, Shanske S, Moraes CT, Silvestri G, Hirano M et al.. MELAS: clinical features, biochemistry and molecular genetics. Ann Neurol. 1992;31(4):391-8. doi:10.1002/ana.410310408
https://doi.org/10.1002/ana.410310408...
,2020 Goto Y, Horai S, Matsuoka T, Koga Y, Nihei K, Kobayashi M et al.. Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS): a correlative study of the clinical features and mitochondrial DNA mutation. Neurology. 1992;42(3):545-50. doi:10.1212/WNL.42.3.545
https://doi.org/10.1212/WNL.42.3.545...
,2525 DiMauro S, Hirano M. MELAS. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH et al., editors. GeneReviews®[Internet]. Seattle (WA): University of Washington, Seattle; 2013 [citado 21 nov 2013]. Disponível em: http://www.ncbi.nlm.nih.gov/books/NBK1233/
http://www.ncbi.nlm.nih.gov/books/NBK123...
.
The methods used to detect each mutation may vary among laboratories (often PCR, PCR/RFLP or direct sequencing), but usually is recommended first screening to targeted mutations to then perform the full sequence analysis.
WHAT ARE THE CLINICAL FEATURES?
Clinical symptoms are highly variable among patients with mitochondrial diseases. Some of these clinical findings may be absent in the early stage of the disease, while in advanced disease patients usually have more uniform clinical manifestations.
Although the onset of clinical manifestation often occurs in childhood and early adulthood, a late onset in adults is not uncommon in patients with MELAS. Moreover, the age of stroke-like episodes may be different among affected members of the same family2626 Bataillard M, Chatzoglou E, Rumbach L, Sternberg D, Tournade A, Laforêt P et al.. Atypical MELAS syndrome associated with a new mitochondrial tRNA glutamine point mutation. Neurology. 2001;56(3):405-7. doi:10.1212/WNL.56.3.405
https://doi.org/10.1212/WNL.56.3.405...
. Children psychomotor development is usually normal in almost all patients with MELAS.
Stroke-like episodes are diagnostic criteria for MELAS and is expected that all patients have this clinical manifestation55 Hirano M, Ricci E, Koenigsberger MR, Defendini R, Pavlakis SG, DeVivo DC et al.. Melas: an original case and clinical criteria for diagnosis. Neuromuscul Disord.1992;2(2):125-35. doi:10.1016/0960-8966(92)90045-8
https://doi.org/10.1016/0960-8966(92)900...
,66 Yatsuga S, Povalko N, Nishioka J, Katayama K, Kakimoto N, Matsuishi T et al.. MELAS: a nationwide prospective cohort study of 96 patients in Japan. Biochim Biophys Acta. 2012;1820(5):619-24. doi:10.1016/j.bbagen.2011.03.015
https://doi.org/10.1016/j.bbagen.2011.03...
. The stroke-like episode may occur alone or in association with signs of the encephalopathy, such as seizures55 Hirano M, Ricci E, Koenigsberger MR, Defendini R, Pavlakis SG, DeVivo DC et al.. Melas: an original case and clinical criteria for diagnosis. Neuromuscul Disord.1992;2(2):125-35. doi:10.1016/0960-8966(92)90045-8
https://doi.org/10.1016/0960-8966(92)900...
,66 Yatsuga S, Povalko N, Nishioka J, Katayama K, Kakimoto N, Matsuishi T et al.. MELAS: a nationwide prospective cohort study of 96 patients in Japan. Biochim Biophys Acta. 2012;1820(5):619-24. doi:10.1016/j.bbagen.2011.03.015
https://doi.org/10.1016/j.bbagen.2011.03...
. The clinical outcome of stroke-like episodes is more benign, with improvement of symptoms in a few months, but the symptoms related encephalopathy, such as dementia and seizures, may progressively worsen66 Yatsuga S, Povalko N, Nishioka J, Katayama K, Kakimoto N, Matsuishi T et al.. MELAS: a nationwide prospective cohort study of 96 patients in Japan. Biochim Biophys Acta. 2012;1820(5):619-24. doi:10.1016/j.bbagen.2011.03.015
https://doi.org/10.1016/j.bbagen.2011.03...
,1212 Iizuka T, Sakai F, Suzuki N, Hata T, Tsukahara S, Fukuda M et al.. Neuronal hyperexcitability in stroke-like episodes of MELAS syndrome. Neurology. 2002;59(6):816-24. doi:10.1212/WNL.59.6.816
https://doi.org/10.1212/WNL.59.6.816...
,1414 Iizuka T, Sakai F. Pathogenesis of stroke-like episodes in MELAS: analysis neurovascular cellular mechanisms. Curr Neurovasc Res. 2005;2(1):29-45. doi:10.2174/1567202052773544
https://doi.org/10.2174/1567202052773544...
,2727 Kaufmann P, Engelstad K, Wei Y, Kulikova R, Oskoui M, Sproule DM et al.. Natural history of MELAS associated with mitochondrial DNA m.3243A>G genotype. Neurology. 2011;77(22):1965-71. doi:10.1212/WNL.0b013e31823a0c7f
https://doi.org/10.1212/WNL.0b013e31823a...
.
The presence of progressive dementia has also been found associated with changes in cerebral perfusion even in the absence of stroke-like episode, but with marked cortical atrophy in the chronic phase of the disease characterized neuronal loss, similar to what occurs in vascular dementia2727 Kaufmann P, Engelstad K, Wei Y, Kulikova R, Oskoui M, Sproule DM et al.. Natural history of MELAS associated with mitochondrial DNA m.3243A>G genotype. Neurology. 2011;77(22):1965-71. doi:10.1212/WNL.0b013e31823a0c7f
https://doi.org/10.1212/WNL.0b013e31823a...
,2828 Nishioka J, Akita Y, Yatsuga S, Katayama K, Matsuishi T, Ishibashi M et al.. Inappropriate intracranial hemodynamics in the natural course of MELAS. Brain Dev. 2008;30(2):100-5. doi:10.1016/j.braindev.2007.06.008
https://doi.org/10.1016/j.braindev.2007....
. Partial seizures are commonly reported in MELAS associated with mutations in the tRNALeugene, similar other mitochondrial diseases as MERRF or PEO, but generalized seizures also occurs in MELAS2929 Lorenzoni PJ, Scola RH, Kay CS, Arndt RC, Freund AA, Bruck I et al.. MELAS: clinical features, muscle biopsy and molecular genetics. Arq Neuropsiquiatr.. 2009;67(3A):668-76. doi:10.1590/S0004-282X2009000400018
https://doi.org/10.1590/S0004-282X200900...
,3030 Lorenzoni PJ, Scola RH, Kay CS, Arndt RC, Silvado CE, Werneck LC. MERRF: clinical features, muscle biopsy and molecular genetics in Brazilian patients. Mitochondrion. 2011;11(3):528-32. doi:10.1016/j.mito.2011.01.003
https://doi.org/10.1016/j.mito.2011.01.0...
.
Headaches occur in the majority of affected individuals and are often severe during the acute phase of the stroke-like2525 DiMauro S, Hirano M. MELAS. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH et al., editors. GeneReviews®[Internet]. Seattle (WA): University of Washington, Seattle; 2013 [citado 21 nov 2013]. Disponível em: http://www.ncbi.nlm.nih.gov/books/NBK1233/
http://www.ncbi.nlm.nih.gov/books/NBK123...
,2929 Lorenzoni PJ, Scola RH, Kay CS, Arndt RC, Freund AA, Bruck I et al.. MELAS: clinical features, muscle biopsy and molecular genetics. Arq Neuropsiquiatr.. 2009;67(3A):668-76. doi:10.1590/S0004-282X2009000400018
https://doi.org/10.1590/S0004-282X200900...
,3131 Conforto AB, Yamamoto FI, Oba-Shinjo SM, Pinto JG, Hoshino M, Scaff M et al.. Screening for MELAS mutations in young patients with stroke of undetermined origin. Arq Neuropsiquiatr. 2007;65(2B):371-6. doi:10.1590/S0004-282X2007000300001
https://doi.org/10.1590/S0004-282X200700...
.
Other multisystemic dysfunctions could be find in MELAS patients: psychiatric manifestations, cerebellar ataxia, myoclonus, neuropathy, ocular motility abnormalities, exercise intolerance, pigmentary retinopathy, optic atrophy, deafness, short stature, diabetes mellitus, alteration of cardiac conduction, cardiomyopathy, gastrointestinal changes, among others55 Hirano M, Ricci E, Koenigsberger MR, Defendini R, Pavlakis SG, DeVivo DC et al.. Melas: an original case and clinical criteria for diagnosis. Neuromuscul Disord.1992;2(2):125-35. doi:10.1016/0960-8966(92)90045-8
https://doi.org/10.1016/0960-8966(92)900...
,66 Yatsuga S, Povalko N, Nishioka J, Katayama K, Kakimoto N, Matsuishi T et al.. MELAS: a nationwide prospective cohort study of 96 patients in Japan. Biochim Biophys Acta. 2012;1820(5):619-24. doi:10.1016/j.bbagen.2011.03.015
https://doi.org/10.1016/j.bbagen.2011.03...
,2020 Goto Y, Horai S, Matsuoka T, Koga Y, Nihei K, Kobayashi M et al.. Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS): a correlative study of the clinical features and mitochondrial DNA mutation. Neurology. 1992;42(3):545-50. doi:10.1212/WNL.42.3.545
https://doi.org/10.1212/WNL.42.3.545...
,2727 Kaufmann P, Engelstad K, Wei Y, Kulikova R, Oskoui M, Sproule DM et al.. Natural history of MELAS associated with mitochondrial DNA m.3243A>G genotype. Neurology. 2011;77(22):1965-71. doi:10.1212/WNL.0b013e31823a0c7f
https://doi.org/10.1212/WNL.0b013e31823a...
,2525 DiMauro S, Hirano M. MELAS. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH et al., editors. GeneReviews®[Internet]. Seattle (WA): University of Washington, Seattle; 2013 [citado 21 nov 2013]. Disponível em: http://www.ncbi.nlm.nih.gov/books/NBK1233/
http://www.ncbi.nlm.nih.gov/books/NBK123...
,2929 Lorenzoni PJ, Scola RH, Kay CS, Arndt RC, Freund AA, Bruck I et al.. MELAS: clinical features, muscle biopsy and molecular genetics. Arq Neuropsiquiatr.. 2009;67(3A):668-76. doi:10.1590/S0004-282X2009000400018
https://doi.org/10.1590/S0004-282X200900...
. Cardiac involvement can occur in up to 50% of patients with MELAS3232 Hirano M, Pavlakis SG. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS): current concepts. J Child Neurol. 1994;9(1):4-13. doi:10.1177/088307389400900102
https://doi.org/10.1177/0883073894009001...
,3333 Okajima Y, Tanabe Y, Takayanagi M, Aotsuka H. A follow up study of myocardial involvement in patients with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS). Heart. 1998;80(3):292-5. doi:10.1136/hrt.80.3.292
https://doi.org/10.1136/hrt.80.3.292...
. However, these disorders are not the main clinical manifestation of the disease.
MELAS patients should be followed at regular intervals to monitor disease progression and the appearance of new symptoms. Annual ophthalmologic, audiologic, cardiologic (electrocardiogram and echocardiogram), and endocrinologic (fasting blood sugar and TSH) assessments also are recommended2525 DiMauro S, Hirano M. MELAS. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH et al., editors. GeneReviews®[Internet]. Seattle (WA): University of Washington, Seattle; 2013 [citado 21 nov 2013]. Disponível em: http://www.ncbi.nlm.nih.gov/books/NBK1233/
http://www.ncbi.nlm.nih.gov/books/NBK123...
.
Children of woman with mtDNA mutation can inherit the mutation3434 Richardson J, Irving L, Hyslop LA, Choudhary M, Murdoch A, Turnbull DM et al.. Concise reviews: assisted reproductive technologies to prevent transmission of mitochondrial DNA disease. Stem Cells. 2015;33(3):639-45. doi:10.1002/stem.1887
https://doi.org/10.1002/stem.1887...
. The risk to other family members depends on the genetic status of the mother2525 DiMauro S, Hirano M. MELAS. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH et al., editors. GeneReviews®[Internet]. Seattle (WA): University of Washington, Seattle; 2013 [citado 21 nov 2013]. Disponível em: http://www.ncbi.nlm.nih.gov/books/NBK1233/
http://www.ncbi.nlm.nih.gov/books/NBK123...
,3434 Richardson J, Irving L, Hyslop LA, Choudhary M, Murdoch A, Turnbull DM et al.. Concise reviews: assisted reproductive technologies to prevent transmission of mitochondrial DNA disease. Stem Cells. 2015;33(3):639-45. doi:10.1002/stem.1887
https://doi.org/10.1002/stem.1887...
. However, it is appropriate to evaluate other family members because the phenotype manifestation results from a combination of factors, due to the different inherit percentages of mutant mtDNA, and therefore, can have a wide range of clinical symptoms from asymptomatic to full manifestation.
WHAT ARE THE IMAGING FEATURES?
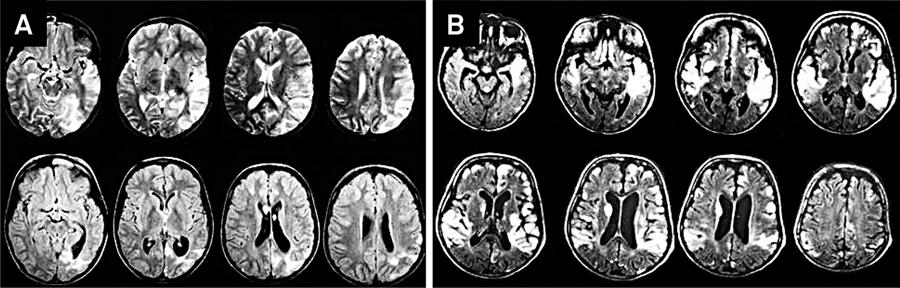
Brain conventional radiological studies, such as CT or MRI, in patients with MELAS reveal changes in gray matter more than in white matter, predominate in the occipital, parietal and temporal lobes, which simulate ischemic stroke (Figure 2)1414 Iizuka T, Sakai F. Pathogenesis of stroke-like episodes in MELAS: analysis neurovascular cellular mechanisms. Curr Neurovasc Res. 2005;2(1):29-45. doi:10.2174/1567202052773544
https://doi.org/10.2174/1567202052773544...
,3535 Haas R, Dietrich R. Neuroimaging of mitochondrial disorders. Mitochondrion. 2004;4(5-6):471-90. doi:10.1016/j.mito.2004.07.008
https://doi.org/10.1016/j.mito.2004.07.0...
,3636 Tschampa HJ, Urbach H, Greschus S, Kunz WS, Kornblum C. Neuroimaging characteristics in mitochondrial encephalopathies associated with the m.3243A>G MTTL1 mutation. J Neurol. 2013;260(4):1071-80. doi:10.1007/s00415-012-6763-4
https://doi.org/10.1007/s00415-012-6763-...
. Most injuries occur in the cortical region of the cerebral hemispheres and more rarely in the cerebellum or basal ganglia3232 Hirano M, Pavlakis SG. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS): current concepts. J Child Neurol. 1994;9(1):4-13. doi:10.1177/088307389400900102
https://doi.org/10.1177/0883073894009001...
,3636 Tschampa HJ, Urbach H, Greschus S, Kunz WS, Kornblum C. Neuroimaging characteristics in mitochondrial encephalopathies associated with the m.3243A>G MTTL1 mutation. J Neurol. 2013;260(4):1071-80. doi:10.1007/s00415-012-6763-4
https://doi.org/10.1007/s00415-012-6763-...
. These brain lesions can be unilateral or bilateral (Figure 2). However, these areas do not occur in a specific vascular territory and angiographic studies show that the vessels in the affected regions have blood flow and sometimes are dilated55 Hirano M, Ricci E, Koenigsberger MR, Defendini R, Pavlakis SG, DeVivo DC et al.. Melas: an original case and clinical criteria for diagnosis. Neuromuscul Disord.1992;2(2):125-35. doi:10.1016/0960-8966(92)90045-8
https://doi.org/10.1016/0960-8966(92)900...
,3232 Hirano M, Pavlakis SG. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS): current concepts. J Child Neurol. 1994;9(1):4-13. doi:10.1177/088307389400900102
https://doi.org/10.1177/0883073894009001...
,3636 Tschampa HJ, Urbach H, Greschus S, Kunz WS, Kornblum C. Neuroimaging characteristics in mitochondrial encephalopathies associated with the m.3243A>G MTTL1 mutation. J Neurol. 2013;260(4):1071-80. doi:10.1007/s00415-012-6763-4
https://doi.org/10.1007/s00415-012-6763-...
. Brain MRI revealed that the lesion in the acute phase begins in the temporal region on focal form, but in 2 to 3 weeks may progress to the parietal and occipital regions, in one third of patients3737 Iizuka T, Sakai F, Kan S, Suzuki N. Slowly progressive spread of the stroke-like lesions in MELAS. Neurology. 2003;61(9):1238-44. doi:10.1212/01.WNL.0000091888.26232.FE
https://doi.org/10.1212/01.WNL.000009188...
. This shows that even after the stroke episode the disease process continues to progress3737 Iizuka T, Sakai F, Kan S, Suzuki N. Slowly progressive spread of the stroke-like lesions in MELAS. Neurology. 2003;61(9):1238-44. doi:10.1212/01.WNL.0000091888.26232.FE
https://doi.org/10.1212/01.WNL.000009188...
. The changing pattern of lactic acid concentration between these regions during the clinical outcome of stroke-like episodes, was also observed in patients with MELAS3838 Kamada K, Takeuchi F, Houkin K, Kitagawa M, Kuriki S, Ogata A et al.. Reversible brain dysfunction in MELAS: MEG, and (1)H MRS analysis. J Neurol Neurosurg Psychiatry. 2001;70(5):675-8. doi:10.1136/jnnp.70.5.675
https://doi.org/10.1136/jnnp.70.5.675...
. These findings may mean that the vulnerability of neurons to mitochondrial dysfunction may be higher in these regions. The exact mechanism of the disease predilection for certain locations in the central nervous system has not been elucidated, but the possibility of heteroplasmy in brain tissue was ruled-out by genetic studies when comparing tissue from different brain areas1111 Gilchrist JM, Sikirica M, Stopa E, Shanske S. Adult-onset MELAS. Evidence for involvement of neurons as well as cerebral vasculature in strokelike episodes. Stroke. 1996;27(8):1420-3. doi:10.1161/01.STR.27.8.1420
https://doi.org/10.1161/01.STR.27.8.1420...
,1414 Iizuka T, Sakai F. Pathogenesis of stroke-like episodes in MELAS: analysis neurovascular cellular mechanisms. Curr Neurovasc Res. 2005;2(1):29-45. doi:10.2174/1567202052773544
https://doi.org/10.2174/1567202052773544...
,3939 Betts J, Jaros E, Perry RH, Schaefer AM, Taylor RW, Abdel-All Z et al.. Molecular neuropathology of MELAS: level of heteroplasmy in individual neurones and evidence of extensive vascular involvement. Neuropathol Appl Neurobiol. 2006;32(4):359-73. doi:10.1111/j.1365-2990.2006.00731.x
https://doi.org/10.1111/j.1365-2990.2006...
. Studies using spectroscopy show increased lactic acid in acute lesions while the use of magnetic resonance diffusion can reveal increased coefficient of diffusion3535 Haas R, Dietrich R. Neuroimaging of mitochondrial disorders. Mitochondrion. 2004;4(5-6):471-90. doi:10.1016/j.mito.2004.07.008
https://doi.org/10.1016/j.mito.2004.07.0...
,4040 Abe K, Yoshimura H, Tanaka H, Fujita N, Hikita T, Sakoda S. Comparison of conventional and diffusion-weighted MRI and proton MR spectroscopy in patients with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like events. Neuroradiology. 2004;46(2):113-7. doi:10.1007/s00234-003-1138-2
https://doi.org/10.1007/s00234-003-1138-...
,4141 Oppenheim C, Galanaud D, Samson Y, Sahel M, Dormont D, Wechsler B et al.. Can diffusion weighted magnetic resonance imaging help differentiate stroke from stroke-like events in MELAS? J Neurol Neurosurg Psychiatry. 2000;69(2):248-50. doi:10.1136/jnnp.69.2.248
https://doi.org/10.1136/jnnp.69.2.248...
. Both methods being more sensitive than conventional in the acute phase of the injury3535 Haas R, Dietrich R. Neuroimaging of mitochondrial disorders. Mitochondrion. 2004;4(5-6):471-90. doi:10.1016/j.mito.2004.07.008
https://doi.org/10.1016/j.mito.2004.07.0...
,4040 Abe K, Yoshimura H, Tanaka H, Fujita N, Hikita T, Sakoda S. Comparison of conventional and diffusion-weighted MRI and proton MR spectroscopy in patients with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like events. Neuroradiology. 2004;46(2):113-7. doi:10.1007/s00234-003-1138-2
https://doi.org/10.1007/s00234-003-1138-...
,4141 Oppenheim C, Galanaud D, Samson Y, Sahel M, Dormont D, Wechsler B et al.. Can diffusion weighted magnetic resonance imaging help differentiate stroke from stroke-like events in MELAS? J Neurol Neurosurg Psychiatry. 2000;69(2):248-50. doi:10.1136/jnnp.69.2.248
https://doi.org/10.1136/jnnp.69.2.248...
. The MRI images are compatible with subcortical laminar necrosis in the subacute phase of episodes3636 Tschampa HJ, Urbach H, Greschus S, Kunz WS, Kornblum C. Neuroimaging characteristics in mitochondrial encephalopathies associated with the m.3243A>G MTTL1 mutation. J Neurol. 2013;260(4):1071-80. doi:10.1007/s00415-012-6763-4
https://doi.org/10.1007/s00415-012-6763-...
,3737 Iizuka T, Sakai F, Kan S, Suzuki N. Slowly progressive spread of the stroke-like lesions in MELAS. Neurology. 2003;61(9):1238-44. doi:10.1212/01.WNL.0000091888.26232.FE
https://doi.org/10.1212/01.WNL.000009188...
.
Imaging features in brain MRI showing unilateral (A) or bilateral (B) lesions in MELAS patients (with permission from Arq Neuropsiquiatr 2009;67:668-676) 29.
The presence of multiple focal areas of cortical necrosis associated with diffuse cortical atrophy in both cerebral hemispheres and cerebellum, are the most frequent pathological findings in the brain of patients with MELAS, while the brainstem is rarely affected3232 Hirano M, Pavlakis SG. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS): current concepts. J Child Neurol. 1994;9(1):4-13. doi:10.1177/088307389400900102
https://doi.org/10.1177/0883073894009001...
,3636 Tschampa HJ, Urbach H, Greschus S, Kunz WS, Kornblum C. Neuroimaging characteristics in mitochondrial encephalopathies associated with the m.3243A>G MTTL1 mutation. J Neurol. 2013;260(4):1071-80. doi:10.1007/s00415-012-6763-4
https://doi.org/10.1007/s00415-012-6763-...
,4242 Tanahashi C, Nakayama A, Yoshida M, Ito M, Mori N, Hashizume Y. MELAS with the mitochondrial DNA 3243 point mutation: a neuropathological study. Acta Neuropathol. 2000;99(1):31-8. doi:10.1007/PL00007403
https://doi.org/10.1007/PL00007403...
. The presence of cortical atrophy and calcifications in the basal ganglia are also found in the progression of some patients3636 Tschampa HJ, Urbach H, Greschus S, Kunz WS, Kornblum C. Neuroimaging characteristics in mitochondrial encephalopathies associated with the m.3243A>G MTTL1 mutation. J Neurol. 2013;260(4):1071-80. doi:10.1007/s00415-012-6763-4
https://doi.org/10.1007/s00415-012-6763-...
,4242 Tanahashi C, Nakayama A, Yoshida M, Ito M, Mori N, Hashizume Y. MELAS with the mitochondrial DNA 3243 point mutation: a neuropathological study. Acta Neuropathol. 2000;99(1):31-8. doi:10.1007/PL00007403
https://doi.org/10.1007/PL00007403...
.
WHAT ARE THE LABORATORIAL AND BIOCHEMICAL FEATURES?
The concentration of cerebrospinal fluid (CSF) protein may be elevated but rarely surpasses 100 mg/dL2525 DiMauro S, Hirano M. MELAS. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH et al., editors. GeneReviews®[Internet]. Seattle (WA): University of Washington, Seattle; 2013 [citado 21 nov 2013]. Disponível em: http://www.ncbi.nlm.nih.gov/books/NBK1233/
http://www.ncbi.nlm.nih.gov/books/NBK123...
,2929 Lorenzoni PJ, Scola RH, Kay CS, Arndt RC, Freund AA, Bruck I et al.. MELAS: clinical features, muscle biopsy and molecular genetics. Arq Neuropsiquiatr.. 2009;67(3A):668-76. doi:10.1590/S0004-282X2009000400018
https://doi.org/10.1590/S0004-282X200900...
. The lactic acid level in blood and CSF is elevated during, or shortly after, the stroke-like episodes in virtually all patients with MELAS2020 Goto Y, Horai S, Matsuoka T, Koga Y, Nihei K, Kobayashi M et al.. Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS): a correlative study of the clinical features and mitochondrial DNA mutation. Neurology. 1992;42(3):545-50. doi:10.1212/WNL.42.3.545
https://doi.org/10.1212/WNL.42.3.545...
,2525 DiMauro S, Hirano M. MELAS. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH et al., editors. GeneReviews®[Internet]. Seattle (WA): University of Washington, Seattle; 2013 [citado 21 nov 2013]. Disponível em: http://www.ncbi.nlm.nih.gov/books/NBK1233/
http://www.ncbi.nlm.nih.gov/books/NBK123...
. The level of ventricular lactic acid measured by spectroscopy, is usually increased in most patients with MELAS and their families, and this increase is associated with the severity of neurological symptoms4343 Kaufmann P, Shungu DC, Sano MC, Jhung S, Engelstad K, Mitsis E et al.. Cerebral lactic acidosis correlates with neurological impairment in MELAS. Neurology. 2004;62(8):1297-302. doi:10.1212/01.WNL.0000120557.83907.A8
https://doi.org/10.1212/01.WNL.000012055...
. Other tests may be used in MELAS investigation, such as serum levels of creatine kinase (CK), but their findings are not specific for MELAS, even though helps especially in the differential diagnosis with other diseases2929 Lorenzoni PJ, Scola RH, Kay CS, Arndt RC, Freund AA, Bruck I et al.. MELAS: clinical features, muscle biopsy and molecular genetics. Arq Neuropsiquiatr.. 2009;67(3A):668-76. doi:10.1590/S0004-282X2009000400018
https://doi.org/10.1590/S0004-282X200900...
. Serum CK may be slightly increased in some patients, usually during or after stroke-like episodes, helping to demonstrate the muscular involvement in these patients, although nonspecific to demonstrate mitochondrial myopathy2020 Goto Y, Horai S, Matsuoka T, Koga Y, Nihei K, Kobayashi M et al.. Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS): a correlative study of the clinical features and mitochondrial DNA mutation. Neurology. 1992;42(3):545-50. doi:10.1212/WNL.42.3.545
https://doi.org/10.1212/WNL.42.3.545...
,2929 Lorenzoni PJ, Scola RH, Kay CS, Arndt RC, Freund AA, Bruck I et al.. MELAS: clinical features, muscle biopsy and molecular genetics. Arq Neuropsiquiatr.. 2009;67(3A):668-76. doi:10.1590/S0004-282X2009000400018
https://doi.org/10.1590/S0004-282X200900...
.
Biochemical studies have showed that several complex of mitochondrial respiratory chain may be deficient, isolated or in combination, but the complex I appears to be more involved, typically associated with changes to other respiratory chain complexes, while the complex II seems least affected55 Hirano M, Ricci E, Koenigsberger MR, Defendini R, Pavlakis SG, DeVivo DC et al.. Melas: an original case and clinical criteria for diagnosis. Neuromuscul Disord.1992;2(2):125-35. doi:10.1016/0960-8966(92)90045-8
https://doi.org/10.1016/0960-8966(92)900...
,1919 Ciafaloni E, Ricci E, Shanske S, Moraes CT, Silvestri G, Hirano M et al.. MELAS: clinical features, biochemistry and molecular genetics. Ann Neurol. 1992;31(4):391-8. doi:10.1002/ana.410310408
https://doi.org/10.1002/ana.410310408...
,2020 Goto Y, Horai S, Matsuoka T, Koga Y, Nihei K, Kobayashi M et al.. Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS): a correlative study of the clinical features and mitochondrial DNA mutation. Neurology. 1992;42(3):545-50. doi:10.1212/WNL.42.3.545
https://doi.org/10.1212/WNL.42.3.545...
,3232 Hirano M, Pavlakis SG. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS): current concepts. J Child Neurol. 1994;9(1):4-13. doi:10.1177/088307389400900102
https://doi.org/10.1177/0883073894009001...
,4444 Kobayashi M, Morishita H, Sugiyama N, Yokochi K, Nakano M, Wada Y et al.. Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes syndrome and NADH-CoQ reductase deficiency. J Inherit Metab Dis. 1986;9(3):301-4. doi:10.1007/BF01799670
https://doi.org/10.1007/BF01799670...
. This can also be observed in MELAS cases with A3243G mutation who present RRF with normal cytochrome c oxidase (COX) staining in muscle biopsy77 Goto Y. Clinical features of MELAS and mitochondrial DNA mutations. Muscle Nerve. 1995;18(Suppl 3):S107-12. doi:10.1002/mus.880181422
https://doi.org/10.1002/mus.880181422...
. In contrast to patients with PEO with A3243G mutation, who show a high incidence of RRF with deficiency of COX activity77 Goto Y. Clinical features of MELAS and mitochondrial DNA mutations. Muscle Nerve. 1995;18(Suppl 3):S107-12. doi:10.1002/mus.880181422
https://doi.org/10.1002/mus.880181422...
. The relationship between the defects of complex I of the respiratory chain and MELAS phenotype is also suggested by the identification of mutations of the ND genes of mtDNA, coding subunits of complex I, in patients with MELAS. Complex II seems less involved in patients with MELAS, possibly by being encoded by nDNA55 Hirano M, Ricci E, Koenigsberger MR, Defendini R, Pavlakis SG, DeVivo DC et al.. Melas: an original case and clinical criteria for diagnosis. Neuromuscul Disord.1992;2(2):125-35. doi:10.1016/0960-8966(92)90045-8
https://doi.org/10.1016/0960-8966(92)900...
,1919 Ciafaloni E, Ricci E, Shanske S, Moraes CT, Silvestri G, Hirano M et al.. MELAS: clinical features, biochemistry and molecular genetics. Ann Neurol. 1992;31(4):391-8. doi:10.1002/ana.410310408
https://doi.org/10.1002/ana.410310408...
. The biochemical analysis of respiratory chain complexes may be normal in some cases.
WHAT ARE THE HISTOLOGICAL FEATURES?
Initially the muscle biopsy may show only muscle fibers with subsarcolemmal accumulation of mitochondria without typical RRF, but all patients with MELAS have RRF in the course of the disease, and its frequency is usually higher in the histochemical reaction for succinic dehydrogenase (SDH) staining than by modified Gomori Trichrome (TGM), similar to what occurs in other mitochondrial diseases (Figure 3)77 Goto Y. Clinical features of MELAS and mitochondrial DNA mutations. Muscle Nerve. 1995;18(Suppl 3):S107-12. doi:10.1002/mus.880181422
https://doi.org/10.1002/mus.880181422...
,2020 Goto Y, Horai S, Matsuoka T, Koga Y, Nihei K, Kobayashi M et al.. Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS): a correlative study of the clinical features and mitochondrial DNA mutation. Neurology. 1992;42(3):545-50. doi:10.1212/WNL.42.3.545
https://doi.org/10.1212/WNL.42.3.545...
,2929 Lorenzoni PJ, Scola RH, Kay CS, Arndt RC, Freund AA, Bruck I et al.. MELAS: clinical features, muscle biopsy and molecular genetics. Arq Neuropsiquiatr.. 2009;67(3A):668-76. doi:10.1590/S0004-282X2009000400018
https://doi.org/10.1590/S0004-282X200900...
. The degree of heteroplasmy (proportion of normal and mutant mtDNA in each tissue) is also an important factor influencing the variability of the muscle biopsy findings4545 Tokunaga M, Mita S, Sakuta R, Nonaka I, Araki S. Increased mitochondrial DNA in blood vessels and ragged-red fibers in mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS). Ann Neurol. 1993;33(3):275-80. doi:10.1002/ana.410330308
https://doi.org/10.1002/ana.410330308...
,4646 Tokunaga M, Mita S, Murakami T, Kumamoto T, Uchino M, Nonaka I et al.. Single muscle fiber analysis of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS). Ann Neurol 1994;35(4):413-9. doi:10.1002/ana.410350407
https://doi.org/10.1002/ana.410350407...
.
Classic features in the muscle biopsy of MELAS: ragged-red fibers (RRF) on modified Gomori trichrome stain (A, B); muscle fibers with normal activity of the cytochrome c oxidase (COX) (C); RRF on succinic dehydrogenase (SDH) stain (D, E); and SDH strongly reactive blood vessel (F). Bar = 50 µm.
However, the two morphological abnormalities in muscle biopsy that can help distinguish MELAS from other mitochondrial diseases are: a large proportion of RRF with normal activity of COX and the presence of vessels with strong reaction for SDH (Figure 3)2929 Lorenzoni PJ, Scola RH, Kay CS, Arndt RC, Freund AA, Bruck I et al.. MELAS: clinical features, muscle biopsy and molecular genetics. Arq Neuropsiquiatr.. 2009;67(3A):668-76. doi:10.1590/S0004-282X2009000400018
https://doi.org/10.1590/S0004-282X200900...
,3232 Hirano M, Pavlakis SG. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS): current concepts. J Child Neurol. 1994;9(1):4-13. doi:10.1177/088307389400900102
https://doi.org/10.1177/0883073894009001...
,4747 Rossmanith W, Freilinger M, Roka J, Raffelsberger T, Moser-Thier K, Prayer D et al.. Isolated cytochrome c oxidase deficiency as a cause of MELAS. J Med Genet. 2008;45(2):117-21. doi:10.1136/jmg.2007.052076
https://doi.org/10.1136/jmg.2007.052076...
,4848 Tanji K, Kaufmann P, Naini AB, Lu J, Parsons TC, Wang D et al.. A novel tRNA(Val) mitochondrial DNA mutation causing MELAS. J Neurol Sci. 2008;270(1-2):23-7. doi:10.1016/j.jns.2008.01.016
https://doi.org/10.1016/j.jns.2008.01.01...
. The quantitative analysis showed that 80-90% of the muscle fibers have greater amount of mtDNA (normal and mutant), and the ratio of mtDNA mutant RRF is extremely high in comparison with the other fibers which do not form RRF4545 Tokunaga M, Mita S, Sakuta R, Nonaka I, Araki S. Increased mitochondrial DNA in blood vessels and ragged-red fibers in mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS). Ann Neurol. 1993;33(3):275-80. doi:10.1002/ana.410330308
https://doi.org/10.1002/ana.410330308...
,4646 Tokunaga M, Mita S, Murakami T, Kumamoto T, Uchino M, Nonaka I et al.. Single muscle fiber analysis of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS). Ann Neurol 1994;35(4):413-9. doi:10.1002/ana.410350407
https://doi.org/10.1002/ana.410350407...
. However, as mentioned earlier, the presence of COX negative fibers in patients with MELAS is lower than that found in other mitochondrial diseases, as MERRF or PEO77 Goto Y. Clinical features of MELAS and mitochondrial DNA mutations. Muscle Nerve. 1995;18(Suppl 3):S107-12. doi:10.1002/mus.880181422
https://doi.org/10.1002/mus.880181422...
,2020 Goto Y, Horai S, Matsuoka T, Koga Y, Nihei K, Kobayashi M et al.. Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS): a correlative study of the clinical features and mitochondrial DNA mutation. Neurology. 1992;42(3):545-50. doi:10.1212/WNL.42.3.545
https://doi.org/10.1212/WNL.42.3.545...
,3030 Lorenzoni PJ, Scola RH, Kay CS, Arndt RC, Silvado CE, Werneck LC. MERRF: clinical features, muscle biopsy and molecular genetics in Brazilian patients. Mitochondrion. 2011;11(3):528-32. doi:10.1016/j.mito.2011.01.003
https://doi.org/10.1016/j.mito.2011.01.0...
. Some authors report that the COX activity in the muscle fibers is variable, and the presence of COX negative areas is segmentally along the same fiber muscle in patients with MELAS, similar to what occurs in other mitochondrial disorders, suggesting that the alteration of complex IV is not the main change in this group of patients77 Goto Y. Clinical features of MELAS and mitochondrial DNA mutations. Muscle Nerve. 1995;18(Suppl 3):S107-12. doi:10.1002/mus.880181422
https://doi.org/10.1002/mus.880181422...
,4848 Tanji K, Kaufmann P, Naini AB, Lu J, Parsons TC, Wang D et al.. A novel tRNA(Val) mitochondrial DNA mutation causing MELAS. J Neurol Sci. 2008;270(1-2):23-7. doi:10.1016/j.jns.2008.01.016
https://doi.org/10.1016/j.jns.2008.01.01...
,4949 Matsuoka T, Goto Y, Hasegawa H, Nonaka I. Segmental cytochrome c-oxidase deficiency in CPEO: teased muscle fiber analysis. Muscle Nerve. 1992;15(2):209-13. doi:10.1002/mus.880150213
https://doi.org/10.1002/mus.880150213...
,5050 Petruzzella V, Moraes CT, Sano MC, Bonilla E, DiMauro S, Schon EA. Extremely high levels of mutant mtDNAs co-localize with cytochrome c oxidase-negative ragged-red fibers in patients harboring a point mutation at nt 3243. Hum Mol Genet. 1994;3(3):449-54. doi:10.1093/hmg/3.3.449
https://doi.org/10.1093/hmg/3.3.449...
. Thus, it may occur in a single muscle fiber portions well demarcated with COX positive and COX negative4949 Matsuoka T, Goto Y, Hasegawa H, Nonaka I. Segmental cytochrome c-oxidase deficiency in CPEO: teased muscle fiber analysis. Muscle Nerve. 1992;15(2):209-13. doi:10.1002/mus.880150213
https://doi.org/10.1002/mus.880150213...
,5050 Petruzzella V, Moraes CT, Sano MC, Bonilla E, DiMauro S, Schon EA. Extremely high levels of mutant mtDNAs co-localize with cytochrome c oxidase-negative ragged-red fibers in patients harboring a point mutation at nt 3243. Hum Mol Genet. 1994;3(3):449-54. doi:10.1093/hmg/3.3.449
https://doi.org/10.1093/hmg/3.3.449...
,5151 Moraes CT, Ricci E, Petruzzella V, Shanske S, DiMauro S, Schon EA et al.. Molecular analysis of the muscle pathology associated with mitochondrial DNA deletions. Nature Genet. 1992;1(5):359-67. doi:10.1038/ng0892-359
https://doi.org/10.1038/ng0892-359...
. These muscle fibers might have the ratio of mtDNA normal and mutant in those regions influence the functional impairment of complex IV, even in patients with mutations without involvement of genes encoding the COX subunits, for example, with A3243G mutation5050 Petruzzella V, Moraes CT, Sano MC, Bonilla E, DiMauro S, Schon EA. Extremely high levels of mutant mtDNAs co-localize with cytochrome c oxidase-negative ragged-red fibers in patients harboring a point mutation at nt 3243. Hum Mol Genet. 1994;3(3):449-54. doi:10.1093/hmg/3.3.449
https://doi.org/10.1093/hmg/3.3.449...
,5151 Moraes CT, Ricci E, Petruzzella V, Shanske S, DiMauro S, Schon EA et al.. Molecular analysis of the muscle pathology associated with mitochondrial DNA deletions. Nature Genet. 1992;1(5):359-67. doi:10.1038/ng0892-359
https://doi.org/10.1038/ng0892-359...
,5252 Sciacco M, Bonilla E, Schon EA, DiMauro S, Moraes CT. Distribution of wild-type and common deletion forms of mtDNA in normal and respiration-deficient muscle fibers from patients with mitochondrial myopathy. Hum Mol Genet. 1994;3(1):13-9. doi:10.1093/hmg/3.1.13
https://doi.org/10.1093/hmg/3.1.13...
.
The presence of vessels in the muscle biopsy, usually arterioles, with strong reaction to SDH (SDH+) is a common finding in patients with MELAS or MERRF, but rarely found in patients with PEO77 Goto Y. Clinical features of MELAS and mitochondrial DNA mutations. Muscle Nerve. 1995;18(Suppl 3):S107-12. doi:10.1002/mus.880181422
https://doi.org/10.1002/mus.880181422...
,2020 Goto Y, Horai S, Matsuoka T, Koga Y, Nihei K, Kobayashi M et al.. Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS): a correlative study of the clinical features and mitochondrial DNA mutation. Neurology. 1992;42(3):545-50. doi:10.1212/WNL.42.3.545
https://doi.org/10.1212/WNL.42.3.545...
,2929 Lorenzoni PJ, Scola RH, Kay CS, Arndt RC, Freund AA, Bruck I et al.. MELAS: clinical features, muscle biopsy and molecular genetics. Arq Neuropsiquiatr.. 2009;67(3A):668-76. doi:10.1590/S0004-282X2009000400018
https://doi.org/10.1590/S0004-282X200900...
,3030 Lorenzoni PJ, Scola RH, Kay CS, Arndt RC, Silvado CE, Werneck LC. MERRF: clinical features, muscle biopsy and molecular genetics in Brazilian patients. Mitochondrion. 2011;11(3):528-32. doi:10.1016/j.mito.2011.01.003
https://doi.org/10.1016/j.mito.2011.01.0...
,5353 Hasegawa H, Matsuoka T, Goto Y, Nonaka I. Strongly succinate dehydrogenase-reactive blood vessels in muscles from patients with mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. Ann Neurol. 1991;29(6):601-5. doi:10.1002/ana.410290606
https://doi.org/10.1002/ana.410290606...
. These vessels may also occur in other tissues such as brain and gastrointenstinal tract77 Goto Y. Clinical features of MELAS and mitochondrial DNA mutations. Muscle Nerve. 1995;18(Suppl 3):S107-12. doi:10.1002/mus.880181422
https://doi.org/10.1002/mus.880181422...
,2020 Goto Y, Horai S, Matsuoka T, Koga Y, Nihei K, Kobayashi M et al.. Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS): a correlative study of the clinical features and mitochondrial DNA mutation. Neurology. 1992;42(3):545-50. doi:10.1212/WNL.42.3.545
https://doi.org/10.1212/WNL.42.3.545...
,5353 Hasegawa H, Matsuoka T, Goto Y, Nonaka I. Strongly succinate dehydrogenase-reactive blood vessels in muscles from patients with mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. Ann Neurol. 1991;29(6):601-5. doi:10.1002/ana.410290606
https://doi.org/10.1002/ana.410290606...
. Electron microscopy has been observed that these vessels have mitochondria in number and size similar to what occurs in the central nervous system1111 Gilchrist JM, Sikirica M, Stopa E, Shanske S. Adult-onset MELAS. Evidence for involvement of neurons as well as cerebral vasculature in strokelike episodes. Stroke. 1996;27(8):1420-3. doi:10.1161/01.STR.27.8.1420
https://doi.org/10.1161/01.STR.27.8.1420...
,5353 Hasegawa H, Matsuoka T, Goto Y, Nonaka I. Strongly succinate dehydrogenase-reactive blood vessels in muscles from patients with mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. Ann Neurol. 1991;29(6):601-5. doi:10.1002/ana.410290606
https://doi.org/10.1002/ana.410290606...
. Similarly to what occurs in RRF, the study of these vessels with strong reaction to SDH shows that the ratio of mtDNA mutant is extremely high in these vessels when compared to vessels with normal reaction to SDH4545 Tokunaga M, Mita S, Sakuta R, Nonaka I, Araki S. Increased mitochondrial DNA in blood vessels and ragged-red fibers in mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS). Ann Neurol. 1993;33(3):275-80. doi:10.1002/ana.410330308
https://doi.org/10.1002/ana.410330308...
,4646 Tokunaga M, Mita S, Murakami T, Kumamoto T, Uchino M, Nonaka I et al.. Single muscle fiber analysis of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS). Ann Neurol 1994;35(4):413-9. doi:10.1002/ana.410350407
https://doi.org/10.1002/ana.410350407...
. This type of vascular involvement in patients with MELAS has no meaning pathogenic fully known, however, some researchers still believe that this change in the blood vessels indicates that MELAS is a systemic angiopathy3232 Hirano M, Pavlakis SG. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS): current concepts. J Child Neurol. 1994;9(1):4-13. doi:10.1177/088307389400900102
https://doi.org/10.1177/0883073894009001...
,4545 Tokunaga M, Mita S, Sakuta R, Nonaka I, Araki S. Increased mitochondrial DNA in blood vessels and ragged-red fibers in mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS). Ann Neurol. 1993;33(3):275-80. doi:10.1002/ana.410330308
https://doi.org/10.1002/ana.410330308...
,4646 Tokunaga M, Mita S, Murakami T, Kumamoto T, Uchino M, Nonaka I et al.. Single muscle fiber analysis of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS). Ann Neurol 1994;35(4):413-9. doi:10.1002/ana.410350407
https://doi.org/10.1002/ana.410350407...
.
For some authors, even in the absence of RRF in muscle biopsy of patients with suspected, MELAS can be supported by the histological diagnosis when is found a strong reaction of these vessels in SDH77 Goto Y. Clinical features of MELAS and mitochondrial DNA mutations. Muscle Nerve. 1995;18(Suppl 3):S107-12. doi:10.1002/mus.880181422
https://doi.org/10.1002/mus.880181422...
,2020 Goto Y, Horai S, Matsuoka T, Koga Y, Nihei K, Kobayashi M et al.. Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS): a correlative study of the clinical features and mitochondrial DNA mutation. Neurology. 1992;42(3):545-50. doi:10.1212/WNL.42.3.545
https://doi.org/10.1212/WNL.42.3.545...
.
Thus, the muscle biopsy of MELAS patients, especially those with the A3243G mutation, usually has vessels with strong reaction for SDH and RRF with normal COX activity2929 Lorenzoni PJ, Scola RH, Kay CS, Arndt RC, Freund AA, Bruck I et al.. MELAS: clinical features, muscle biopsy and molecular genetics. Arq Neuropsiquiatr.. 2009;67(3A):668-76. doi:10.1590/S0004-282X2009000400018
https://doi.org/10.1590/S0004-282X200900...
,4545 Tokunaga M, Mita S, Sakuta R, Nonaka I, Araki S. Increased mitochondrial DNA in blood vessels and ragged-red fibers in mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS). Ann Neurol. 1993;33(3):275-80. doi:10.1002/ana.410330308
https://doi.org/10.1002/ana.410330308...
,4646 Tokunaga M, Mita S, Murakami T, Kumamoto T, Uchino M, Nonaka I et al.. Single muscle fiber analysis of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS). Ann Neurol 1994;35(4):413-9. doi:10.1002/ana.410350407
https://doi.org/10.1002/ana.410350407...
,5050 Petruzzella V, Moraes CT, Sano MC, Bonilla E, DiMauro S, Schon EA. Extremely high levels of mutant mtDNAs co-localize with cytochrome c oxidase-negative ragged-red fibers in patients harboring a point mutation at nt 3243. Hum Mol Genet. 1994;3(3):449-54. doi:10.1093/hmg/3.3.449
https://doi.org/10.1093/hmg/3.3.449...
.
WHAT ARE THE MAIN DIFFERENTIAL DIAGNOSES?
Many differential diagnoses have been published, but three categories should be especially considerate in investigation of MELAS patients:
First, MELAS should be considered in the differential diagnosis of all acute stroke in young people along with heart disease, carotid or vertebral diseases, sickle cell disease, vasculopathies, lipoprotein dyscrasias, venous thrombosis, Moyamoya disease, complicated migraine (as familial hemiplegic migraine), Fabry disease, homocystinuria caused by cystathionine beta-synthase deficiency, and other2525 DiMauro S, Hirano M. MELAS. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH et al., editors. GeneReviews®[Internet]. Seattle (WA): University of Washington, Seattle; 2013 [citado 21 nov 2013]. Disponível em: http://www.ncbi.nlm.nih.gov/books/NBK1233/
http://www.ncbi.nlm.nih.gov/books/NBK123...
. Previous studies not support screening for mtDNA mutations to diagnose oligosymptomatic forms of MELAS in cryptogenic strokes in the absence of other features of the disease3131 Conforto AB, Yamamoto FI, Oba-Shinjo SM, Pinto JG, Hoshino M, Scaff M et al.. Screening for MELAS mutations in young patients with stroke of undetermined origin. Arq Neuropsiquiatr. 2007;65(2B):371-6. doi:10.1590/S0004-282X2007000300001
https://doi.org/10.1590/S0004-282X200700...
. Besides appropriate specific tests, a maternal history of other problems suggesting mitochondrial dysfunction (short stature, migraine, hearing loss, diabetes mellitus, cardiac involvement), clinical manifestation (improvement of the stroke symptoms but worsening, or start, seizures and/or encephalopathy) and brain imaging (stroke not corresponding to a vascular distribution or change in stroke pattern after acute ictus) can help orient the clinician toward the correct diagnosis and guide mitochondrial histological and/or genetic testing.
Second, stroke-like episodes can also be rarely associated with a variety of other mitochondrial disorders including PEO, Kearn-Sayre syndrome, MERRF, Leigh syndrome, optic neuropathy, maternally inherited diabetes mellitus with or without deafness, cardiomyopathy, deafness, and other. In addition, some patients also present MELAS with mutations in the nuclear DNA genes, asPOLG, and typical phenotype can suffer changes. The family history, clinical features and laboratorial data can help orient the clinician toward the correct diagnosis.
Third, the inborn errors of metabolism which causes progressive encephalopathies, especially when onset is in childhood and early adulthood, as X-linked adrenoleukodystrophy, metachromatic leukodystrophy, Krabbe disease, GM1 gangliosidosis, GM2 gangliosidosis, Fabry disease, Niemann Pick type C, amino acidopathies, organic acid disorders, among other diseases5454 Vanderver A. Tools for diagnosis of leukodystrophies and other disorders presenting with white matter disease. Curr Neurol Neurosci Rep. 2005;5(2):110-8. doi:10.1007/s11910-005-0008-1. Because of the multiplicity of conditions, many different diagnostic tests are used for screening. An abnormal result is often followed by a subsequent “definitive test” to confirm the suspected diagnosis.
WHAT IS THE TREATMENT?
Similar to occurs in other mitochondrial diseases, there is no specific treatment for MELAS. Therapeutic compounds may ameliorate symptoms in individual cases; however, the available therapeutic interventions are not able to affect the essential progression of this disease.
Many therapeutic strategies have been adopted based in the result of isolated case reports or limited clinical studies that have included a heterogeneous population of patients with MELAS or other mitochondrial disorders. The therapeutic compounds were used in the treatment of MELAS to improve respiratory chain or to reduce the levels of reactive oxygen species arising from disrupted mitochondrial metabolism. Some of the most frequently prescribed agents include ubidecarenone (coenzyme Q10, CoQ), idebenone, edaravone, levoarginine (L-arginine), complex B vitamins, vitamin C, vitamin E, and levocarnitine (L-carnitine) in various combinations and doses2525 DiMauro S, Hirano M. MELAS. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH et al., editors. GeneReviews®[Internet]. Seattle (WA): University of Washington, Seattle; 2013 [citado 21 nov 2013]. Disponível em: http://www.ncbi.nlm.nih.gov/books/NBK1233/
http://www.ncbi.nlm.nih.gov/books/NBK123...
,5555 Finsterer J. Management of mitochondrial stroke-like-episodes. Eur J Neurol. 2009;16(11):1178-84. doi:10.1111/j.1468-1331.2009.02789.x
https://doi.org/10.1111/j.1468-1331.2009...
,5656 Koga Y, Povalko N, Nishioka J, Katayama K, Yatsuga S, Matsuishi T. Molecular pathology of MELAS and L-arginine effects. Biochim Biophys Acta. 2012;1820(5):608-14. doi:10.1016/j.bbagen.2011.09.005
https://doi.org/10.1016/j.bbagen.2011.09...
,5757 Finsterer J. Stroke and stroke-like episodes in muscle disease. Open Neurol J. 2012;6(1):26-36. doi:10.2174/1874205X01206010026
https://doi.org/10.2174/1874205X01206010...
,5858 Horvath R, Gorman G, Chinnery PF. How can we treat mitochondrial encephalomyopathies? Approaches to therapy. Neurotherapeutics 2008;5(4):558-68. doi:10.1016/j.nurt.2008.07.002
https://doi.org/10.1016/j.nurt.2008.07.0...
. The concomitant administration of these different medications can be useful and has been benefit to some MELAS patients.
There is no standardized treatment of stroke-like episodes but there is increasing evidence that these patients benefit from the administration of L-arginine and consequent antiepileptic treatment if the stroke-like episodes are associated with epileptic activity2525 DiMauro S, Hirano M. MELAS. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH et al., editors. GeneReviews®[Internet]. Seattle (WA): University of Washington, Seattle; 2013 [citado 21 nov 2013]. Disponível em: http://www.ncbi.nlm.nih.gov/books/NBK1233/
http://www.ncbi.nlm.nih.gov/books/NBK123...
,5555 Finsterer J. Management of mitochondrial stroke-like-episodes. Eur J Neurol. 2009;16(11):1178-84. doi:10.1111/j.1468-1331.2009.02789.x
https://doi.org/10.1111/j.1468-1331.2009...
. Although the underlying mechanisms are not completely understood, in acute phase of the stroke-like episodes in MELAS, seems that L-arginine therapy improve microcirculation and endothelial dysfunction, and improve almost all symptoms associated with stroke-like, with the exception of migraine headaches and visual fields5555 Finsterer J. Management of mitochondrial stroke-like-episodes. Eur J Neurol. 2009;16(11):1178-84. doi:10.1111/j.1468-1331.2009.02789.x
https://doi.org/10.1111/j.1468-1331.2009...
,5656 Koga Y, Povalko N, Nishioka J, Katayama K, Yatsuga S, Matsuishi T. Molecular pathology of MELAS and L-arginine effects. Biochim Biophys Acta. 2012;1820(5):608-14. doi:10.1016/j.bbagen.2011.09.005
https://doi.org/10.1016/j.bbagen.2011.09...
,5757 Finsterer J. Stroke and stroke-like episodes in muscle disease. Open Neurol J. 2012;6(1):26-36. doi:10.2174/1874205X01206010026
https://doi.org/10.2174/1874205X01206010...
,5959 Koga Y, Akita Y, Nishioka J, Yatsuga S, Povalko N, Katayama K et al.. MELAS and L-arginine therapy. Mitochondrion. 2007;7(1-2):133-9. doi:10.1016/j.mito.2006.11.006
https://doi.org/10.1016/j.mito.2006.11.0...
. According to these studies L-arginine is applied in a dosage until 0.5 g/kg body weight intravenously during the acute phase, followed by oral administration thereafter5555 Finsterer J. Management of mitochondrial stroke-like-episodes. Eur J Neurol. 2009;16(11):1178-84. doi:10.1111/j.1468-1331.2009.02789.x
https://doi.org/10.1111/j.1468-1331.2009...
,5656 Koga Y, Povalko N, Nishioka J, Katayama K, Yatsuga S, Matsuishi T. Molecular pathology of MELAS and L-arginine effects. Biochim Biophys Acta. 2012;1820(5):608-14. doi:10.1016/j.bbagen.2011.09.005
https://doi.org/10.1016/j.bbagen.2011.09...
,5757 Finsterer J. Stroke and stroke-like episodes in muscle disease. Open Neurol J. 2012;6(1):26-36. doi:10.2174/1874205X01206010026
https://doi.org/10.2174/1874205X01206010...
. Occasionally, L-arginine is also given together with other drugs5555 Finsterer J. Management of mitochondrial stroke-like-episodes. Eur J Neurol. 2009;16(11):1178-84. doi:10.1111/j.1468-1331.2009.02789.x
https://doi.org/10.1111/j.1468-1331.2009...
. Symptomatic drug treatment of stroke-like episode include antiepileptic treatment if it was accompanied by seizures, analgesic treatment if it was accompanied by headache, or anti-psychotic or sedative therapy if it was dominated by confusion, agitation, anxiety, hyperactivity, or psychosis5757 Finsterer J. Stroke and stroke-like episodes in muscle disease. Open Neurol J. 2012;6(1):26-36. doi:10.2174/1874205X01206010026
https://doi.org/10.2174/1874205X01206010...
. Physical therapy should be implemented in individuals after stroke-like2525 DiMauro S, Hirano M. MELAS. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH et al., editors. GeneReviews®[Internet]. Seattle (WA): University of Washington, Seattle; 2013 [citado 21 nov 2013]. Disponível em: http://www.ncbi.nlm.nih.gov/books/NBK1233/
http://www.ncbi.nlm.nih.gov/books/NBK123...
.
MELAS management also includes additional therapy for its complications, such as, cardiac disease (standard pharmacologic therapy), diabetes mellitus (dietary modification, oral hypoglycemic agents and/or insulin therapy), deafness (hearing devices and cochlear implantation) or epilepsy (traditional antiepileptic treatment)2525 DiMauro S, Hirano M. MELAS. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH et al., editors. GeneReviews®[Internet]. Seattle (WA): University of Washington, Seattle; 2013 [citado 21 nov 2013]. Disponível em: http://www.ncbi.nlm.nih.gov/books/NBK1233/
http://www.ncbi.nlm.nih.gov/books/NBK123...
,5858 Horvath R, Gorman G, Chinnery PF. How can we treat mitochondrial encephalomyopathies? Approaches to therapy. Neurotherapeutics 2008;5(4):558-68. doi:10.1016/j.nurt.2008.07.002
https://doi.org/10.1016/j.nurt.2008.07.0...
,6060 Finsterer J, Zarrouk Mahjoub S. Mitochondrial toxicity of antiepileptic drugs and their tolerability in mitochondrial disorders. Expert Opin Drug Metab Toxicol. 2012;8(1):71-9. doi:10.1517/17425255.2012.644535
https://doi.org/10.1517/17425255.2012.64...
. Because febrile illnesses may trigger acute exacerbations, MELAS patients should receive standard childhood vaccinations, flu vaccine, and pneumococcal vaccine2525 DiMauro S, Hirano M. MELAS. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH et al., editors. GeneReviews®[Internet]. Seattle (WA): University of Washington, Seattle; 2013 [citado 21 nov 2013]. Disponível em: http://www.ncbi.nlm.nih.gov/books/NBK1233/
http://www.ncbi.nlm.nih.gov/books/NBK123...
,5555 Finsterer J. Management of mitochondrial stroke-like-episodes. Eur J Neurol. 2009;16(11):1178-84. doi:10.1111/j.1468-1331.2009.02789.x
https://doi.org/10.1111/j.1468-1331.2009...
. In addition, toxins or drugs that had potential to cause mitochondrial dysfunction or lesion, such as aminoglycoside antibiotics, linezolid, aspirin, Zidovudine, cigarettes or alcohol consumption, should be recognized and avoided5555 Finsterer J. Management of mitochondrial stroke-like-episodes. Eur J Neurol. 2009;16(11):1178-84. doi:10.1111/j.1468-1331.2009.02789.x
https://doi.org/10.1111/j.1468-1331.2009...
,5858 Horvath R, Gorman G, Chinnery PF. How can we treat mitochondrial encephalomyopathies? Approaches to therapy. Neurotherapeutics 2008;5(4):558-68. doi:10.1016/j.nurt.2008.07.002
https://doi.org/10.1016/j.nurt.2008.07.0...
. Valproic acid for seizure treatment (high risk by carnitine uptake inhibition) and dichloroacetate for acute stroke-like episodes (high risk for peripheral neuropathy) are not recommended5858 Horvath R, Gorman G, Chinnery PF. How can we treat mitochondrial encephalomyopathies? Approaches to therapy. Neurotherapeutics 2008;5(4):558-68. doi:10.1016/j.nurt.2008.07.002
https://doi.org/10.1016/j.nurt.2008.07.0...
,6060 Finsterer J, Zarrouk Mahjoub S. Mitochondrial toxicity of antiepileptic drugs and their tolerability in mitochondrial disorders. Expert Opin Drug Metab Toxicol. 2012;8(1):71-9. doi:10.1517/17425255.2012.644535
https://doi.org/10.1517/17425255.2012.64...
. Exercise (endurance and resistance training) is helpful in MELAS as well other mitochondrial diseases5858 Horvath R, Gorman G, Chinnery PF. How can we treat mitochondrial encephalomyopathies? Approaches to therapy. Neurotherapeutics 2008;5(4):558-68. doi:10.1016/j.nurt.2008.07.002
https://doi.org/10.1016/j.nurt.2008.07.0...
.
It is appropriate to offer genetic counseling (including discussion of potential risks to children and reproductive options) to young women in fertile age who are affected or at risk5858 Horvath R, Gorman G, Chinnery PF. How can we treat mitochondrial encephalomyopathies? Approaches to therapy. Neurotherapeutics 2008;5(4):558-68. doi:10.1016/j.nurt.2008.07.002
https://doi.org/10.1016/j.nurt.2008.07.0...
. Prenatal testing and preimplantation genetic diagnosis may be an option for some families in which the disease-causing mutations have been identified but their interpretation is complex yet2525 DiMauro S, Hirano M. MELAS. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH et al., editors. GeneReviews®[Internet]. Seattle (WA): University of Washington, Seattle; 2013 [citado 21 nov 2013]. Disponível em: http://www.ncbi.nlm.nih.gov/books/NBK1233/
http://www.ncbi.nlm.nih.gov/books/NBK123...
,3434 Richardson J, Irving L, Hyslop LA, Choudhary M, Murdoch A, Turnbull DM et al.. Concise reviews: assisted reproductive technologies to prevent transmission of mitochondrial DNA disease. Stem Cells. 2015;33(3):639-45. doi:10.1002/stem.1887
https://doi.org/10.1002/stem.1887...
,5858 Horvath R, Gorman G, Chinnery PF. How can we treat mitochondrial encephalomyopathies? Approaches to therapy. Neurotherapeutics 2008;5(4):558-68. doi:10.1016/j.nurt.2008.07.002
https://doi.org/10.1016/j.nurt.2008.07.0...
. The transfer of nuclear DNA from fertilized oocytes or zygotes harboring a mtDNA mutation to an enucleated recipient cells could theoretically prevent transmission of mtDNA diseases and this promising therapy is under investigation3434 Richardson J, Irving L, Hyslop LA, Choudhary M, Murdoch A, Turnbull DM et al.. Concise reviews: assisted reproductive technologies to prevent transmission of mitochondrial DNA disease. Stem Cells. 2015;33(3):639-45. doi:10.1002/stem.1887
https://doi.org/10.1002/stem.1887...
,5858 Horvath R, Gorman G, Chinnery PF. How can we treat mitochondrial encephalomyopathies? Approaches to therapy. Neurotherapeutics 2008;5(4):558-68. doi:10.1016/j.nurt.2008.07.002
https://doi.org/10.1016/j.nurt.2008.07.0...
. In the pregnancy, MELAS women should be monitored, mainly for diabetes mellitus and respiratory failure, which may require specific therapeutic interventions2525 DiMauro S, Hirano M. MELAS. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH et al., editors. GeneReviews®[Internet]. Seattle (WA): University of Washington, Seattle; 2013 [citado 21 nov 2013]. Disponível em: http://www.ncbi.nlm.nih.gov/books/NBK1233/
http://www.ncbi.nlm.nih.gov/books/NBK123...
.
References
-
1Koenigsberger MR, Pellock JM, DiMauro S, Eastwood A.. Juvenile mitochondrial myopathy, short stature and lactic acidosis: a clinical, biochemical, and ultrastructural study (Abstract #80). In: Fifth Annual Meeting of the Child Neurological Society; 1976 October 28-30; Monterey, CA.
-
2Shapira Y, Cederbaum SD, Cancilla PA, Nielsen D, Lippe BM. Familial poliodystrophy, mitochondrial myopathy, and lactate acidemia. Neurology. 1975;25(7):614-21. doi:10.1212/WNL.25.7.614
» https://doi.org/10.1212/WNL.25.7.614 -
3Pavlakis SG, Phillips PC, DiMauro S, De Vivo DC, Rowland LP. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: a distinctive clinical syndrome. Ann Neurol.1984;16(4):481-8. doi:10.1002/ana.410160409
» https://doi.org/10.1002/ana.410160409 -
4Werneck LC, Abdalla H, Lohr A. [MELAS (mitochondrial encephalopathy, lactic acidosis and stroke like episodes): case report]. Arq Neuropsiquiatr. 1987;45(3) 288-94. Portuguese. doi:10.1590/S0004-282X1987000300009
» https://doi.org/10.1590/S0004-282X1987000300009 -
5Hirano M, Ricci E, Koenigsberger MR, Defendini R, Pavlakis SG, DeVivo DC et al.. Melas: an original case and clinical criteria for diagnosis. Neuromuscul Disord.1992;2(2):125-35. doi:10.1016/0960-8966(92)90045-8
» https://doi.org/10.1016/0960-8966(92)90045-8 -
6Yatsuga S, Povalko N, Nishioka J, Katayama K, Kakimoto N, Matsuishi T et al.. MELAS: a nationwide prospective cohort study of 96 patients in Japan. Biochim Biophys Acta. 2012;1820(5):619-24. doi:10.1016/j.bbagen.2011.03.015
» https://doi.org/10.1016/j.bbagen.2011.03.015 -
7Goto Y. Clinical features of MELAS and mitochondrial DNA mutations. Muscle Nerve. 1995;18(Suppl 3):S107-12. doi:10.1002/mus.880181422
» https://doi.org/10.1002/mus.880181422 -
8Mizukami K, Sasaki M, Suzuki T, Shiraishi H, Koizumi J, Ohkoshi N et al.. Central nervous system changes in mitochondrial encephalomyopathy: light and electron microscopic study. Acta Neuropathol. 1992;83(4):449-52. doi:10.1007/BF00713541
» https://doi.org/10.1007/BF00713541 -
9Ohama E, Ohara S, Ikuta F, Tanaka K, Nishizawa M, Miyatake T. Mitochondrial angiopathy in cerebral blood vessels of mitochondrial encephalomyopathy. Acta Neuropathol. 1987;74(3):226-33. doi:10.1007/BF00688185
» https://doi.org/10.1007/BF00688185 -
10Sakuta R, Nonaka I. Vascular involvement in mitochondrial myopathy. Ann Neurol. 1989;25(6):594-601. doi:10.1002/ana.410250611
» https://doi.org/10.1002/ana.410250611 -
11Gilchrist JM, Sikirica M, Stopa E, Shanske S. Adult-onset MELAS. Evidence for involvement of neurons as well as cerebral vasculature in strokelike episodes. Stroke. 1996;27(8):1420-3. doi:10.1161/01.STR.27.8.1420
» https://doi.org/10.1161/01.STR.27.8.1420 -
12Iizuka T, Sakai F, Suzuki N, Hata T, Tsukahara S, Fukuda M et al.. Neuronal hyperexcitability in stroke-like episodes of MELAS syndrome. Neurology. 2002;59(6):816-24. doi:10.1212/WNL.59.6.816
» https://doi.org/10.1212/WNL.59.6.816 -
13Sparaco M, Bonilla E, DiMauro S, Powers JM. Neuropathology of mitochondrial encephalomyopathies due to mitochondrial DNA defects. J Neuropathol Exp Neurol. 1993;52(1):1-10. doi:10.1097/00005072-199301000-00001
» https://doi.org/10.1097/00005072-199301000-00001 -
14Iizuka T, Sakai F. Pathogenesis of stroke-like episodes in MELAS: analysis neurovascular cellular mechanisms. Curr Neurovasc Res. 2005;2(1):29-45. doi:10.2174/1567202052773544
» https://doi.org/10.2174/1567202052773544 -
15Goto YI, Nonaka I, Horai S. A mutation in the tRNA(Leu)(UUR gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990;348(6302):651-3. doi:10.1038/348651a0
» https://doi.org/10.1038/348651a0 -
16Kobayashi Y, Momoi MY, Tominaga K, Momoi T, Nihei K, Yanagisawa M et al.. A point mutation in the mitochondrial tRNA(Leu)(UUR) gene in MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes). Biochem Biophys Res Comm. 1990;173(3):816-22. doi:10.1016/S0006-291X(05)80860-5
» https://doi.org/10.1016/S0006-291X(05)80860-5 -
17Finsterer J. Genetic, pathogenetic, and phenotypic implications of the mitochondrial A3243G tRNALeu(UUR) mutation. Acta Neurol Scand. 2007;116(1):1-14. doi:10.1111/j.1600-0404.2007.00836.x
» https://doi.org/10.1111/j.1600-0404.2007.00836.x -
18Hao R, Yao YN, Zheng YG, Xu MG, Wang ED. Reduction of mitochondrial tRNALeu(UUR) aminoacylation by some MELAS-associated mutations. FEBS Lett. 2004;578(1-2):135-9. doi:10.1016/j.febslet.2004.11.004
» https://doi.org/10.1016/j.febslet.2004.11.004 -
19Ciafaloni E, Ricci E, Shanske S, Moraes CT, Silvestri G, Hirano M et al.. MELAS: clinical features, biochemistry and molecular genetics. Ann Neurol. 1992;31(4):391-8. doi:10.1002/ana.410310408
» https://doi.org/10.1002/ana.410310408 -
20Goto Y, Horai S, Matsuoka T, Koga Y, Nihei K, Kobayashi M et al.. Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS): a correlative study of the clinical features and mitochondrial DNA mutation. Neurology. 1992;42(3):545-50. doi:10.1212/WNL.42.3.545
» https://doi.org/10.1212/WNL.42.3.545 -
21Hammans SR, Sweeney MG, Brockington M, Morgan-Hughes JA, Harding AE. Mitochondrial encephalopathies: molecular genetic diagnosis from blood samples. Lancet. 1991;337(8753):1311-3. doi:10.1016/0140-6736(91)92981-7
» https://doi.org/10.1016/0140-6736(91)92981-7 -
22Goto Y, Nonaka I, Horai S. A new mtDNA mutation associated with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS). Biochim Biophys Acta. 1991;1097(3): 238-40. doi:10.1016/0925-4439(91)90042-8
» https://doi.org/10.1016/0925-4439(91)90042-8 -
23Zhao D, Hong D, Zhang W, et al.. Mutations in mitochondrially encoded complex I enzyme as the second common cause in a cohort of Chinese patients with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes. J Hum Genet. 2011;56(11):759-64. doi:10.1038/jhg.2011.96
» https://doi.org/10.1038/jhg.2011.96 -
24Zeviani M, Di Donato S. Mitochondrial disorders. Brain. 2004;127(10):2153-72. doi:10.1093/brain/awh259
» https://doi.org/10.1093/brain/awh259 -
25DiMauro S, Hirano M. MELAS. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH et al., editors. GeneReviews®[Internet]. Seattle (WA): University of Washington, Seattle; 2013 [citado 21 nov 2013]. Disponível em: http://www.ncbi.nlm.nih.gov/books/NBK1233/
» http://www.ncbi.nlm.nih.gov/books/NBK1233/ -
26Bataillard M, Chatzoglou E, Rumbach L, Sternberg D, Tournade A, Laforêt P et al.. Atypical MELAS syndrome associated with a new mitochondrial tRNA glutamine point mutation. Neurology. 2001;56(3):405-7. doi:10.1212/WNL.56.3.405
» https://doi.org/10.1212/WNL.56.3.405 -
27Kaufmann P, Engelstad K, Wei Y, Kulikova R, Oskoui M, Sproule DM et al.. Natural history of MELAS associated with mitochondrial DNA m.3243A>G genotype. Neurology. 2011;77(22):1965-71. doi:10.1212/WNL.0b013e31823a0c7f
» https://doi.org/10.1212/WNL.0b013e31823a0c7f -
28Nishioka J, Akita Y, Yatsuga S, Katayama K, Matsuishi T, Ishibashi M et al.. Inappropriate intracranial hemodynamics in the natural course of MELAS. Brain Dev. 2008;30(2):100-5. doi:10.1016/j.braindev.2007.06.008
» https://doi.org/10.1016/j.braindev.2007.06.008 -
29Lorenzoni PJ, Scola RH, Kay CS, Arndt RC, Freund AA, Bruck I et al.. MELAS: clinical features, muscle biopsy and molecular genetics. Arq Neuropsiquiatr.. 2009;67(3A):668-76. doi:10.1590/S0004-282X2009000400018
» https://doi.org/10.1590/S0004-282X2009000400018 -
30Lorenzoni PJ, Scola RH, Kay CS, Arndt RC, Silvado CE, Werneck LC. MERRF: clinical features, muscle biopsy and molecular genetics in Brazilian patients. Mitochondrion. 2011;11(3):528-32. doi:10.1016/j.mito.2011.01.003
» https://doi.org/10.1016/j.mito.2011.01.003 -
31Conforto AB, Yamamoto FI, Oba-Shinjo SM, Pinto JG, Hoshino M, Scaff M et al.. Screening for MELAS mutations in young patients with stroke of undetermined origin. Arq Neuropsiquiatr. 2007;65(2B):371-6. doi:10.1590/S0004-282X2007000300001
» https://doi.org/10.1590/S0004-282X2007000300001 -
32Hirano M, Pavlakis SG. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS): current concepts. J Child Neurol. 1994;9(1):4-13. doi:10.1177/088307389400900102
» https://doi.org/10.1177/088307389400900102 -
33Okajima Y, Tanabe Y, Takayanagi M, Aotsuka H. A follow up study of myocardial involvement in patients with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS). Heart. 1998;80(3):292-5. doi:10.1136/hrt.80.3.292
» https://doi.org/10.1136/hrt.80.3.292 -
34Richardson J, Irving L, Hyslop LA, Choudhary M, Murdoch A, Turnbull DM et al.. Concise reviews: assisted reproductive technologies to prevent transmission of mitochondrial DNA disease. Stem Cells. 2015;33(3):639-45. doi:10.1002/stem.1887
» https://doi.org/10.1002/stem.1887 -
35Haas R, Dietrich R. Neuroimaging of mitochondrial disorders. Mitochondrion. 2004;4(5-6):471-90. doi:10.1016/j.mito.2004.07.008
» https://doi.org/10.1016/j.mito.2004.07.008 -
36Tschampa HJ, Urbach H, Greschus S, Kunz WS, Kornblum C. Neuroimaging characteristics in mitochondrial encephalopathies associated with the m.3243A>G MTTL1 mutation. J Neurol. 2013;260(4):1071-80. doi:10.1007/s00415-012-6763-4
» https://doi.org/10.1007/s00415-012-6763-4 -
37Iizuka T, Sakai F, Kan S, Suzuki N. Slowly progressive spread of the stroke-like lesions in MELAS. Neurology. 2003;61(9):1238-44. doi:10.1212/01.WNL.0000091888.26232.FE
» https://doi.org/10.1212/01.WNL.0000091888.26232.FE -
38Kamada K, Takeuchi F, Houkin K, Kitagawa M, Kuriki S, Ogata A et al.. Reversible brain dysfunction in MELAS: MEG, and (1)H MRS analysis. J Neurol Neurosurg Psychiatry. 2001;70(5):675-8. doi:10.1136/jnnp.70.5.675
» https://doi.org/10.1136/jnnp.70.5.675 -
39Betts J, Jaros E, Perry RH, Schaefer AM, Taylor RW, Abdel-All Z et al.. Molecular neuropathology of MELAS: level of heteroplasmy in individual neurones and evidence of extensive vascular involvement. Neuropathol Appl Neurobiol. 2006;32(4):359-73. doi:10.1111/j.1365-2990.2006.00731.x
» https://doi.org/10.1111/j.1365-2990.2006.00731.x -
40Abe K, Yoshimura H, Tanaka H, Fujita N, Hikita T, Sakoda S. Comparison of conventional and diffusion-weighted MRI and proton MR spectroscopy in patients with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like events. Neuroradiology. 2004;46(2):113-7. doi:10.1007/s00234-003-1138-2
» https://doi.org/10.1007/s00234-003-1138-2 -
41Oppenheim C, Galanaud D, Samson Y, Sahel M, Dormont D, Wechsler B et al.. Can diffusion weighted magnetic resonance imaging help differentiate stroke from stroke-like events in MELAS? J Neurol Neurosurg Psychiatry. 2000;69(2):248-50. doi:10.1136/jnnp.69.2.248
» https://doi.org/10.1136/jnnp.69.2.248 -
42Tanahashi C, Nakayama A, Yoshida M, Ito M, Mori N, Hashizume Y. MELAS with the mitochondrial DNA 3243 point mutation: a neuropathological study. Acta Neuropathol. 2000;99(1):31-8. doi:10.1007/PL00007403
» https://doi.org/10.1007/PL00007403 -
43Kaufmann P, Shungu DC, Sano MC, Jhung S, Engelstad K, Mitsis E et al.. Cerebral lactic acidosis correlates with neurological impairment in MELAS. Neurology. 2004;62(8):1297-302. doi:10.1212/01.WNL.0000120557.83907.A8
» https://doi.org/10.1212/01.WNL.0000120557.83907.A8 -
44Kobayashi M, Morishita H, Sugiyama N, Yokochi K, Nakano M, Wada Y et al.. Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes syndrome and NADH-CoQ reductase deficiency. J Inherit Metab Dis. 1986;9(3):301-4. doi:10.1007/BF01799670
» https://doi.org/10.1007/BF01799670 -
45Tokunaga M, Mita S, Sakuta R, Nonaka I, Araki S. Increased mitochondrial DNA in blood vessels and ragged-red fibers in mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS). Ann Neurol. 1993;33(3):275-80. doi:10.1002/ana.410330308
» https://doi.org/10.1002/ana.410330308 -
46Tokunaga M, Mita S, Murakami T, Kumamoto T, Uchino M, Nonaka I et al.. Single muscle fiber analysis of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS). Ann Neurol 1994;35(4):413-9. doi:10.1002/ana.410350407
» https://doi.org/10.1002/ana.410350407 -
47Rossmanith W, Freilinger M, Roka J, Raffelsberger T, Moser-Thier K, Prayer D et al.. Isolated cytochrome c oxidase deficiency as a cause of MELAS. J Med Genet. 2008;45(2):117-21. doi:10.1136/jmg.2007.052076
» https://doi.org/10.1136/jmg.2007.052076 -
48Tanji K, Kaufmann P, Naini AB, Lu J, Parsons TC, Wang D et al.. A novel tRNA(Val) mitochondrial DNA mutation causing MELAS. J Neurol Sci. 2008;270(1-2):23-7. doi:10.1016/j.jns.2008.01.016
» https://doi.org/10.1016/j.jns.2008.01.016 -
49Matsuoka T, Goto Y, Hasegawa H, Nonaka I. Segmental cytochrome c-oxidase deficiency in CPEO: teased muscle fiber analysis. Muscle Nerve. 1992;15(2):209-13. doi:10.1002/mus.880150213
» https://doi.org/10.1002/mus.880150213 -
50Petruzzella V, Moraes CT, Sano MC, Bonilla E, DiMauro S, Schon EA. Extremely high levels of mutant mtDNAs co-localize with cytochrome c oxidase-negative ragged-red fibers in patients harboring a point mutation at nt 3243. Hum Mol Genet. 1994;3(3):449-54. doi:10.1093/hmg/3.3.449
» https://doi.org/10.1093/hmg/3.3.449 -
51Moraes CT, Ricci E, Petruzzella V, Shanske S, DiMauro S, Schon EA et al.. Molecular analysis of the muscle pathology associated with mitochondrial DNA deletions. Nature Genet. 1992;1(5):359-67. doi:10.1038/ng0892-359
» https://doi.org/10.1038/ng0892-359 -
52Sciacco M, Bonilla E, Schon EA, DiMauro S, Moraes CT. Distribution of wild-type and common deletion forms of mtDNA in normal and respiration-deficient muscle fibers from patients with mitochondrial myopathy. Hum Mol Genet. 1994;3(1):13-9. doi:10.1093/hmg/3.1.13
» https://doi.org/10.1093/hmg/3.1.13 -
53Hasegawa H, Matsuoka T, Goto Y, Nonaka I. Strongly succinate dehydrogenase-reactive blood vessels in muscles from patients with mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. Ann Neurol. 1991;29(6):601-5. doi:10.1002/ana.410290606
» https://doi.org/10.1002/ana.410290606 -
54Vanderver A. Tools for diagnosis of leukodystrophies and other disorders presenting with white matter disease. Curr Neurol Neurosci Rep. 2005;5(2):110-8. doi:10.1007/s11910-005-0008-1
-
55Finsterer J. Management of mitochondrial stroke-like-episodes. Eur J Neurol. 2009;16(11):1178-84. doi:10.1111/j.1468-1331.2009.02789.x
» https://doi.org/10.1111/j.1468-1331.2009.02789.x -
56Koga Y, Povalko N, Nishioka J, Katayama K, Yatsuga S, Matsuishi T. Molecular pathology of MELAS and L-arginine effects. Biochim Biophys Acta. 2012;1820(5):608-14. doi:10.1016/j.bbagen.2011.09.005
» https://doi.org/10.1016/j.bbagen.2011.09.005 -
57Finsterer J. Stroke and stroke-like episodes in muscle disease. Open Neurol J. 2012;6(1):26-36. doi:10.2174/1874205X01206010026
» https://doi.org/10.2174/1874205X01206010026 -
58Horvath R, Gorman G, Chinnery PF. How can we treat mitochondrial encephalomyopathies? Approaches to therapy. Neurotherapeutics 2008;5(4):558-68. doi:10.1016/j.nurt.2008.07.002
» https://doi.org/10.1016/j.nurt.2008.07.002 -
59Koga Y, Akita Y, Nishioka J, Yatsuga S, Povalko N, Katayama K et al.. MELAS and L-arginine therapy. Mitochondrion. 2007;7(1-2):133-9. doi:10.1016/j.mito.2006.11.006
» https://doi.org/10.1016/j.mito.2006.11.006 -
60Finsterer J, Zarrouk Mahjoub S. Mitochondrial toxicity of antiepileptic drugs and their tolerability in mitochondrial disorders. Expert Opin Drug Metab Toxicol. 2012;8(1):71-9. doi:10.1517/17425255.2012.644535
» https://doi.org/10.1517/17425255.2012.644535
Publication Dates
-
Publication in this collection
Nov 2015
History
-
Received
02 June 2015 -
Accepted
23 June 2015