ABSTRACT
Autoimmune encephalitis (AIE) is one of the most common causes of noninfectious encephalitis. It can be triggered by tumors, infections, or it may be cryptogenic. The neurological manifestations can be either acute or subacute and usually develop within six weeks. There are a variety of clinical manifestations including behavioral and psychiatric symptoms, autonomic disturbances, movement disorders, and seizures. We reviewed common forms of AIE and discuss their diagnostic approach and treatment.
encephalitis; antibodies, neoplasm; status epilepticus; anti-N-Methyl-D-Aspartate receptor encephalitis; immunoglobulin; rituximab
RESUMO
As encefalites autoimunes (EAI) são a principal causa de encefalite não-infecciosa. As manifestações neurológicas são variadas, incluindo alterações comportamentais ou psiquiátricas, disautonomia, transtornos do movimento e epilepsia. Habitualmente a instalação dos sintomas ocorre em até 6 semanas, de forma aguda ou subaguda. As EAI podem ser desencadeadas por tumores, quadros infecciosos virais ou ainda apresentar etiologia criptogênica. Este artigo revisa as principais EAI, estratégias de diagnóstico e tratamento.
encefalites; anticorpos antineoplásicos; status epiléptico; encefalite antirreceptor de N-Metil-D-Aspartato; imunoglobulinas; rituximab
Autoimmune encephalitis (AIE) is considered one of the most common causes of noninfectious acute encephalitis. It is estimated that 20% of all encephalitis cases in northern Europe are immune-mediated11. Granerod J, Ambrose HE, Davies NW, Clewley JP, Walsh AL, Morgan D et al. Causes of encephalitis and differences in their clinical presentations in England: A multicentre, population-based prospective study. Lancet Infect Dis. 2010;10(12):835-44. https://doi.org/10.1016/S1473-3099(10)70222-X
https://doi.org/10.1016/S1473-3099(10)70...
. The California Encephalitis Project found that anti-N-methyl-D-aspartate receptor (anti-NMDAR) encephalitis occurred in 47% of patients under 30 years of age22. Gable MS, Sheriff H, Dalmau J, Tilley DH, Glaser CA. The frequency of autoimmune N-methyl-D-aspartate receptor encephalitis surpasses that of individual viral etiologies in young individuals enrolled in the California Encephalitis Project. Clin Infect Dis. 2012;54(7):899-904. https://doi.org/10.1093/cid/cir1038
https://doi.org/10.1093/cid/cir1038...
. Autoimmune encephalitis is typically an acute or subacute onset and that may become chronic later33. Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016;15(4):391-404. https://doi.org/10.1016/S1474-4422(15)00401-9
https://doi.org/10.1016/S1474-4422(15)00...
. Suggested mechanisms that may trigger AIE include tumors (paraneoplastic), infections (parainfectious), or it may be cryptogenic44. Linnoila JJ, Binnicker MJ, Majed M, Klein CJ, McKeon A. CSF herpes virus and autoantibody profiles in the evaluation of encephalitis. Neurol - Neuroimmunol Neuroinflammation. 2016;3(4):e245. https://doi.org/10.1212/NXI.0000000000000245
https://doi.org/10.1212/NXI.000000000000...
.
Autoimmune encephalitis has a wide variety of clinical manifestations including behavioral and psychiatric symptoms, autonomic disturbances, movement disorders and seizures33. Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016;15(4):391-404. https://doi.org/10.1016/S1474-4422(15)00401-9
https://doi.org/10.1016/S1474-4422(15)00...
,55. Sabater L, Gaig C, Gelpi E, Bataller L, Lewerenz J, Torres-Vega E et al. A novel non-rapid-eye movement and rapid-eye-movement parasomnia with sleep breathing disorder associated with antibodies to IgLON5: a case series, characterisation of the antigen, and post-mortem study. Lancet Neurol. 2014;13(6):575-86. https://doi.org/10.1016/S1474-4422(14)70051-1
https://doi.org/10.1016/S1474-4422(14)70...
. We reviewed common causes of AIE and discuss their pathophysiology, diagnostic approach and management.
Pathophysiology of autoimmune encephalitis: an overview
Autoimmune encephalitis presents an immune response against neuronal autoantigens with production of antibodies66. Lancaster E, Dalmau J. Neuronal autoantigens: pathogenesis, associated disorders and antibody testing. Nat Rev Neurol. 2012;8(7):380-90. https://doi.org/10.1038/nrneurol.2012.99
https://doi.org/10.1038/nrneurol.2012.99...
. Anti-neuronal antibodies are classified into antibodies against cell surface antigens (CSAab), antibodies against synaptic antigens (SyAab) and antibodies against intraneuronal antigens (INAab), also known as onconeural antibodies66. Lancaster E, Dalmau J. Neuronal autoantigens: pathogenesis, associated disorders and antibody testing. Nat Rev Neurol. 2012;8(7):380-90. https://doi.org/10.1038/nrneurol.2012.99
https://doi.org/10.1038/nrneurol.2012.99...
,77. Coevorden-Hameete MH, de Graaff E, Titulaer MJ, Hoogenraad CC, Sillevis Smitt PE. Molecular and cellular mechanisms underlying anti-neuronal antibody mediated disorders of the central nervous system. Autoimmun Rev. 2014;13(3):299-312. https://doi.org/10.1016/j.autrev.2013.10.016
https://doi.org/10.1016/j.autrev.2013.10...
.
The CSAab (i.e., anti-NMDAR antibodies) target molecules involved in neurotransmission leading to neuronal dysfunction77. Coevorden-Hameete MH, de Graaff E, Titulaer MJ, Hoogenraad CC, Sillevis Smitt PE. Molecular and cellular mechanisms underlying anti-neuronal antibody mediated disorders of the central nervous system. Autoimmun Rev. 2014;13(3):299-312. https://doi.org/10.1016/j.autrev.2013.10.016
https://doi.org/10.1016/j.autrev.2013.10...
. They may have agonistic or antagonistic effects on the receptors, block ion channel pores or disrupt the interaction with neighboring molecules. They could also alter receptor localization at the membrane or cause receptor internalization, thus reducing cell surface expression of receptors77. Coevorden-Hameete MH, de Graaff E, Titulaer MJ, Hoogenraad CC, Sillevis Smitt PE. Molecular and cellular mechanisms underlying anti-neuronal antibody mediated disorders of the central nervous system. Autoimmun Rev. 2014;13(3):299-312. https://doi.org/10.1016/j.autrev.2013.10.016
https://doi.org/10.1016/j.autrev.2013.10...
. Moreover, they could lead to complement deposition and activation of natural killer cells resulting in cell death77. Coevorden-Hameete MH, de Graaff E, Titulaer MJ, Hoogenraad CC, Sillevis Smitt PE. Molecular and cellular mechanisms underlying anti-neuronal antibody mediated disorders of the central nervous system. Autoimmun Rev. 2014;13(3):299-312. https://doi.org/10.1016/j.autrev.2013.10.016
https://doi.org/10.1016/j.autrev.2013.10...
.
The SyAab are believed to contribute to alteration of neurotransmitter release. In contrast, INAab (i.e., anti-Hu, anti-Yo, anti-Ma) are most likely not directly pathogenic and probably an epiphenomenon of T-cell-mediated immune response77. Coevorden-Hameete MH, de Graaff E, Titulaer MJ, Hoogenraad CC, Sillevis Smitt PE. Molecular and cellular mechanisms underlying anti-neuronal antibody mediated disorders of the central nervous system. Autoimmun Rev. 2014;13(3):299-312. https://doi.org/10.1016/j.autrev.2013.10.016
https://doi.org/10.1016/j.autrev.2013.10...
.
The term AIE is used to describe a group of neurological disorders with symptoms of limbic and extra-limbic dysfunction in association with CSAab or SyAab88. Leypoldt F, Wandinger K-P, Bien CG, Dalmau J. Autoimmune Encephalitis. Eur Neurol Rev. 2013;8(1):31-7. https://doi.org/10.17925/ENR.2013.08.01.31
https://doi.org/10.17925/ENR.2013.08.01....
. Patients may have antibodies of more than one type, and CSAab and SyAab may be present together with anti-INAab, especially in paraneoplastic AIE.
Patients with AIE associated with CSAab have a more favorable prognosis. In contrast, those with AIE associated with INAab often show limited response to immunotherapy and their symptoms are mostly irreversible due to T-cell-mediated neuronal damage66. Lancaster E, Dalmau J. Neuronal autoantigens: pathogenesis, associated disorders and antibody testing. Nat Rev Neurol. 2012;8(7):380-90. https://doi.org/10.1038/nrneurol.2012.99
https://doi.org/10.1038/nrneurol.2012.99...
,77. Coevorden-Hameete MH, de Graaff E, Titulaer MJ, Hoogenraad CC, Sillevis Smitt PE. Molecular and cellular mechanisms underlying anti-neuronal antibody mediated disorders of the central nervous system. Autoimmun Rev. 2014;13(3):299-312. https://doi.org/10.1016/j.autrev.2013.10.016
https://doi.org/10.1016/j.autrev.2013.10...
.
Clinical manifestations
Patients with AIE may present with a variety of movement disorders such as ataxia, dystonia, myoclonus, and orofacial dyskinesia. Seizures are the most common symptom and different types of seizures may be seen, including refractory status epilepticus99. Davis R, Dalmau J. Autoimmunity, seizures, and status epilepticus. Epilepsia. 2013;54(6 Suppl 6):46-9. https://doi.org/10.1111/epi.12276
https://doi.org/10.1111/epi.12276...
. Autonomic disturbances are also frequently reported such as sudoresis, hypertension, tachycardia and hypoventilation. Some patients may show involvement of the myenteric plexus and develop gastrointestinal manifestations (diarrhea, gastroparesis, and constipation). Sleep disturbances such as agrypnia excitata, insomnia, abnormal sleep movements and behaviors, sleep apnea, and hypersomnia are also found1010. Tobin WO, Lennon VA, Komorowski L, Probst C, Clardy SL, Aksamit AJ et al. DPPX potassium channel antibody: frequency, clinical accompaniments, and outcomes in 20 patients. Neurology. 2014; 83(20):1797-803. https://doi.org/10.1212/WNL.0000000000000991
https://doi.org/10.1212/WNL.000000000000...
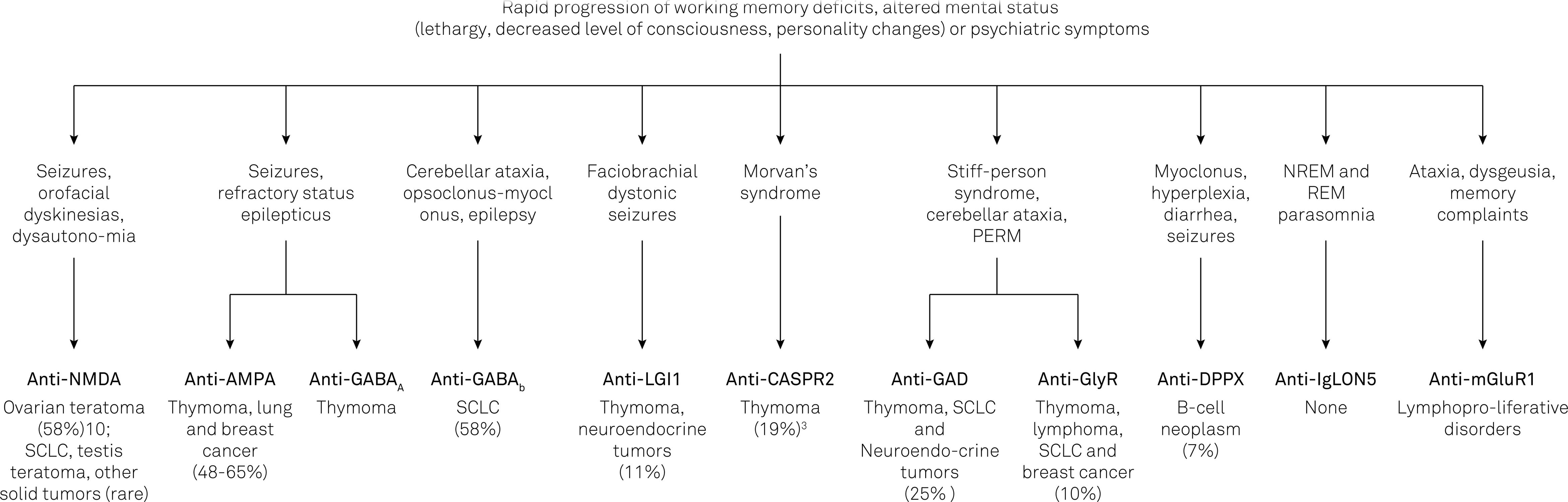
. Some of these findings are suggestive of a certain type of encephalitis and may indicate a specific underlying antibody and tumor (Figure 1).
Table 1 summarizes the diagnostic criteria for limbic encephalitis. These include working memory deficits, mood changes, and often seizures within three months from onset. Many patients with AIE present with a broader spectrum of neurological symptoms. For improving the recognition of immune-mediated cases, diagnostic criteria for suspected AIE were recently established including criteria for seronegative AIE (Table 2). Definite soropositive AIE requires typical clinical picture and positive anti-neuronal antibodies
Anti-NMDAR encephalitis
Anti-N-methyl-D-aspartate receptor encephalitis is one of the most common causes of AIE and was originally described in 2007 in a cohort of 12 patients, 11 of them with ovarian teratomas1111. Dalmau J, Tüzün E, Wu HY, Masjuan J, Rossi JE, Voloschin A et al. Paraneoplastic anti- N -methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61(1):25-36. https://doi.org/10.1002/ana.21050
https://doi.org/10.1002/ana.21050...
. This condition predominantly affects children and young female patients33. Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016;15(4):391-404. https://doi.org/10.1016/S1474-4422(15)00401-9
https://doi.org/10.1016/S1474-4422(15)00...
. Underlying malignancies are found mainly in patients between the age of 12–45 years; most of them are ovarian teratomas (94%), followed by extraovarian teratomas (2%), and other tumors (4%). Herpes simplex virus-1 encephalitis appears to be a trigger for anti-NMDAR encephalitis; most postherpetic AIE cases are now believed to be anti-NMDAR encephalitis.
Approximately 70% of patients present with prodromal symptoms such as fever, headache, nausea, vomiting, diarrhea, and flu-like symptoms, two weeks before the onset of neurological manifestations. Behavioral complaints, psychosis, delusions, hallucinations and paranoia, accompanied with memory deficits and language disturbance, are frequently found at an early stage33. Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016;15(4):391-404. https://doi.org/10.1016/S1474-4422(15)00401-9
https://doi.org/10.1016/S1474-4422(15)00...
,1212. Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol. 2011;10(1):63-74. https://doi.org/10.1016/S1474-4422(10)70253-2PMID:21163445
https://doi.org/10.1016/S1474-4422(10)70...
. The most common movement disorders are orofacial dyskinesias, choreoathethosis, and dystonia1212. Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol. 2011;10(1):63-74. https://doi.org/10.1016/S1474-4422(10)70253-2PMID:21163445
https://doi.org/10.1016/S1474-4422(10)70...
. Patients may progress to catatonia or mutism, followed by an altered level of consciousness and autonomic instability. An important differential diagnosis of anti-NMDAR encephalitis is neuroleptic malignant syndrome, because many patients are initially treated with neuroleptics for behavioral symptoms33. Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016;15(4):391-404. https://doi.org/10.1016/S1474-4422(15)00401-9
https://doi.org/10.1016/S1474-4422(15)00...
,1313. Titulaer MJ, McCracken L, Gabilondo I, Armangué T, Glaser C, Iizuka T et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. 2013; 12(2):157-65. https://doi.org/10.1016/S1474-4422(12)70310-1
https://doi.org/10.1016/S1474-4422(12)70...
. Table 3 summarizes the diagnostic criteria for anti-NMDAR encephalitis.
Seizures are seen in 16% of female and 34% of male patients; they can be focal seizures or patients may present with status epilepticus. Extreme delta brush is a specific electroencephalogram (EEG) pattern found in 30% of patients with anti-NMDAR encephalitis1414. Schmitt SE, Pargeon K, Frechette ES, Hirsch LJ. Extreme delta brush: a unique EEG pattern in adults with anti-NMDA receptor encephalitis. Neurology. 2012;79:1094-100. https://doi.org/10.1212/WNL.0b013e3182698cd8
https://doi.org/10.1212/WNL.0b013e318269...
. Children more frequently present with behavioral symptoms and movement disorders, whereas adults present with psychiatric symptoms and seizures.
Brain magnetic resonance imaging (MRI) is abnormal in 35% of patients at disease onset, but it may show late abnormalities in 50%, mainly nonspecific hyperintense lesions in the grey and white matter. Rare cases show lesions suggestive of demyelination and overlap with demyelinating syndromes such as neuromyelitis optica spectrum disorders (NMOSD) associated with anti-aquaporin-4 antibodies or demyelinating diseases associated with myelin oligodendrocyte glycoprotein antibodies(MOG-ab)1313. Titulaer MJ, McCracken L, Gabilondo I, Armangué T, Glaser C, Iizuka T et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. 2013; 12(2):157-65. https://doi.org/10.1016/S1474-4422(12)70310-1
https://doi.org/10.1016/S1474-4422(12)70...
.
Anti-AMPAR encephalitis
Patients with anti-α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (anti-AMPAR) encephalitis characteristically present with seizures, memory impairment and psychosis. Some may develop sleep disturbances and movement disorders. Anti-AMPAR encephalitis is paraneoplastic in etiology in 64% of cases, mostly associated with thymoma, ovarian teratoma and lung and breast cancer. Brain MRI shows T2 and FLAIR hyperintensities, particularly in the medial temporal lobe. Subcortical and cortical lesions, which are sometimes suggestive of demyelination, may also be found. Cerebrospinal fluid (CSF) examination may show pleocytosis and oligoclonal bands1515. Höftberger R, Sonderen A, Leypoldt F, Houghton D, Geschwind M, Gelfand J et al. Encephalitis and AMPA receptor antibodies: novel findings in a case series of 22 patients. Neurology. 2015;84(24):2403-12. https://doi.org/10.1212/WNL.0000000000001682
https://doi.org/10.1212/WNL.000000000000...
.
Anti-GABA-AR encephalitis
Anti-gamma-aminobutyric acid A receptor (anti-GABA-AR) encephalitis was first reported in 2014 in six patients (two male children, one female teenager and three male adults)1616. Petit-Pedrol M, Armangue T, Peng X, Bataller L, Cellucci T, Davis R et al. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: a case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol. 2014;13(3):276-86. https://doi.org/10.1016/S1474-4422(13)70299-0
https://doi.org/10.1016/S1474-4422(13)70...
. They developed a rapidly progressive encephalopathy with early behavioral or cognitive changes that evolved with refractory seizures and multifocal lesions as seen on brain MRI1616. Petit-Pedrol M, Armangue T, Peng X, Bataller L, Cellucci T, Davis R et al. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: a case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol. 2014;13(3):276-86. https://doi.org/10.1016/S1474-4422(13)70299-0
https://doi.org/10.1016/S1474-4422(13)70...
. In most of these patients, CSF analysis showed lymphocytic pleocytosis with or without oligoclonal bands. A recent study identified an underlying neoplasia in 27% of these patients, mostly thymomas1717. Spatola M, Petit-Pedrol M, Simabukuro MM, Armangue T, Castro FJ, Barcelo Artigues MI et al. Investigations in GABAA receptor antibody-associated encephalitis. Neurology. 2017;88 (11):1012-20. https://doi.org/10.1212/WNL.0000000000003713
https://doi.org/10.1212/WNL.000000000000...
. Similar to that seen in patients with anti-gamma-aminobutyric acid B receptor (GABA-BR) and anti-AMPAR antibodies, they may also present with coexisting autoimmune disorders such as thyroiditis or myasthenia1818. Ohkawa T, Satake S, Yokoi N, Miyazaki Y, Ohshita T, Sobue G et al. Identification and characterization of GABA(A) receptor autoantibodies in autoimmune encephalitis. J Neurosci. 2014;34(24):8151-63. https://doi.org/10.1523/JNEUROSCI.4415-13.2014
https://doi.org/10.1523/JNEUROSCI.4415-1...
.
Anti-GABA-BR encephalitis
Anti-GABA-BR encephalitis is characterized by cognitive symptoms with severe seizures or status epilepticus1919. Lancaster E, Lai M, Peng X, Hughes E, Constantinescu R, Raizer J et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol. 2010;9(1):67-76. https://doi.org/10.1016/S1474-4422(09)70324-2
https://doi.org/10.1016/S1474-4422(09)70...
. Other presentations include ataxia and opsoclonus-myoclonus. In a small series of 20 patients with anti-GABA-BR, about 50% were found to have small-cell lung cancer2020. Höftberger R, Titulaer MJ, Sabater L, Dome B, Rózsás A, Hegedus B et al. Encephalitis and GABAB receptor antibodies: novel findings in a new case series of 20 patients. Neurology. 2013;81(17):1500-6. https://doi.org/10.1212/WNL.0b013e3182a9585f
https://doi.org/10.1212/WNL.0b013e3182a9...
. Males and females appear to be equally affected. The long-term prognosis in anti-GABA-BR encephalitis is determined by the presence of an underlying malignancy2121. Lancaster E, Martinez-Hernandez E, Dalmau J. Encephalitis and antibodies to synaptic and neuronal cell surface proteins. Neurology. 2011;77(2):179-89. https://doi.org/10.1212/WNL.0b013e318224afde
https://doi.org/10.1212/WNL.0b013e318224...
.
Anti-LGI1 and anti-CASPR2 encephalitis (formerly known as anti-VGKC-associated encephalitis)
The first reports of anti-voltage-gated potassium channel-complex antibodies (anti-VGKC) date back to 2001 and described patients with neuromyotonia, Morvan’s syndrome and limbic encephalitis2222. Sonderen A, Schreurs MW, Bruijn MA, Boukhrissi S, Nagtzaam MM, Hulsenboom ES et al. The relevance of VGKC positivity in the absence of LGI1 and Caspr2 antibodies. Neurology. 2016;86(18):1692-9. https://doi.org/10.1212/WNL.0000000000002637
https://doi.org/10.1212/WNL.000000000000...
. Other rare phenotypes included epilepsy and painful polyneuropathy2323. Lilleker JB, Jones MS, Mohanraj R. VGKC complex antibodies in epilepsy: diagnostic yield and therapeutic implications. Seizure. 2013;22(9):776-9. https://doi.org/10.1016/j.seizure.2013.06.004
https://doi.org/10.1016/j.seizure.2013.0...
. Anti-VGKC antibodies, in fact, later turned out to be directed against proteins that form a complex with VGKC called leucine-rich glioma-inactivated 1 (LGI1 and contactin-associated protein-like 2 (CASPR-2)2424. Irani SR, Pettingill P, Kleopa KA, Schiza N, Waters P, Mazia C et al. Morvan syndrome: clinical and serological observations in 29 cases. Ann Neurol. 2012;72(2):241-55. https://doi.org/10.1002/ana.23577
https://doi.org/10.1002/ana.23577...
,2525. van Sonderen A, Schreurs MWJ, Wirtz PW, Sillevis Smitt PAE, Titulaer MJ. From VGKC to LGI1 and Caspr2 encephalitis: the evolution of a disease entity over time. Autoimmun Rev. 2016;15(10):970-4. https://doi.org/10.1016/j.autrev.2016.07.018
https://doi.org/10.1016/j.autrev.2016.07...
. Each of these antibodies presents with specific clinical symptoms.
The LGI1 is a secreted synaptic protein that interacts with transmembrane proteins ADAM22 and ADAM23 to form a trans-synaptic complex involving potassium channels and AMPAR. Genetic disruption of LGI1 protein in humans causes autosomal dominant lateral temporal lobe epilepsy2626. Lancaster E, Huijbers MG, Bar V, Boronat A, Wong A, Martinez-Hernandez E et al. Investigations of caspr2, an autoantigen of encephalitis and neuromyotonia. Ann Neurol. 2011;69(2):303-11. https://doi.org/10.1002/ana.22297
https://doi.org/10.1002/ana.22297...
. The clinical spectrum of anti-LGI1 encephalitis usually comprises limbic encephalitis, hyponatremia and seizures. Half of the patients develop faciobrachial dystonic seizures, which are characterized by brief unilateral contractions of the arm (often evolving into the ipsilateral face or leg) that are shorter than three seconds and occur several times a day. Two-thirds of patients present with brain MRI hyperintensities in the medial temporal lobe (Figure 2). Paraneoplastic anti-LGI1 encephalitis is uncommon; however, patients should be screened for lung cancer2727. Sonderen A, Thijs RD, Coenders EC, Jiskoot LC, Sanchez E, Bruijn MA et al. Anti-LGI1 encephalitis: clinical syndrome and long-term follow-up. Neurology. 2016;87(14):1449-56. https://doi.org/10.1212/WNL.0000000000003173
https://doi.org/10.1212/WNL.000000000000...
. Relapses may occur in up to 20% of patients2828. Poliak S, Salomon D, Elhanany H, Sabanay H, Kiernan B, Pevny L et al. Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J Cell Biol. 2003;162(6):1149-60. https://doi.org/10.1083/jcb.200305018
https://doi.org/10.1083/jcb.200305018...
.
The CASPR-2 is a juxtaparanodal adhesion molecule that interacts with contactin 2 and the cytoskeleton, and is involved in clustering of potassium channels in myelinated axons2828. Poliak S, Salomon D, Elhanany H, Sabanay H, Kiernan B, Pevny L et al. Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J Cell Biol. 2003;162(6):1149-60. https://doi.org/10.1083/jcb.200305018
https://doi.org/10.1083/jcb.200305018...
. Anti-CASPR2 antibodies are associated with peripheral nerve hyperexcitability (myokymia, fasciculations, cramps) and encephalitis. Other symptoms include dysautonomia and insomnia (agrypnia excitata). Nearly one-third of patients develop Morvan’s syndrome, a complex disorder affecting the peripheral and central nervous system that is characterized by distal movement disorders of the upper limbs, peripheral nerve hyperexcitability, dysautonomia, pain, and encephalitis2424. Irani SR, Pettingill P, Kleopa KA, Schiza N, Waters P, Mazia C et al. Morvan syndrome: clinical and serological observations in 29 cases. Ann Neurol. 2012;72(2):241-55. https://doi.org/10.1002/ana.23577
https://doi.org/10.1002/ana.23577...
. Most individuals affected are male and one-third of them present with paraneoplastic manifestations, usually associated with thymoma, lung cancer or endometrial carcinoma2727. Sonderen A, Thijs RD, Coenders EC, Jiskoot LC, Sanchez E, Bruijn MA et al. Anti-LGI1 encephalitis: clinical syndrome and long-term follow-up. Neurology. 2016;87(14):1449-56. https://doi.org/10.1212/WNL.0000000000003173
https://doi.org/10.1212/WNL.000000000000...
,2626. Lancaster E, Huijbers MG, Bar V, Boronat A, Wong A, Martinez-Hernandez E et al. Investigations of caspr2, an autoantigen of encephalitis and neuromyotonia. Ann Neurol. 2011;69(2):303-11. https://doi.org/10.1002/ana.22297
https://doi.org/10.1002/ana.22297...
.
The clinical relevance of anti-VGKC antibodies against unknown target antigens is unclear. About half of patients with anti-VGKC encephalitis do not present antibodies against LGI1 or CASPR-2. These specific groups of patients develop a wide variety of clinical syndromes raising the question whether anti-VGKC antibodies are truly a marker of disease in these patients2525. van Sonderen A, Schreurs MWJ, Wirtz PW, Sillevis Smitt PAE, Titulaer MJ. From VGKC to LGI1 and Caspr2 encephalitis: the evolution of a disease entity over time. Autoimmun Rev. 2016;15(10):970-4. https://doi.org/10.1016/j.autrev.2016.07.018
https://doi.org/10.1016/j.autrev.2016.07...
. Anti-VGKC antibodies have been described, for example, in patients with Creutzfeldt-Jakob disease, suggesting that these antibodies might not be pathogenic. Anti-VGKC titers with negative anti-LGI1 and anti-CASPR-2 antibodies are usually low (< 0.3 pM)—although there is no clear cutoff value to differentiate between patients with and without AIE—thus it is not recommended to use them as evidence of immune-mediated pathogenesis2222. Sonderen A, Schreurs MW, Bruijn MA, Boukhrissi S, Nagtzaam MM, Hulsenboom ES et al. The relevance of VGKC positivity in the absence of LGI1 and Caspr2 antibodies. Neurology. 2016;86(18):1692-9. https://doi.org/10.1212/WNL.0000000000002637
https://doi.org/10.1212/WNL.000000000000...
,2929. Sonderen A, Petit-Pedrol M, Dalmau J, Titulaer MJ. The value of LGI1, Caspr2 and voltage-gated potassium channel antibodies in encephalitis. Nat Rev Neurol. 2017;13(5):290-301. https://doi.org/10.1038/nrneurol.2017.43
https://doi.org/10.1038/nrneurol.2017.43...
.
Anti-GAD encephalitis
Glutamic acid decarboxylase (GAD) is an enzyme that catalyzes the conversion of glutamic acid to the neurotransmitter GABA. Anti-GAD antibodies have been associated with other autoimmune disorders such as insulin-dependent diabetes mellitus. The main neurological syndromes associated with anti-GAD antibodies include stiff-person syndrome, cerebellar ataxia, epilepsy and limbic encephalitis3030. Vale TC, Pedroso JL, Alquéres RA, Dutra LA, Barsottini OGP. Spontaneous downbeat nystagmus as a clue for the diagnosis of ataxia associated with anti-GAD antibodies. J Neurol Sci. 2015;359(1-2):21-3. https://doi.org/10.1016/j.jns.2015.10.024
https://doi.org/10.1016/j.jns.2015.10.02...
.
Stiff-person syndrome is a neurological disorder frequently associated with anti-GAD antibodies characterized by muscle stiffness resulting from co-contractions of agonist and antagonist muscles and painful spasms3131. Alexopoulos H, Akrivou S, Dalakas MC. Glycine receptor antibodies in stiff-person syndrome and other GAD-positive CNS disorders. Neurology. 2013;81(22):1962-4. https://doi.org/10.1212/01.wnl.0000436617.40779.65
https://doi.org/10.1212/01.wnl.000043661...
. Another classic finding in this syndrome is the patient’s pronounced startle responses.
Ataxia associated with anti-GAD antibodies is usually slowly progressive and evolves over months or years. Abnormal ocular movements with spontaneous downbeat nystagmus have also been described3030. Vale TC, Pedroso JL, Alquéres RA, Dutra LA, Barsottini OGP. Spontaneous downbeat nystagmus as a clue for the diagnosis of ataxia associated with anti-GAD antibodies. J Neurol Sci. 2015;359(1-2):21-3. https://doi.org/10.1016/j.jns.2015.10.024
https://doi.org/10.1016/j.jns.2015.10.02...
. Nearly 7% of patients with anti-GAD antibodies present with temporal lobe epilepsy or status epilepticus, and 5% develop limbic encephalitis3232. Khawaja AM, Vines BL, Miller DW, Szaflarski JP, Amara AW. Refractory status epilepticus and glutamic acid decarboxylase antibodies in adults: presentation, treatment and outcomes. Epileptic Disord. 2016;18(1):34-43. https://doi.org/10.1684/epd.2016.0797
https://doi.org/10.1684/epd.2016.0797...
,3333. Saiz A, Blanco Y, Sabater L, González F, Bataller L, Casamitjana R et al. Spectrum of neurological syndromes associated with glutamic acid decarboxylase antibodies: diagnostic clues for this association. Brain. 2008;131(10):2553-63. https://doi.org/10.1093/brain/awn183
https://doi.org/10.1093/brain/awn183...
.
Anti-GAD antibodies are rarely of paraneoplastic origin. Patients with stiff-person syndrome, cerebellar ataxia or other neurological syndromes typically associated with anti-GAD antibodies do not need to be aggressively or repeatedly screened for cancer. However, the presence of anti-GAD antibodies in patients with limbic encephalitis or other classic paraneoplastic syndromes (paraneoplastic cerebellar degeneration, opsoclonus-myoclonus syndrome or paraneoplastic encephalomyelitis) is associated with a 10-fold increase in the risk of cancer and tumor screening is thus recommended3434. Ariño H, Höftberger R, Gresa-Arribas N, Martínez-Hernández E, Armangue T, Kruer MC et al. Paraneoplastic Neurological Syndromes and Glutamic Acid Decarboxylase Antibodies. JAMA Neurol. 2015;72(8):1-8. https://doi.org/10.1001/jamaneurol.2015.0749
https://doi.org/10.1001/jamaneurol.2015....
.
Anti-GlyR encephalitis
Glycine receptors (GlyR) are chloride channels that facilitate inhibitory neurotransmission in the brain and spinal cord3535. Hutchinson M, Waters P, McHugh J, Gorman G, O’Riordan S, Connolly S et al. Progressive encephalomyelitis, rigidity, and myoclonus: a novel glycine receptor antibody. Neurology. 2008;71(16):1291-2. https://doi.org/10.1212/01.wnl.0000327606.50322.f0
https://doi.org/10.1212/01.wnl.000032760...
. Anti-GlyR antibodies were first described in patients with progressive encephalomyelitis with rigidity and myoclonus and later in patients with stiff-person syndrome3131. Alexopoulos H, Akrivou S, Dalakas MC. Glycine receptor antibodies in stiff-person syndrome and other GAD-positive CNS disorders. Neurology. 2013;81(22):1962-4. https://doi.org/10.1212/01.wnl.0000436617.40779.65
https://doi.org/10.1212/01.wnl.000043661...
,3535. Hutchinson M, Waters P, McHugh J, Gorman G, O’Riordan S, Connolly S et al. Progressive encephalomyelitis, rigidity, and myoclonus: a novel glycine receptor antibody. Neurology. 2008;71(16):1291-2. https://doi.org/10.1212/01.wnl.0000327606.50322.f0
https://doi.org/10.1212/01.wnl.000032760...
,3636. McKeon A, Martinez-Hernandez E, Lancaster E, Matsumoto JY, Harvey RJ, McEvoy KM et al. Glycine receptor autoimmune spectrum with stiff-man syndrome phenotype. JAMA Neurol. 2013;70(1):44-50. https://doi.org/10.1001/jamaneurol.2013.574
https://doi.org/10.1001/jamaneurol.2013....
. Recently, anti-GlyR antibodies have also been reported in patients with cerebellar ataxia and anti-GAD antibodies and patients with demyelinating diseases including optic neuritis and multiple sclerosis, but their clinical significance remains unclear3737. Ariño H, Gresa-Arribas N, Blanco Y, Martínez-Hernández E, Sabater L, Petit-Pedrol M et al. Cerebellar ataxia and glutamic acid decarboxylase antibodies. JAMA Neurol. 2014;71(8):1009-16. https://doi.org/10.1001/jamaneurol.2014.1011
https://doi.org/10.1001/jamaneurol.2014....
,3838. Martinez-Hernandez E, Sepulveda M, Rostásy K, Höftberger R, Graus F, Harvey RJ et al. Antibodies to aquaporin 4, myelin-oligodendrocyte glycoprotein, and the glycine receptor α1 subunit in patients with isolated optic neuritis. JAMA Neurol. 2015;72(2):187-93. https://doi.org/10.1001/jamaneurol.2014.3602
https://doi.org/10.1001/jamaneurol.2014....
. Anti-GlyR antibodies are usually not associated with tumors, although there have been reports of patients with underlying thymoma, small-cell lung cancer, breast cancer and chronic lymphocytic leukemia.
Anti-DPPX encephalitis
Dipeptidyl peptidase-like protein 6 (DPPX) is a subunit of Kv4.2 potassium channels expressed in the hippocampus, cerebellum, striatum, and myenteric plexus. Patients with anti-DPPX antibodies show neuropsychiatric symptoms (agitation and confusion), myoclonus, tremor, startle reflex, seizures, stiff-person syndrome and prodromal diarrhea of unknown etiology. In addition, they may have symptoms of dysautonomia including arrhythmias, thermodysregulation, diaphoresis, urinary symptoms and sleep disorders3939. Piepgras J, Höltje M, Michel K, Li Q, Otto C, Drenckhahn C et al. Anti-DPPX encephalitis: pathogenic effects of antibodies on gut and brain neurons. Neurology. 2015;85(10):890-7. https://doi.org/10.1212/WNL.0000000000001907
https://doi.org/10.1212/WNL.000000000000...
,4040. Boronat A, Gelfand JM, Gresa-Arribas N, Jeong HY, Walsh M, Roberts K et al. Encephalitis and antibodies to dipeptidyl-peptidase-like protein-6, a subunit of Kv4.2 potassium channels. Ann Neurol. 2013;73(1):120-8. https://doi.org/10.1002/ana.23756
https://doi.org/10.1002/ana.23756...
.
The CSF analysis usually shows pleocytosis and increased protein concentrations. Functional tests in the serum of a DPPX-positive patient demonstrated increased excitability of enteric neurons and downregulation of DPPX and Kv4.2 from hippocampal neuron membranes, which is suggestive of the pathogenic effects of anti-DPPX antibodies in anti-DPPX encephalitis3939. Piepgras J, Höltje M, Michel K, Li Q, Otto C, Drenckhahn C et al. Anti-DPPX encephalitis: pathogenic effects of antibodies on gut and brain neurons. Neurology. 2015;85(10):890-7. https://doi.org/10.1212/WNL.0000000000001907
https://doi.org/10.1212/WNL.000000000000...
.
Encephalopathy associated with anti-IgLON5 antibodies
The IgLON family member 5 (IgLON5) is a neuronal cell adhesion molecule of the immunoglobulin superfamily. Patients with anti-IgLON5 antibodies present with a unique non-REM (rapid eye movement) and REM parasomnia with obstructive sleep apnea, stridor, episodic central hypoventilation, dementia, gait instability, chorea, dysarthria, dysphagia, dysautonomia and supranuclear gaze palsy resembling that seen in classic tauopathy55. Sabater L, Gaig C, Gelpi E, Bataller L, Lewerenz J, Torres-Vega E et al. A novel non-rapid-eye movement and rapid-eye-movement parasomnia with sleep breathing disorder associated with antibodies to IgLON5: a case series, characterisation of the antigen, and post-mortem study. Lancet Neurol. 2014;13(6):575-86. https://doi.org/10.1016/S1474-4422(14)70051-1
https://doi.org/10.1016/S1474-4422(14)70...
,4040. Boronat A, Gelfand JM, Gresa-Arribas N, Jeong HY, Walsh M, Roberts K et al. Encephalitis and antibodies to dipeptidyl-peptidase-like protein-6, a subunit of Kv4.2 potassium channels. Ann Neurol. 2013;73(1):120-8. https://doi.org/10.1002/ana.23756
https://doi.org/10.1002/ana.23756...
. All published cases reported the presence of the alleles HLA-DQB1*0501 and HLA-DRB1*1001 suggesting genetic susceptibility to this disease. Neuropathological postmortem studies have shown a novel tauopathy with extensive neuronal deposits of hyperphosphorylated tau mainly involving the tegmentum of the brainstem and hypothalamus4141. Gelpi E, Höftberger R, Graus F, Ling H, Holton JL, Dawson T et al. Neuropathological criteria of anti-IgLON5-related tauopathy. Acta Neuropathol. 2016;132(4):531-43. https://doi.org/10.1007/s00401-016-1591-8
https://doi.org/10.1007/s00401-016-1591-...
. This novel encephalopathy provides an intriguing link between neurodegeneration and cell-surface autoimmunity. A recent study has shown that anti-IgLON5 antibodies recognize Ig-like domain 2 as an immunogenic region and causes irreversible internalization of IgLON5 from the neuronal membrane. These findings support a potential pathogenic role of anti-IgLON5 antibodies in the associated encephalopathy4242. Gaig C, Graus F, Compta Y, Högl B, Bataller L, Brüggemann N et al. Clinical manifestations of the anti-IgLON5 disease. Neurology. 2017;88(18):1736-43. https://doi.org/10.1212/WNL.0000000000003887
https://doi.org/10.1212/WNL.000000000000...
.
Anti-mGluR1 and anti-mGluR5 encephalitis
Metabotropic glutamate receptor 1 (mGluR1) and metabotropic glutamate receptor 5 (mGluR5) are both G-protein-coupled receptors that share an 85% amino acid sequence homology. Both receptors are involved in modulating synaptic functions including long-term depression. While mGluR1 facilitates long-term depression at parallel fiber Purkinje cell synapses, which are critical for cerebellar motor learning, mGluR5 is more relevant for long-term depression in the hippocampus.
All patients with anti-mGluR1 antibodies develop cerebellar ataxia of subacute onset, and some may present with additional symptoms such as paranoia, dysgeusia, diplopia and cognitive deficits. Common tumors found to be associated with anti-mGluR1 antibodies are hematologic malignancies and prostate adenocarcinoma4343. Lopez-Chiriboga AS, Komorowski L, Kümpfel T, Probst C, Hinson SR, Pittock SJ et al. Metabotropic glutamate receptor type 1 autoimmunity. Neurology. 2016;86(11):1009-13. https://doi.org/10.1212/WNL.0000000000002476
https://doi.org/10.1212/WNL.000000000000...
. Response to immunotherapy is variable.
Patients with anti-mGluR5-abs present with a form of encephalitis named “Ophelia syndrome”, a clinical syndrome that includes memory loss and psychosis in association with Hodgkin’s lymphoma4444. Lancaster E, Martinez-Hernandez E, Titulaer MJ, Boulos M, Weaver S, Antoine JC et al. Antibodies to metabotropic glutamate receptor 5 in the Ophelia syndrome. Neurology. 2011;77(18):1698-701. https://doi.org/10.1212/WNL.0b013e3182364a44
https://doi.org/10.1212/WNL.0b013e318236...
. The outcome of reported cases is generally good after treatment of the lymphoma and immunotherapy4444. Lancaster E, Martinez-Hernandez E, Titulaer MJ, Boulos M, Weaver S, Antoine JC et al. Antibodies to metabotropic glutamate receptor 5 in the Ophelia syndrome. Neurology. 2011;77(18):1698-701. https://doi.org/10.1212/WNL.0b013e3182364a44
https://doi.org/10.1212/WNL.0b013e318236...
.
Considerations on specific diagnostic methods for AIE
Four different techniques are used to detect antibodies against cell surface antigens: cell-based assay (CBA) with HEK293 cells; tissue-based assay (TBA) on brain tissue of rodents using indirect immunohistochemistry or indirect immunofluorescence, and culture of dissociated hippocampal neurons from rats.
Cell-based assays are highly sensitive and robust signals are diagnostic of specific antigens4545. Gresa-Arribas N, Titulaer MJ, Torrents A, Aguilar E, McCracken L, Leypoldt F et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol. 2014;13(2):167-77. https://doi.org/10.1016/S1474-4422(13)70282-5
https://doi.org/10.1016/S1474-4422(13)70...
. The TBA provides an excellent screening approach as it can detect most currently-known antibodies and reveal new antibodies. Staining of live neuronal cell cultures are performed mainly in research laboratories and can be useful when TBAs and CBAs show conflicting results. It confirms that antibodies directed against neuronal surface antigens are present (Figure 3).
Techniques for the detection of anti-neuronal antibodies. Indirect immunohistochemistry on rat brain tissue with a serum of a patient with anti-NMDAR encephalitis shows strong labeling of hippocampal neurons (A) whereas a serum of a healthy individual is negative (B). In the cell-based assay, antibodies are identified on HEK293 cells transfected with related antigens. (C) red: CSF from a patient with AMPAR antibodies, green: commercial antibody against the GluA1/2 subunits of AMPARs). (D) Live hippocampal neuron culture; strong dot-like labeling of the neuronal membrane indicates antibodies against cell surface antigens.
Testing for surface receptor antibodies should always be performed in both serum and CSF of the patients. This has several reasons: (1) in some specific syndromes (e.g. anti-NMDAR encephalitis) anti-neuronal antibodies may be found only in CSF (in a recent study 13% of anti-NMDAR encephalitis patients did not have detectable anti-NMDAR antibodies in serum4646. Dahm L, Ott C, Steiner J, Stepniak B, Teegen B, Saschenbrecker S et al. Seroprevalence of autoantibodies against brain antigens in health and disease. Ann Neurol. 2014;76(1):82-94. https://doi.org/10.1002/ana.24189
https://doi.org/10.1002/ana.24189...
) while other antibodies (e.g. LGI1) may, in rare instances, be detectable only in serum. (2) Some patients may have a different antibody spectrum in serum and CSF, for example, some patients may have anti-NMDAR encephalitis in serum and CSF and, in addition, GABA-AR antibodies only in serum. In these cases, the antibody present in the CSF determines the clinical phenotype. (3) Antibody-titers in CSF correlate better with the clinical presentation than serum titers, and (4) serum testing harbors the risk of background reactivity that gives rise to false positive results. Recently, serum testing with CBA identified anti-NMDAR antibodies mainly of the IgA and IgM subclass in schizophrenia, Creutzfeldt-Jakob disease, depression, Parkinson’s disease, and healthy individuals4646. Dahm L, Ott C, Steiner J, Stepniak B, Teegen B, Saschenbrecker S et al. Seroprevalence of autoantibodies against brain antigens in health and disease. Ann Neurol. 2014;76(1):82-94. https://doi.org/10.1002/ana.24189
https://doi.org/10.1002/ana.24189...
. However, presence of these antibodies could not be shown in CSF and, therefore, the clinical significance of serum IgA and IgM anti-NMDAR antibodies remains to be clarified.
There are diagnostic challenges to testing for AIE in Brazil as commercial CBA and TBA kits are not approved for use by national regulatory authorities. Partner or research laboratories overseas perform most testing and some of these laboratories perform CBAs only. We currently recommend that samples from patients with clinical manifestations suggestive of AIE who tested negative by CBA should be referred to research laboratories that use other assessment methods.
Negative test results do not rule out immune-mediated disorders and nonspecific background signals may cause false positive test results. Steroid use may also interfere with the diagnostic test. Hence, test results should be interpreted with caution and put into the context of the clinical presentation. Because antibodies may remain positive despite clinical features, clinicians should focus on patient treatment rather than antibody titers.
Differential diagnosis
Differential diagnosis in anti-neuronal AIE includes Hashimoto’s encephalopathy and other steroid-responsive encephalopathies, acute disseminated encephalomyelitis, neuromyelitis optica spectrum disorders, central nervous system vasculitis, neuropsychiatric lupus, angiocentric lymphoma, Rasmussen’s encephalitis and febrile infection-related epilepsy syndrome. It is important to rule out HIV infection, syphilis, Creutzfeldt-Jakob disease, as well as human herpes virus-6-associated encephalitis in immunocompromised patients11. Granerod J, Ambrose HE, Davies NW, Clewley JP, Walsh AL, Morgan D et al. Causes of encephalitis and differences in their clinical presentations in England: A multicentre, population-based prospective study. Lancet Infect Dis. 2010;10(12):835-44. https://doi.org/10.1016/S1473-3099(10)70222-X
https://doi.org/10.1016/S1473-3099(10)70...
,33. Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016;15(4):391-404. https://doi.org/10.1016/S1474-4422(15)00401-9
https://doi.org/10.1016/S1474-4422(15)00...
,88. Leypoldt F, Wandinger K-P, Bien CG, Dalmau J. Autoimmune Encephalitis. Eur Neurol Rev. 2013;8(1):31-7. https://doi.org/10.17925/ENR.2013.08.01.31
https://doi.org/10.17925/ENR.2013.08.01....
,3333. Saiz A, Blanco Y, Sabater L, González F, Bataller L, Casamitjana R et al. Spectrum of neurological syndromes associated with glutamic acid decarboxylase antibodies: diagnostic clues for this association. Brain. 2008;131(10):2553-63. https://doi.org/10.1093/brain/awn183
https://doi.org/10.1093/brain/awn183...
.
Viral infections are known triggers for AIE33. Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016;15(4):391-404. https://doi.org/10.1016/S1474-4422(15)00401-9
https://doi.org/10.1016/S1474-4422(15)00...
,44. Linnoila JJ, Binnicker MJ, Majed M, Klein CJ, McKeon A. CSF herpes virus and autoantibody profiles in the evaluation of encephalitis. Neurol - Neuroimmunol Neuroinflammation. 2016;3(4):e245. https://doi.org/10.1212/NXI.0000000000000245
https://doi.org/10.1212/NXI.000000000000...
. It is believed that virus-mediated cerebral tissue damage may lead to antigen exposition that triggers the development of anti-neuronal antibodies. Herpes viruses may trigger autoimmunity to NMDAR, dopamine D2 receptor, GABA-AR and other synaptic proteins that have are not yet been characterized.
Tumor investigation
All patients with AIE should be screened for tumors at disease onset. The nature of the antibody, and to a lesser extent the clinical syndrome, determines the risk and type of an underlying malignancy4747. Titulaer MJ, Soffietti R, Dalmau J, Gilhus NE, Giometto B, Graus F et al. Screening for tumours in paraneoplastic syndromes: report of an EFNS Task Force. Eur J Neurol. 2011;18(1):19-e3. https://doi.org/10.1111/j.1468-1331.2010.03220.x
https://doi.org/10.1111/j.1468-1331.2010...
. Due to the low frequency of tumor association in patients with anti-LGI1 and anti-GAD antibodies, patient screening should be considered at disease onset, with no need for periodic screenings88. Leypoldt F, Wandinger K-P, Bien CG, Dalmau J. Autoimmune Encephalitis. Eur Neurol Rev. 2013;8(1):31-7. https://doi.org/10.17925/ENR.2013.08.01.31
https://doi.org/10.17925/ENR.2013.08.01....
. Tumor treatment is essential for neurological improvement2525. van Sonderen A, Schreurs MWJ, Wirtz PW, Sillevis Smitt PAE, Titulaer MJ. From VGKC to LGI1 and Caspr2 encephalitis: the evolution of a disease entity over time. Autoimmun Rev. 2016;15(10):970-4. https://doi.org/10.1016/j.autrev.2016.07.018
https://doi.org/10.1016/j.autrev.2016.07...
.
If initial tumor screening is negative but the patient has antibodies that are typically paraneoplastic (e.g. anti-NMDAR in young adult women, anti-CASPR2, anti-AMPAR and anti-GABA-BR), screening should be repeated after three to six months, followed by screenings every six months for four years4848. Dalmau J, Rosenfeld MR. Paraneoplastic syndromes of the CNS. Lancet Neurol. 2008;7(4):327-40. https://doi.org/10.1016/S1474-4422(08)70060-7
https://doi.org/10.1016/S1474-4422(08)70...
.
Computed tomography (CT) of the chest should be performed; if negative, fluorodeoxyglucose positron emission tomography (FDG-PET) is indicated as it increases cancer detection by 20%4949. McKeon A, Apiwattanakul M, Lachance DH, Lennon VA, Mandrekar JN, Boeve BF. Positron emission tomography-computed tomography in paraneoplastic neurologic disorders. Arch Neurol. 2010;67(3):322. https://doi.org/10.1001/archneurol.2009.336
https://doi.org/10.1001/archneurol.2009....
. Mammograms followed by MRI are used for breast cancer screening. For the pelvic region and testes, ultrasound is the investigation of first choice followed by pelvic CT4747. Titulaer MJ, Soffietti R, Dalmau J, Gilhus NE, Giometto B, Graus F et al. Screening for tumours in paraneoplastic syndromes: report of an EFNS Task Force. Eur J Neurol. 2011;18(1):19-e3. https://doi.org/10.1111/j.1468-1331.2010.03220.x
https://doi.org/10.1111/j.1468-1331.2010...
. It is important to note that ovarian teratomas are not seen in FDG-PET scans.
Treatment and prognosis
Various treatment approaches including corticosteroids, intravenous immunoglobulin, plasma exchange, rituximab and cyclophosphamide are currently used1313. Titulaer MJ, McCracken L, Gabilondo I, Armangué T, Glaser C, Iizuka T et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. 2013; 12(2):157-65. https://doi.org/10.1016/S1474-4422(12)70310-1
https://doi.org/10.1016/S1474-4422(12)70...
. Data on treatment response and prognosis are mostly available for anti-NMDAR encephalitis5050. Vincent A, Bien CG, Irani SR, Waters P. Autoantibodies associated with diseases of the CNS: new developments and future challenges. Lancet Neurol. 2011;10(8):759-72. https://doi.org/10.1016/S1474-4422(11)70096-5
https://doi.org/10.1016/S1474-4422(11)70...
.
Evidence suggests that early immunotherapy improves outcome33. Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016;15(4):391-404. https://doi.org/10.1016/S1474-4422(15)00401-9
https://doi.org/10.1016/S1474-4422(15)00...
, thus treatment for AIE should not be delayed. Patients should receive either methylprednisolone 1 g IV for 3–5 days and intravenous immunoglobulin (0.4 g/kg/day for five days) or methylprednisolone and plasmapheresis. We favor the use of plasmapheresis in patients with refractory seizures and severe dysautonomia, although there is no compelling evidence of superiority of any approach. If an associated tumor is detected, oncologic management (chemotherapy or tumor resection) is important for improvement.
Autoimmune encephalitis patients who fail to improve after 10–14 days should receive second-line therapies such as rituximab or cyclophosphamide, or both33. Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016;15(4):391-404. https://doi.org/10.1016/S1474-4422(15)00401-9
https://doi.org/10.1016/S1474-4422(15)00...
,1313. Titulaer MJ, McCracken L, Gabilondo I, Armangué T, Glaser C, Iizuka T et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. 2013; 12(2):157-65. https://doi.org/10.1016/S1474-4422(12)70310-1
https://doi.org/10.1016/S1474-4422(12)70...
. Specialized centers usually prescribe rituximab (375 mg/m22. Gable MS, Sheriff H, Dalmau J, Tilley DH, Glaser CA. The frequency of autoimmune N-methyl-D-aspartate receptor encephalitis surpasses that of individual viral etiologies in young individuals enrolled in the California Encephalitis Project. Clin Infect Dis. 2012;54(7):899-904. https://doi.org/10.1093/cid/cir1038
https://doi.org/10.1093/cid/cir1038...
) weekly for four weeks and cyclophosphamide (750 mg/m22. Gable MS, Sheriff H, Dalmau J, Tilley DH, Glaser CA. The frequency of autoimmune N-methyl-D-aspartate receptor encephalitis surpasses that of individual viral etiologies in young individuals enrolled in the California Encephalitis Project. Clin Infect Dis. 2012;54(7):899-904. https://doi.org/10.1093/cid/cir1038
https://doi.org/10.1093/cid/cir1038...
) for six months in patients older than 16 years. Younger patients should receive rituximab alone33. Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016;15(4):391-404. https://doi.org/10.1016/S1474-4422(15)00401-9
https://doi.org/10.1016/S1474-4422(15)00...
.
Better prognosis has been associated with early treatment, no requirement for intensive care admission and non-paraneoplastic AIE2020. Höftberger R, Titulaer MJ, Sabater L, Dome B, Rózsás A, Hegedus B et al. Encephalitis and GABAB receptor antibodies: novel findings in a new case series of 20 patients. Neurology. 2013;81(17):1500-6. https://doi.org/10.1212/WNL.0b013e3182a9585f
https://doi.org/10.1212/WNL.0b013e3182a9...
. The treatment response and relapse rate vary among patients with AIE. Half of the patients with anti-NMDAR encephalitis fail initial immunotherapy and may require second-line treatment options, with relapses occurring in 12%. Relapses may occur in 31% of patients with anti-LGI1 encephalitis and 10% of those with anti-CASPR2 encephalitis, sometimes years after the first episode. About 33% of the patients with anti-LGI1 encephalitis are left disabled, mostly due to memory problems.
FINAL REMARKS
Autoimmune encephalitis may present with a wide variety of symptoms of either acute or subacute onset. The diagnosis should not rely solely on antibody testing as patients with AIE may be seronegative. Clinicians must be aware of the different laboratory assessment methods available as well as proper interpretation of results. Clinical presentation and physical examination are of extreme importance in AIE. Patients benefit from early aggressive treatment and relapses may occur.
References
-
1Granerod J, Ambrose HE, Davies NW, Clewley JP, Walsh AL, Morgan D et al. Causes of encephalitis and differences in their clinical presentations in England: A multicentre, population-based prospective study. Lancet Infect Dis. 2010;10(12):835-44. https://doi.org/10.1016/S1473-3099(10)70222-X
» https://doi.org/10.1016/S1473-3099(10)70222-X -
2Gable MS, Sheriff H, Dalmau J, Tilley DH, Glaser CA. The frequency of autoimmune N-methyl-D-aspartate receptor encephalitis surpasses that of individual viral etiologies in young individuals enrolled in the California Encephalitis Project. Clin Infect Dis. 2012;54(7):899-904. https://doi.org/10.1093/cid/cir1038
» https://doi.org/10.1093/cid/cir1038 -
3Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016;15(4):391-404. https://doi.org/10.1016/S1474-4422(15)00401-9
» https://doi.org/10.1016/S1474-4422(15)00401-9 -
4Linnoila JJ, Binnicker MJ, Majed M, Klein CJ, McKeon A. CSF herpes virus and autoantibody profiles in the evaluation of encephalitis. Neurol - Neuroimmunol Neuroinflammation. 2016;3(4):e245. https://doi.org/10.1212/NXI.0000000000000245
» https://doi.org/10.1212/NXI.0000000000000245 -
5Sabater L, Gaig C, Gelpi E, Bataller L, Lewerenz J, Torres-Vega E et al. A novel non-rapid-eye movement and rapid-eye-movement parasomnia with sleep breathing disorder associated with antibodies to IgLON5: a case series, characterisation of the antigen, and post-mortem study. Lancet Neurol. 2014;13(6):575-86. https://doi.org/10.1016/S1474-4422(14)70051-1
» https://doi.org/10.1016/S1474-4422(14)70051-1 -
6Lancaster E, Dalmau J. Neuronal autoantigens: pathogenesis, associated disorders and antibody testing. Nat Rev Neurol. 2012;8(7):380-90. https://doi.org/10.1038/nrneurol.2012.99
» https://doi.org/10.1038/nrneurol.2012.99 -
7Coevorden-Hameete MH, de Graaff E, Titulaer MJ, Hoogenraad CC, Sillevis Smitt PE. Molecular and cellular mechanisms underlying anti-neuronal antibody mediated disorders of the central nervous system. Autoimmun Rev. 2014;13(3):299-312. https://doi.org/10.1016/j.autrev.2013.10.016
» https://doi.org/10.1016/j.autrev.2013.10.016 -
8Leypoldt F, Wandinger K-P, Bien CG, Dalmau J. Autoimmune Encephalitis. Eur Neurol Rev. 2013;8(1):31-7. https://doi.org/10.17925/ENR.2013.08.01.31
» https://doi.org/10.17925/ENR.2013.08.01.31 -
9Davis R, Dalmau J. Autoimmunity, seizures, and status epilepticus. Epilepsia. 2013;54(6 Suppl 6):46-9. https://doi.org/10.1111/epi.12276
» https://doi.org/10.1111/epi.12276 -
10Tobin WO, Lennon VA, Komorowski L, Probst C, Clardy SL, Aksamit AJ et al. DPPX potassium channel antibody: frequency, clinical accompaniments, and outcomes in 20 patients. Neurology. 2014; 83(20):1797-803. https://doi.org/10.1212/WNL.0000000000000991
» https://doi.org/10.1212/WNL.0000000000000991 -
11Dalmau J, Tüzün E, Wu HY, Masjuan J, Rossi JE, Voloschin A et al. Paraneoplastic anti- N -methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61(1):25-36. https://doi.org/10.1002/ana.21050
» https://doi.org/10.1002/ana.21050 -
12Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol. 2011;10(1):63-74. https://doi.org/10.1016/S1474-4422(10)70253-2PMID:21163445
» https://doi.org/10.1016/S1474-4422(10)70253-2PMID:21163445 -
13Titulaer MJ, McCracken L, Gabilondo I, Armangué T, Glaser C, Iizuka T et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. 2013; 12(2):157-65. https://doi.org/10.1016/S1474-4422(12)70310-1
» https://doi.org/10.1016/S1474-4422(12)70310-1 -
14Schmitt SE, Pargeon K, Frechette ES, Hirsch LJ. Extreme delta brush: a unique EEG pattern in adults with anti-NMDA receptor encephalitis. Neurology. 2012;79:1094-100. https://doi.org/10.1212/WNL.0b013e3182698cd8
» https://doi.org/10.1212/WNL.0b013e3182698cd8 -
15Höftberger R, Sonderen A, Leypoldt F, Houghton D, Geschwind M, Gelfand J et al. Encephalitis and AMPA receptor antibodies: novel findings in a case series of 22 patients. Neurology. 2015;84(24):2403-12. https://doi.org/10.1212/WNL.0000000000001682
» https://doi.org/10.1212/WNL.0000000000001682 -
16Petit-Pedrol M, Armangue T, Peng X, Bataller L, Cellucci T, Davis R et al. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: a case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol. 2014;13(3):276-86. https://doi.org/10.1016/S1474-4422(13)70299-0
» https://doi.org/10.1016/S1474-4422(13)70299-0 -
17Spatola M, Petit-Pedrol M, Simabukuro MM, Armangue T, Castro FJ, Barcelo Artigues MI et al. Investigations in GABAA receptor antibody-associated encephalitis. Neurology. 2017;88 (11):1012-20. https://doi.org/10.1212/WNL.0000000000003713
» https://doi.org/10.1212/WNL.0000000000003713 -
18Ohkawa T, Satake S, Yokoi N, Miyazaki Y, Ohshita T, Sobue G et al. Identification and characterization of GABA(A) receptor autoantibodies in autoimmune encephalitis. J Neurosci. 2014;34(24):8151-63. https://doi.org/10.1523/JNEUROSCI.4415-13.2014
» https://doi.org/10.1523/JNEUROSCI.4415-13.2014 -
19Lancaster E, Lai M, Peng X, Hughes E, Constantinescu R, Raizer J et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol. 2010;9(1):67-76. https://doi.org/10.1016/S1474-4422(09)70324-2
» https://doi.org/10.1016/S1474-4422(09)70324-2 -
20Höftberger R, Titulaer MJ, Sabater L, Dome B, Rózsás A, Hegedus B et al. Encephalitis and GABAB receptor antibodies: novel findings in a new case series of 20 patients. Neurology. 2013;81(17):1500-6. https://doi.org/10.1212/WNL.0b013e3182a9585f
» https://doi.org/10.1212/WNL.0b013e3182a9585f -
21Lancaster E, Martinez-Hernandez E, Dalmau J. Encephalitis and antibodies to synaptic and neuronal cell surface proteins. Neurology. 2011;77(2):179-89. https://doi.org/10.1212/WNL.0b013e318224afde
» https://doi.org/10.1212/WNL.0b013e318224afde -
22Sonderen A, Schreurs MW, Bruijn MA, Boukhrissi S, Nagtzaam MM, Hulsenboom ES et al. The relevance of VGKC positivity in the absence of LGI1 and Caspr2 antibodies. Neurology. 2016;86(18):1692-9. https://doi.org/10.1212/WNL.0000000000002637
» https://doi.org/10.1212/WNL.0000000000002637 -
23Lilleker JB, Jones MS, Mohanraj R. VGKC complex antibodies in epilepsy: diagnostic yield and therapeutic implications. Seizure. 2013;22(9):776-9. https://doi.org/10.1016/j.seizure.2013.06.004
» https://doi.org/10.1016/j.seizure.2013.06.004 -
24Irani SR, Pettingill P, Kleopa KA, Schiza N, Waters P, Mazia C et al. Morvan syndrome: clinical and serological observations in 29 cases. Ann Neurol. 2012;72(2):241-55. https://doi.org/10.1002/ana.23577
» https://doi.org/10.1002/ana.23577 -
25van Sonderen A, Schreurs MWJ, Wirtz PW, Sillevis Smitt PAE, Titulaer MJ. From VGKC to LGI1 and Caspr2 encephalitis: the evolution of a disease entity over time. Autoimmun Rev. 2016;15(10):970-4. https://doi.org/10.1016/j.autrev.2016.07.018
» https://doi.org/10.1016/j.autrev.2016.07.018 -
26Lancaster E, Huijbers MG, Bar V, Boronat A, Wong A, Martinez-Hernandez E et al. Investigations of caspr2, an autoantigen of encephalitis and neuromyotonia. Ann Neurol. 2011;69(2):303-11. https://doi.org/10.1002/ana.22297
» https://doi.org/10.1002/ana.22297 -
27Sonderen A, Thijs RD, Coenders EC, Jiskoot LC, Sanchez E, Bruijn MA et al. Anti-LGI1 encephalitis: clinical syndrome and long-term follow-up. Neurology. 2016;87(14):1449-56. https://doi.org/10.1212/WNL.0000000000003173
» https://doi.org/10.1212/WNL.0000000000003173 -
28Poliak S, Salomon D, Elhanany H, Sabanay H, Kiernan B, Pevny L et al. Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J Cell Biol. 2003;162(6):1149-60. https://doi.org/10.1083/jcb.200305018
» https://doi.org/10.1083/jcb.200305018 -
29Sonderen A, Petit-Pedrol M, Dalmau J, Titulaer MJ. The value of LGI1, Caspr2 and voltage-gated potassium channel antibodies in encephalitis. Nat Rev Neurol. 2017;13(5):290-301. https://doi.org/10.1038/nrneurol.2017.43
» https://doi.org/10.1038/nrneurol.2017.43 -
30Vale TC, Pedroso JL, Alquéres RA, Dutra LA, Barsottini OGP. Spontaneous downbeat nystagmus as a clue for the diagnosis of ataxia associated with anti-GAD antibodies. J Neurol Sci. 2015;359(1-2):21-3. https://doi.org/10.1016/j.jns.2015.10.024
» https://doi.org/10.1016/j.jns.2015.10.024 -
31Alexopoulos H, Akrivou S, Dalakas MC. Glycine receptor antibodies in stiff-person syndrome and other GAD-positive CNS disorders. Neurology. 2013;81(22):1962-4. https://doi.org/10.1212/01.wnl.0000436617.40779.65
» https://doi.org/10.1212/01.wnl.0000436617.40779.65 -
32Khawaja AM, Vines BL, Miller DW, Szaflarski JP, Amara AW. Refractory status epilepticus and glutamic acid decarboxylase antibodies in adults: presentation, treatment and outcomes. Epileptic Disord. 2016;18(1):34-43. https://doi.org/10.1684/epd.2016.0797
» https://doi.org/10.1684/epd.2016.0797 -
33Saiz A, Blanco Y, Sabater L, González F, Bataller L, Casamitjana R et al. Spectrum of neurological syndromes associated with glutamic acid decarboxylase antibodies: diagnostic clues for this association. Brain. 2008;131(10):2553-63. https://doi.org/10.1093/brain/awn183
» https://doi.org/10.1093/brain/awn183 -
34Ariño H, Höftberger R, Gresa-Arribas N, Martínez-Hernández E, Armangue T, Kruer MC et al. Paraneoplastic Neurological Syndromes and Glutamic Acid Decarboxylase Antibodies. JAMA Neurol. 2015;72(8):1-8. https://doi.org/10.1001/jamaneurol.2015.0749
» https://doi.org/10.1001/jamaneurol.2015.0749 -
35Hutchinson M, Waters P, McHugh J, Gorman G, O’Riordan S, Connolly S et al. Progressive encephalomyelitis, rigidity, and myoclonus: a novel glycine receptor antibody. Neurology. 2008;71(16):1291-2. https://doi.org/10.1212/01.wnl.0000327606.50322.f0
» https://doi.org/10.1212/01.wnl.0000327606.50322.f0 -
36McKeon A, Martinez-Hernandez E, Lancaster E, Matsumoto JY, Harvey RJ, McEvoy KM et al. Glycine receptor autoimmune spectrum with stiff-man syndrome phenotype. JAMA Neurol. 2013;70(1):44-50. https://doi.org/10.1001/jamaneurol.2013.574
» https://doi.org/10.1001/jamaneurol.2013.574 -
37Ariño H, Gresa-Arribas N, Blanco Y, Martínez-Hernández E, Sabater L, Petit-Pedrol M et al. Cerebellar ataxia and glutamic acid decarboxylase antibodies. JAMA Neurol. 2014;71(8):1009-16. https://doi.org/10.1001/jamaneurol.2014.1011
» https://doi.org/10.1001/jamaneurol.2014.1011 -
38Martinez-Hernandez E, Sepulveda M, Rostásy K, Höftberger R, Graus F, Harvey RJ et al. Antibodies to aquaporin 4, myelin-oligodendrocyte glycoprotein, and the glycine receptor α1 subunit in patients with isolated optic neuritis. JAMA Neurol. 2015;72(2):187-93. https://doi.org/10.1001/jamaneurol.2014.3602
» https://doi.org/10.1001/jamaneurol.2014.3602 -
39Piepgras J, Höltje M, Michel K, Li Q, Otto C, Drenckhahn C et al. Anti-DPPX encephalitis: pathogenic effects of antibodies on gut and brain neurons. Neurology. 2015;85(10):890-7. https://doi.org/10.1212/WNL.0000000000001907
» https://doi.org/10.1212/WNL.0000000000001907 -
40Boronat A, Gelfand JM, Gresa-Arribas N, Jeong HY, Walsh M, Roberts K et al. Encephalitis and antibodies to dipeptidyl-peptidase-like protein-6, a subunit of Kv4.2 potassium channels. Ann Neurol. 2013;73(1):120-8. https://doi.org/10.1002/ana.23756
» https://doi.org/10.1002/ana.23756 -
41Gelpi E, Höftberger R, Graus F, Ling H, Holton JL, Dawson T et al. Neuropathological criteria of anti-IgLON5-related tauopathy. Acta Neuropathol. 2016;132(4):531-43. https://doi.org/10.1007/s00401-016-1591-8
» https://doi.org/10.1007/s00401-016-1591-8 -
42Gaig C, Graus F, Compta Y, Högl B, Bataller L, Brüggemann N et al. Clinical manifestations of the anti-IgLON5 disease. Neurology. 2017;88(18):1736-43. https://doi.org/10.1212/WNL.0000000000003887
» https://doi.org/10.1212/WNL.0000000000003887 -
43Lopez-Chiriboga AS, Komorowski L, Kümpfel T, Probst C, Hinson SR, Pittock SJ et al. Metabotropic glutamate receptor type 1 autoimmunity. Neurology. 2016;86(11):1009-13. https://doi.org/10.1212/WNL.0000000000002476
» https://doi.org/10.1212/WNL.0000000000002476 -
44Lancaster E, Martinez-Hernandez E, Titulaer MJ, Boulos M, Weaver S, Antoine JC et al. Antibodies to metabotropic glutamate receptor 5 in the Ophelia syndrome. Neurology. 2011;77(18):1698-701. https://doi.org/10.1212/WNL.0b013e3182364a44
» https://doi.org/10.1212/WNL.0b013e3182364a44 -
45Gresa-Arribas N, Titulaer MJ, Torrents A, Aguilar E, McCracken L, Leypoldt F et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol. 2014;13(2):167-77. https://doi.org/10.1016/S1474-4422(13)70282-5
» https://doi.org/10.1016/S1474-4422(13)70282-5 -
46Dahm L, Ott C, Steiner J, Stepniak B, Teegen B, Saschenbrecker S et al. Seroprevalence of autoantibodies against brain antigens in health and disease. Ann Neurol. 2014;76(1):82-94. https://doi.org/10.1002/ana.24189
» https://doi.org/10.1002/ana.24189 -
47Titulaer MJ, Soffietti R, Dalmau J, Gilhus NE, Giometto B, Graus F et al. Screening for tumours in paraneoplastic syndromes: report of an EFNS Task Force. Eur J Neurol. 2011;18(1):19-e3. https://doi.org/10.1111/j.1468-1331.2010.03220.x
» https://doi.org/10.1111/j.1468-1331.2010.03220.x -
48Dalmau J, Rosenfeld MR. Paraneoplastic syndromes of the CNS. Lancet Neurol. 2008;7(4):327-40. https://doi.org/10.1016/S1474-4422(08)70060-7
» https://doi.org/10.1016/S1474-4422(08)70060-7 -
49McKeon A, Apiwattanakul M, Lachance DH, Lennon VA, Mandrekar JN, Boeve BF. Positron emission tomography-computed tomography in paraneoplastic neurologic disorders. Arch Neurol. 2010;67(3):322. https://doi.org/10.1001/archneurol.2009.336
» https://doi.org/10.1001/archneurol.2009.336 -
50Vincent A, Bien CG, Irani SR, Waters P. Autoantibodies associated with diseases of the CNS: new developments and future challenges. Lancet Neurol. 2011;10(8):759-72. https://doi.org/10.1016/S1474-4422(11)70096-5
» https://doi.org/10.1016/S1474-4422(11)70096-5
Publication Dates
-
Publication in this collection
Jan 2018
History
-
Received
18 Mar 2017 -
Reviewed
31 Aug 2017 -
Accepted
18 Sept 2017