ABSTRACT
The expanding therapeutic arsenal in multiple sclerosis (MS) has allowed for more effective and personalized treatment, but the choice and management of disease-modifying therapies (DMTs) is becoming increasingly complex. In this context, experts from the Brazilian Committee on Treatment and Research in Multiple Sclerosis and the Neuroimmunology Scientific Department of the Brazilian Academy of Neurology have convened to establish this Brazilian Consensus for the Treatment of MS, based on their understanding that neurologists should be able to prescribe MS DMTs according to what is better for each patient, based on up-to-date evidence and practice. We herein propose practical recommendations for the treatment of MS, with the main focus on the choice and management of DMTs, as well as present a review of the scientific rationale supporting therapeutic strategies in MS.
Keywords:
multiple sclerosis; drug therapy; consensus
RESUMO
O crescent arsenal terapêutico na esclerose múltipla (EM) tem permitido tratamentos mais efetivos e personalizados, mas a escolha e o manejo das terapias modificadoras da doença (TMDs) tem se tornado cada vez mais complexos. Neste contexto, especialistas do Comitê Brasileiro de Tratamento e Pesquisa em Esclerose Múltipla e do Departamento Científico de Neuroimunologia da Academia Brasileira de Neurologia reuniram-se para estabelecer este Consenso Brasileiro para o Tratamento da EM, baseados no entendimento de que neurologistas devem ter a possibilidade de prescrever TMDs para EM de acordo com o que é melhor para cada paciente, com base em evidências e práticas atualizadas. Por meio deste documento, propomos recomendações práticas para o tratamento da EM, com foco principal na escolha e no manejo das TMDs, e revisamos os argumentos que embasam as estratégias de tratamento na EM.
Palavras-chave:
esclerose múltipla; tratamento farmacológico; consenso
Multiple sclerosis (MS) is a chronic and disabling disease that mostly affects young patients, resulting in huge consequences for their physical and cognitive domains, and impacting on their quality of life and employability. Its incidence and prevalence is increasing worldwide11. Multiple Sclerosis International Federation. Atlas of MS 2013: mapping multiple sclerosis around the world. London: Multiple Sclerosis International Federation; 2013. and still no definite cause is known, the reason why a straight-line delineation of therapeutic recommendation has been hindered and the overall trend has been driven toward a personalized and individualized therapeutic rationale.
Throughout the last decade, the number of disease-modifying therapies (DMTs) approved for the treatment of MS has increased from just a few to more than a dozen. In addition, a number of therapies are often used off-label and several investigational drugs are going through the later stages of development and may soon be approved. While this represents an unprecedented opportunity for personalized therapy in MS, the choice of DMTs is becoming increasingly complex.
Many aspects of the management of MS have not yet been formally assessed by clinical trials. Remarkably, there are few head-to-head comparisons between DMTs, few trials in progressive MS, and no direct comparison between the therapeutic strategies known as escalation (starting with safer but less efficacious drugs, and escalating to more efficacious and often riskier drugs, as needed) and induction (starting with highly efficacious “aggressive” treatments in order to prevent irreversible accumulation of disability, yet with some serious adverse effects)22. Comi G, Radaelli M, Soelberg Sørensen P. Evolving concepts in the treatment of relapsing multiple sclerosis. Lancet. 2017 Apr;389(10076):1347-56. https://doi.org/10.1016/S0140-6736(16)32388-1
https://doi.org/10.1016/S0140-6736(16)32...
. Nevertheless, these are key aspects and must be addressed by guidelines, even if optimal evidence is not available.
The treatment of MS in Brazil is largely limited by the Ministry of Health Clinical Protocols and Therapeutic Guidelines (Protocolos Clínicos e Diretrizes Terapêuticas)33. Ministério da Saúde (Brasil). Secretaria de Atenção à Saúde. Secretaria de Ciência, Tecnologia e Insumos Estratégicos. Portaria Conjunta Nº 10, de 2 de abril de 2018. Aprova o protocolo clínico e diretrizes trapêuticas da esclerose múltipla. Brasília, DF, 2018 [accessed 2018 May 8] Available from: http://portalarquivos2.saude.gov.br/images/pdf/2018/abril/09/PORTARIA-CONJUNTA-N-10-ESCLEROSE-MULTIPLA.09.04.2018.pdf
http://portalarquivos2.saude.gov.br/imag...
, since most patients get their DMTs from the public health system. The current Clinical Protocols and Therapeutic Guidelines determine a one-size-fits-all strategy, with minimal flexibility to tailor therapy to the needs and preferences of individual patients. Moreover, it delays access to DMTs of higher efficacy and fails to include some of the locally-approved DMTs.
In this context, experts from the Brazilian Committee on Treatment and Research in Multiple Sclerosis and the Neuroimmunology Scientific Department of the Brazilian Academy of Neurology have convened to establish this Brazilian Consensus for the Treatment of MS, based on an understanding that neurologists should be able to prescribe MS DMTs according to what is better for each patient, and based on up-to-date evidence and practice.
METHODS
This guideline represents a consensus of Brazilian experts on the management of MS. A panel of 35 representatives from the Brazilian Committee on Treatment and Research in Multiple Sclerosis and the Brazilian Academy of Neurology met on June 6th, 2016. A preliminary version of the proposed protocol, previously drafted by two representatives (JB and VDM), was discussed point-by-point by the panel members and modified as needed until consensus was reached.
Disease-modifying therapies were considered for inclusion in the protocol if at least one phase 3 trial demonstrated their efficacy in MS, plus they were approved by at least one of three regulatory bodies: the Brazilian Health Regulatory Agency (Agência Nacional de Vigilância Sanitária, ANVISA); the United States Food and Drug Administration (FDA); or the European Medicines Agency (EMA). Also considered for inclusion were DMTs for which a positive phase 2 or 3 trial was available, yet without approval for MS by the aforementioned regulatory agencies, but only in the case of them already being available or approved for another indication in Brazil, in order to maximize the utilization of locally-available resources. Previous national recommendations published by the Brazilian Committee on Treatment and Research in Multiple Sclerosis44. Tilbery CP, Moreira MA, Mendes MF, Lana-Peixoto MA. [Recommendations for the use of immunomodulatory drugs in multiple sclerosis: the BCTRIMS consensus]. Arq Neuropsiquiatr. 2000 Sep;58 3A:769-76. Portuguese. https://doi.org/10.1590/S0004-282X2000000400030
https://doi.org/10.1590/S0004-282X200000...
,55. Callegaro D, Lana-Peixoto MA, Moreira MA, Marchiori PE, Bacheschi LA, Arruda WO et al. [The BCTRIMS Expanded Consensus on treatment of multiple sclerosis: I. The evidences for the use of immunosuppressive agents, plasma exchange and autologous hematopoietic stem cell transplantation]. Arq Neuropsiquiatr. 2002 Sep;60 3-B:869-74. Portuguese. https://doi.org/10.1590/S0004-282X2002000500035
https://doi.org/10.1590/S0004-282X200200...
,66. Moreira MA, Lana-Peixoto MA, Callegaro D, Haussen SR, Gama PD, Gabbai AA et al. [The BCTRIMS expanded consensus on treatment of multiple sclerosis: II. The evidences for the use of glucocorticoids and immunomodulatory treatments]. Arq Neuropsiquiatr. 2002 Sep;60 3-B:875-80. Portuguese. https://doi.org/10.1590/S0004-282X2002000500036
https://doi.org/10.1590/S0004-282X200200...
,77. Lana-Peixoto MA, Callegaro D, Moreira MA, Campos GB, Marchiori PE, Gabbai AA et al. [The BCTRIMS Expanded Consensus on treatment of multiple sclerosis: III. Evidence and recommendation-based guidelines]. Arq Neuropsiquiatr. 2002 Sep;60 3-B:881-6. Portuguese. https://doi.org/10.1590/S0004-282X2002000500037
https://doi.org/10.1590/S0004-282X200200...
and the Brazilian Academy of Neurology88. Machado S. Recomendações Esclerose Múltipla - Academia Brasileira de Neurologia. São Paulo: Omnifarma; 2012.,99. Comini-Frota ER, Vasconcelos CCF, Mendes MF. Guideline for multiple sclerosis treatment in Brazil: Consensus from the Neuroimmunology Scientific Department of the Brazilian Academy of Neurology. Arq Neuropsiquiatr. 2017 Jan;75(1):57-65. https://doi.org/10.1590/0004-282X20160185
https://doi.org/10.1590/0004-282X2016018...
were taken into account, as were some key international guidelines1010. Freedman MS, Selchen D, Arnold DL, Prat A, Banwell B, Yeung M et al. Treatment optimization in MS: Canadian MS Working Group updated recommendations. Can J Neurol Sci. 2013 May;40(3):307-23. https://doi.org/10.1017/S0317167100014244
https://doi.org/10.1017/S031716710001424...
,1111. Scolding N, Barnes D, Cader S, Chataway J, Chaudhuri A, Coles A et al. Association of British Neurologists: revised (2015) guidelines for prescribing disease-modifying treatments in multiple sclerosis. Pract Neurol. 2015 Aug;15(4):273-9. https://doi.org/10.1136/practneurol-2015-001139
https://doi.org/10.1136/practneurol-2015...
,22. Comi G, Radaelli M, Soelberg Sørensen P. Evolving concepts in the treatment of relapsing multiple sclerosis. Lancet. 2017 Apr;389(10076):1347-56. https://doi.org/10.1016/S0140-6736(16)32388-1
https://doi.org/10.1016/S0140-6736(16)32...
.
On June 23rd, 2016, the panel-approved protocol was presented at the 17th Brazilian Committee on Treatment and Research in Multiple Sclerosis Annual Meeting, the largest Brazilian conference on MS, where it was endorsed by the attendees. Subsequent minor changes were made due to new information that became available during manuscript preparation. The completed manuscript was subsequently circulated among all panel members for final approval.
DISEASE-RELATED CONCEPTS
The diagnosis of MS must be made according to the McDonald criteria, revised in 20171313. Thompson AJ, Banwell BL, Barkhof F, Carrol WM, Coetzee T, Comi G et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018 Feb;17(2):162-73. https://doi.org/10.1016/S1474-4422(17)30470-2
https://doi.org/10.1016/S1474-4422(17)30...
. The clinical course must be defined according to the classification of Lublin, modified in 2013, which includes four phenotypes: clinically isolated syndrome (CIS), relapsing-remitting MS (RRMS), secondary progressive MS (SPMS), and primary progressive MS (PPMS)1414. Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sørensen PS, Thompson AJ et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014 Jul;83(3):278-86. https://doi.org/10.1212/WNL.0000000000000560
https://doi.org/10.1212/WNL.000000000000...
. The relapsing-remitting and progressive forms may be further stratified by the presence or absence of disease activity, which can be defined as the occurrence of clinical relapses and/or detection of radiological findings (gadolinium-enhancing lesions and/or new or enlarging T2-weighted lesions) on follow-up magnetic resonance imaging (MRI)1414. Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sørensen PS, Thompson AJ et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014 Jul;83(3):278-86. https://doi.org/10.1212/WNL.0000000000000560
https://doi.org/10.1212/WNL.000000000000...
. The progressive phenotypes may be further categorized according to the presence or absence of ongoing progression, which can be assessed by clinical measurements of disability, at least once annually1414. Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sørensen PS, Thompson AJ et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014 Jul;83(3):278-86. https://doi.org/10.1212/WNL.0000000000000560
https://doi.org/10.1212/WNL.000000000000...
.
In order to guide treatment decisions, some additional stratifications are needed: CIS must be classified according to the risk of evolving to MS, and disease activity, when present, must be classified according to its level, even though universally-accepted criteria are lacking. For the purpose of this protocol, we define high-risk CIS as any case wherein one or more typical T2 lesion(s) on MRI is seen, provided both the clinical presentation and MRI lesion(s) are suggestive of central nervous system (CNS) demyelination and not attributable to other diseases. For such patients, the risk of developing MS ranges from 48% to 81%, depending on the number of lesions1515. Tintore M, Rovira À, Río J, Otero-Romero S, Arrambide G, Tur C et al. Defining high, medium and low impact prognostic factors for developing multiple sclerosis. Brain. 2015 Jul;138(Pt 7):1863-74. https://doi.org/10.1093/brain/awv105
https://doi.org/10.1093/brain/awv105...
.
Therapeutic decisions in MS are often based on the degree of disease activity, which can be rated as low, moderate (or average), and high. The definition of highly active MS is still debatable, but we herein adopt this definition: 1) two or more disabling relapses with incomplete resolution and at least one gadolinium-enhancing lesion or significant increase in T2 lesion load in the previous year in treatment-naive patients and 2) breakthrough disease activity in the previous year, under an adequate course of at least one DMT (in the absence of intolerance or nonadherence), presenting with at least one relapse in the previous year while on therapy and at least nine T2 hyperintense lesions or at least one gadolinium-enhancing lesion1616. Fazekas F, Bajenaru O, Berger T, Fabjan TH, Ledinek AH, Jakab G et al. How does fingolimod (Gilenya®) fit in the treatment algorithm for highly active relapsing-remitting multiple sclerosis? Front Neurol. 2013 May;4:10. https://doi.org/10.3389/fneur.2013.00010
https://doi.org/10.3389/fneur.2013.00010...
,1717. Devonshire V, Havrdova E, Radue EW, O’Connor P, Zhang-Auberson L, Agoropoulou C et al. Relapse and disability outcomes in patients with multiple sclerosis treated with fingolimod: subgroup analyses of the double-blind, randomised, placebo-controlled FREEDOMS study. Lancet Neurol. 2012 May;11(5):420-8. https://doi.org/10.1016/S1474-4422(12)70056-X
https://doi.org/10.1016/S1474-4422(12)70...
. For the purpose of this consensus, whenever disease activity is present but does not fulfill the criteria for high activity, it is considered as low or moderate activity.
Aggressive disease can be defined as RRMS with one or more of the following features: 1) Expanded Disability Status Scale score of 4 reached within five years of onset, or early and unexpected acquisition of disability followed by frequent relapses; 2) two or more relapses with incomplete resolution in the past year; 3) two or more MRI studies showing new or enlarging T2 lesions or gadolinium-enhancing lesions despite treatment; 4) no response to therapy with one or more DMTs for up to one year1818. Rush CA, MacLean HJ, Freedman MS. Aggressive multiple sclerosis: proposed definition and treatment algorithm. Nat Rev Neurol. 2015 Jul;11(7):379-89. https://doi.org/10.1038/nrneurol.2015.85
https://doi.org/10.1038/nrneurol.2015.85...
,1919. Menon S, Shirani A, Zhao Y, Oger J, Traboulsee A, Freedman MS et al. Characterising aggressive multiple sclerosis. J Neurol Neurosurg Psychiatry. 2013 Nov;84(11):1192-8. https://doi.org/10.1136/jnnp-2013-304951
https://doi.org/10.1136/jnnp-2013-304951...
.
Nonetheless, the above definitions of high activity and aggressive disease lack the sensitivity to detect many cases with poor prognosis early enough in the disease course. However, there are now known to be several clinical and radiological factors associated with a poorer prognosis (Table 1), which may be present from the onset or become apparent during follow-up and suggest evolution to a more “aggressive” form of the disease1818. Rush CA, MacLean HJ, Freedman MS. Aggressive multiple sclerosis: proposed definition and treatment algorithm. Nat Rev Neurol. 2015 Jul;11(7):379-89. https://doi.org/10.1038/nrneurol.2015.85
https://doi.org/10.1038/nrneurol.2015.85...
. Since there is a clear relationship between the amount of disease activity and final prognosis (presented later in the text), we recommend the parameters of disease activity should always be analyzed alongside the elements concerning prognosis.
The panel understands that these factors (especially if present in association) should prompt the treating neurologist to consider disease activity as high and the prognosis as poor, and to manage DMTs accordingly, even before the patient effectively meets the definition for “aggressive” MS. This approach may allow for the early optimization of DMTs before irreversible accumulation of disability is established.
TREATMENT-RELATED CONCEPTS
This rationale is based on what has been learned over the years from observational studies, controlled clinical trials, pathophysiological concepts and, more recently, real-world registries. In this context, treatment algorithms from many countries1111. Scolding N, Barnes D, Cader S, Chataway J, Chaudhuri A, Coles A et al. Association of British Neurologists: revised (2015) guidelines for prescribing disease-modifying treatments in multiple sclerosis. Pract Neurol. 2015 Aug;15(4):273-9. https://doi.org/10.1136/practneurol-2015-001139
https://doi.org/10.1136/practneurol-2015...
,2020. Ingwersen J, Aktas O, Hartung HP. Advances in and algorithms for the treatment of relapsing-remitting multiple sclerosis. Neurotherapeutics. 2016 Jan;13(1):47-57. https://doi.org/10.1007/s13311-015-0412-4
https://doi.org/10.1007/s13311-015-0412-...
,2121. Wingerchuk DM, Carter JL. Multiple sclerosis: current and emerging disease-modifying therapies and treatment strategies. Mayo Clin Proc. 2014 Feb;89(2):225-40. https://doi.org/10.1016/j.mayocp.2013.11.002
https://doi.org/10.1016/j.mayocp.2013.11...
,2222. Vosoughi R, Freedman MS. Therapy of MS. Clin Neurol Neurosurg. 2010 Jun;112(5):365-85. https://doi.org/10.1016/j.clineuro.2010.03.010
https://doi.org/10.1016/j.clineuro.2010....
,2323. Balabanov P, Haas M, Elferink A, Bakchine S, Broich K. Addressing the regulatory and scientific challenges in multiple sclerosis: a statement from the EU regulators. Mult Scler. 2014 Sep;20(10):1282-7. https://doi.org/10.1177/1352458514546876
https://doi.org/10.1177/1352458514546876...
,2424. Sorensen PS. New management algorithms in multiple sclerosis. Curr Opin Neurol. 2014 Jun;27(3):246-59. https://doi.org/10.1097/WCO.0000000000000096
https://doi.org/10.1097/WCO.000000000000...
,2525. Torkildsen Ø, Myhr KM, Bø L. Disease-modifying treatments for multiple sclerosis - a review of approved medications. Eur J Neurol. 2016 Jan;23 Suppl 1:18-27. https://doi.org/10.1111/ene.12883
https://doi.org/10.1111/ene.12883...
,2626. Cross AH, Naismith RT. Established and novel disease-modifying treatments in multiple sclerosis. J Intern Med. 2014 Apr;275(4):350-63. https://doi.org/10.1111/joim.12203
https://doi.org/10.1111/joim.12203...
,2727. Carrithers MD. Update on disease-modifying treatments for multiple sclerosis. Clin Ther. 2014 Dec;36(12):1938-45. https://doi.org/10.1016/j.clinthera.2014.08.006
https://doi.org/10.1016/j.clinthera.2014...
,2828. Michel L, Larochelle C, Prat A. Update on treatments in multiple sclerosis. Presse Med. 2015 Apr;44(4 Pt 2):e137-51. https://doi.org/10.1016/j.lpm.2015.02.008
https://doi.org/10.1016/j.lpm.2015.02.00...
,2929. Broadley SA, Barnett MH, Boggild M, Brew BJ, Butzkueven H, Heard R et al. Therapeutic approaches to disease modifying therapy for multiple sclerosis in adults: an Australian and New Zealand perspective: part 3 treatment practicalities and recommendations. J Clin Neurosci. 2014 Nov;21(11):1857-65. https://doi.org/10.1016/j.jocn.2014.01.017
https://doi.org/10.1016/j.jocn.2014.01.0...
,3030. García-Merino A, Fernández O, Montalbán X, Andrés C, Oreja-Guevar C, Rodríguez-Antigüedad A et al. Documento del Grupo de Consenso de la Sociedad Española de Neurología sobre el uso de medicamentos en esclerosis múltiple. Neurología. 2013;28(6):375-8. https://doi.org/10.1016/j.nrl.2013.01.009
https://doi.org/10.1016/j.nrl.2013.01.00...
,3131. Limmroth V. Treatment of relapsing-remitting multiple sclerosis: current and future algorithms. Eur Neurol. 2014;72(s1 Suppl 1):35-8. https://doi.org/10.1159/000367624
https://doi.org/10.1159/000367624...
have been guided by what have turned out to be very important concepts: 1) early and effective treatment; 2) the existence of a therapeutic window of opportunity; 3) early optimization of treatment during the therapeutic window; 4) the existence of different clinical and radiological phenotypes; and 5) treatment decisions based on different levels of inflammatory activity and/or specific clinical phenotypes, wherever possible, always favoring the best efficacy/risk ratio.
It is widely accepted that the main aspect to be avoided or minimized in MS is the accumulation and progression of disability, with this representing the central goal in planning therapeutic decisions. However, the immediate therapeutic effect of current DMTs is, in fact, centered upon the prevention of clinical and radiologic activity outbreaks, with these being related to focal inflammation. The accumulated disability in MS follows a two-step process. The first phase is very dependent on focal inflammation due to the impairment of adaptive peripheral immunity, predominating in the initial phase of the disease, and leads to the relapsing-remitting phase in which early epitope spreading occurs and underlies the early pathogenic events3232. Quintana FJ, Patel B, Yeste A, Nyirenda M, Kenison J, Rahbari R et al. Epitope spreading as an early pathogenic event in pediatric multiple sclerosis. Neurology. 2014 Dec;83(24):2219-26. https://doi.org/10.1212/WNL.0000000000001066
https://doi.org/10.1212/WNL.000000000000...
,3333. Hohlfeld R, Dornmair K, Meinl E, Wekerle H. The search for the target antigens of multiple sclerosis, part 2: CD8+ T cells, B cells, and antibodies in the focus of reverse-translational research. Lancet Neurol. 2016 Mar;15(3):317-31. https://doi.org/10.1016/S1474-4422(15)00313-0
https://doi.org/10.1016/S1474-4422(15)00...
,3434. Hohlfeld R, Dornmair K, Meinl E, Wekerle H. The search for the target antigens of multiple sclerosis, part 1: autoreactive CD4+ T lymphocytes as pathogenic effectors and therapeutic targets. Lancet Neurol. 2016 Feb;15(2):198-209. https://doi.org/10.1016/S1474-4422(15)00334-8
https://doi.org/10.1016/S1474-4422(15)00...
. This tends to decline over time as other pathogenic mechanisms, mostly related to innate immunity, lead to the second phase of the disease, which is not as dependent on focal inflammation3535. Leray E, Yaouanq J, Le Page E, Coustans M, Laplaud D, Oger J et al. Evidence for a two-stage disability progression in multiple sclerosis. Brain. 2010 Jul;133(Pt 7):1900-13. https://doi.org/10.1093/brain/awq076
https://doi.org/10.1093/brain/awq076...
, but rather on other more spread mechanisms, like CNS-compartmentalized inflammation inside lymphoid follicles within the meninges3636. Michel L, Touil H, Pikor NB, Gommerman JL, Prat A, Bar-Or A. B Cells in the multiple sclerosis central nervous system: trafficking and contribution to CNS-compartmentalized inflammation. Front Immunol. 2015 Dec;6:636. https://doi.org/10.3389/fimmu.2015.00636
https://doi.org/10.3389/fimmu.2015.00636...
,3737. Pikor NB, Prat A, Bar-Or A, Gommerman JL. Meningeal tertiary lymphoid tissues and multiple sclerosis: a gathering place for diverse types of immune cells during CNS autoimmunity. Front Immunol. 2016 Jan;6:657. https://doi.org/10.3389/fimmu.2015.00657
https://doi.org/10.3389/fimmu.2015.00657...
, underlying the progressive phase3838. Meinl E, Krumbholz M, Derfuss T, Junker A, Hohlfeld R. Compartmentalization of inflammation in the CNS: a major mechanism driving progressive multiple sclerosis. J Neurol Sci. 2008 Nov;274(1-2):42-4. https://doi.org/10.1016/j.jns.2008.06.032
https://doi.org/10.1016/j.jns.2008.06.03...
. The first stage is characterized by the occurrence of clinical relapses and new/enlarging or gadolinium-enhancing lesions on MRI, due to focal demyelinating inflammatory lesions in the early stages of the disease, while the second stage is characterized by the progressive and sustained worsening of functional capacity by mechanisms of axonal degeneration and brain atrophy, which although probably present since the beginning, are more evident in the later stages of the disease or in the so-called secondary progressive phase.
There is much evidence relating the degree of inflammatory activity in the first phase of the disease to the disability grade achieved in the second phase. Large observational cohorts have demonstrated that frequent relapses3939. Jokubaitis VG, Spelman T, Kalincik T, Lorscheider J, Havrdova E, Horakova D et al. Predictors of long-term disability accrual in relapse-onset multiple sclerosis. Ann Neurol. 2016 Jul;80(1):89-100. https://doi.org/10.1002/ana.24682
https://doi.org/10.1002/ana.24682...
in the early years of disease4040. Scalfari A, Neuhaus A, Degenhardt A, Rice GP, Muraro PA, Daumer M et al. The natural history of multiple sclerosis: a geographically based study 10: relapses and long-term disability. Brain. 2010 Jul;133(Pt 7):1914-29. https://doi.org/10.1093/brain/awq118
https://doi.org/10.1093/brain/awq118...
,4141. Scalfari A, Neuhaus A, Daumer M, Muraro PA, Ebers GC. Onset of secondary progressive phase and long-term evolution of multiple sclerosis. J Neurol Neurosurg Psychiatry. 2014 Jan;85(1):67-75. https://doi.org/10.1136/jnnp-2012-304333
https://doi.org/10.1136/jnnp-2012-304333...
, as well as short intervals between relapses4040. Scalfari A, Neuhaus A, Degenhardt A, Rice GP, Muraro PA, Daumer M et al. The natural history of multiple sclerosis: a geographically based study 10: relapses and long-term disability. Brain. 2010 Jul;133(Pt 7):1914-29. https://doi.org/10.1093/brain/awq118
https://doi.org/10.1093/brain/awq118...
,4242. Myhr KM, Riise T, Vedeler C, Nortvedt MW, Grønning R, Midgard R et al. Disability and prognosis in multiple sclerosis: demographic and clinical variables important for the ability to walk and awarding of disability pension. Mult Scler. 2001 Feb;7(1):59-65. https://doi.org/10.1177/135245850100700110
https://doi.org/10.1177/1352458501007001...
,4343. Amato MP, Ponziani G, Bartolozzi ML, Siracusa G. A prospective study on the natural history of multiple sclerosis: clues to the conduct and interpretation of clinical trials. J Neurol Sci. 1999 Oct;168(2):96-106. https://doi.org/10.1016/S0022-510X(99)00143-4
https://doi.org/10.1016/S0022-510X(99)00...
, are predictors of reaching greater levels of disability in shorter time periods, while the number of relapses after five years of disease does not substantially influence the time to achieve levels of permanent incapacity or the degree of severity of this phase4040. Scalfari A, Neuhaus A, Degenhardt A, Rice GP, Muraro PA, Daumer M et al. The natural history of multiple sclerosis: a geographically based study 10: relapses and long-term disability. Brain. 2010 Jul;133(Pt 7):1914-29. https://doi.org/10.1093/brain/awq118
https://doi.org/10.1093/brain/awq118...
. In other words, the early course influences long-term evolution. Relapses with residual sequelae also contribute to the long-term accumulation of disability4343. Amato MP, Ponziani G, Bartolozzi ML, Siracusa G. A prospective study on the natural history of multiple sclerosis: clues to the conduct and interpretation of clinical trials. J Neurol Sci. 1999 Oct;168(2):96-106. https://doi.org/10.1016/S0022-510X(99)00143-4
https://doi.org/10.1016/S0022-510X(99)00...
,4444. Lublin FD, Baier M, Cutter G. Effect of relapses on development of residual deficit in multiple sclerosis. Neurology. 2003 Dec;61(11):1528-32. https://doi.org/10.1212/01.WNL.0000096175.39831.21
https://doi.org/10.1212/01.WNL.000009617...
,4545. Hirst C, Ingram G, Pearson O, Pickersgill T, Scolding N, Robertson N. Contribution of relapses to disability in multiple sclerosis. J Neurol. 2008 Feb;255(2):280-7. https://doi.org/10.1007/s00415-008-0743-8
https://doi.org/10.1007/s00415-008-0743-...
,4646. Vercellino M, Romagnolo A, Mattioda A, Masera S, Piacentino C, Merola A et al. Multiple sclerosis relapses: a multivariable analysis of residual disability determinants. Acta Neurol Scand. 2009 Feb;119(2):126-30. https://doi.org/10.1111/j.1600-0404.2008.01076.x
https://doi.org/10.1111/j.1600-0404.2008...
,4747. Bergamaschi R, Berzuini C, Romani A, Cosi V. Predicting secondary progression in relapsing-remitting multiple sclerosis: a Bayesian analysis. J Neurol Sci. 2001 Aug;189(1-2):13-21. https://doi.org/10.1016/S0022-510X(01)00572-X
https://doi.org/10.1016/S0022-510X(01)00...
. This means that irreversible axonal degeneration occurs very early and is, at least partly, related to inflammation22. Comi G, Radaelli M, Soelberg Sørensen P. Evolving concepts in the treatment of relapsing multiple sclerosis. Lancet. 2017 Apr;389(10076):1347-56. https://doi.org/10.1016/S0140-6736(16)32388-1
https://doi.org/10.1016/S0140-6736(16)32...
,4848. Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998 Jan;338(5):278-85. https://doi.org/10.1056/NEJM199801293380502
https://doi.org/10.1056/NEJM199801293380...
. It has also been observed that an older age at disease onset is related to increased risk of a rapid shift to a secondary progressive course4747. Bergamaschi R, Berzuini C, Romani A, Cosi V. Predicting secondary progression in relapsing-remitting multiple sclerosis: a Bayesian analysis. J Neurol Sci. 2001 Aug;189(1-2):13-21. https://doi.org/10.1016/S0022-510X(01)00572-X
https://doi.org/10.1016/S0022-510X(01)00...
,4949. Riise T, Grønning M, Fernández O, Lauer K, Midgard R, Minderhoud JM et al. Early prognostic factors for disability in multiple sclerosis, a European multicenter study. Acta Neurol Scand. 1992 Mar;85(3):212-8. https://doi.org/10.1111/j.1600-0404.1992.tb04031.x
https://doi.org/10.1111/j.1600-0404.1992...
,5050. Tremlett H, Yinshan Zhao, Devonshire V. Natural history of secondary-progressive multiple sclerosis. Mult Scler. 2008 Apr;14(3):314-24. https://doi.org/10.1177/1352458507084264
https://doi.org/10.1177/1352458507084264...
,5151. Scalfari A, Neuhaus A, Daumer M, Ebers GC, Muraro PA. Age and disability accumulation in multiple sclerosis. Neurology. 2011 Sep;77(13):1246-52. https://doi.org/10.1212/WNL.0b013e318230a17d
https://doi.org/10.1212/WNL.0b013e318230...
,5252. Gholipour T, Healy B, Baruch NF, Weiner HL, Chitnis T. Demographic and clinical characteristics of malignant multiple sclerosis. Neurology. 2011 Jun;76(23):1996-2001. https://doi.org/10.1212/WNL.0b013e31821e559d
https://doi.org/10.1212/WNL.0b013e31821e...
,5353. Cossburn M, Ingram G, Hirst C, Ben-Shlomo Y, Pickersgill TP, Robertson NP. Age at onset as a determinant of presenting phenotype and initial relapse recovery in multiple sclerosis. Mult Scler. 2012 Jan;18(1):45-54. https://doi.org/10.1177/1352458511417479
https://doi.org/10.1177/1352458511417479...
, probably due to the progressive decline with aging of the functional reserves, neuroplasticity and recovery mechanisms, including remyelination. The lesion load on MRI5454. Bermel RA, You X, Foulds P, Hyde R, Simon JH, Fisher E et al. Predictors of long-term outcome in multiple sclerosis patients treated with interferon β. Ann Neurol. 2013 Jan;73(1):95-103. https://doi.org/10.1002/ana.23758
https://doi.org/10.1002/ana.23758...
,5555. Río J, Rovira A, Tintoré M, Huerga E, Nos C, Tellez N et al. Relationship between MRI lesion activity and response to IFN-β in relapsing–remitting multiple sclerosis patients. Mult. Scler. J. 2008;14(4):479-84. https://doi.org/10.1177/1352458507085555
https://doi.org/10.1177/1352458507085555...
,5656. Losseff NA, Miller DH, Kidd D, Thompson AJ. The predictive value of gadolinium enhancement for long term disability in relapsing-remitting multiple sclerosis: preliminary results. Mult Scler. 2001 Feb;7(1):23-5.,5757. Fisniku LK, Brex PA, Altmann DR, Miszkiel KA, Benton CE, Lanyon R et al. Disability and T2 MRI lesions: a 20-year follow-up of patients with relapse onset of multiple sclerosis. Brain. 2008 Mar;131(Pt 3):808-17. https://doi.org/10.1093/brain/awm329
https://doi.org/10.1093/brain/awm329...
,5858. Maghzi AH, Revirajan N, Julian LJ, Spain R, Mowry EM, Liu S et al. Magnetic resonance imaging correlates of clinical outcomes in early multiple sclerosis. Mult Scler Relat Disord. 2014 Nov;3(6):720-7. https://doi.org/10.1016/j.msard.2014.07.003
https://doi.org/10.1016/j.msard.2014.07....
, as well as evidence of cerebral atrophy5959. Sormani MP, Arnold DL, De Stefano N. Treatment effect on brain atrophy correlates with treatment effect on disability in multiple sclerosis. Ann Neurol. 2014 Jan;75(1):43-9. https://doi.org/10.1002/ana.24018
https://doi.org/10.1002/ana.24018...
,6060. De Stefano N, Narayanan S, Francis GS, Arnaoutelis R, Tartaglia MC, Antel JP et al. Evidence of axonal damage in the early stages of multiple sclerosis and its relevance to disability. Arch Neurol. 2001 Jan;58(1):65-70. https://doi.org/10.1001/archneur.58.1.65 PMID:11176938
https://doi.org/10.1001/archneur.58.1.65...
,6161. von Gumberz J, Mahmoudi M, Young K, Schippling S, Martin R, Heesen C et al. Short-term MRI measurements as predictors of EDSS progression in relapsing-remitting multiple sclerosis: grey matter atrophy but not lesions are predictive in a real-life setting. PeerJ. 2016 Sep;4:e2442. https://doi.org/10.7717/peerj.2442
https://doi.org/10.7717/peerj.2442...
,6262. Kalincik T, Vaneckova M, Tyblova M, Krasensky J, Seidl Z, Havrdova E, et al. Volumetric MRI markers and predictors of disease activity in early multiple sclerosis: a longitudinal cohort study. PLoS One. 2012;7(11):e50101. https://doi.org/10.1371/journal.pone.0050101
https://doi.org/10.1371/journal.pone.005...
,6363. Rocca MA, Comi G, Filippi M. The role of T1-weighted derived measures of neurodegeneration for assessing disability progression in multiple sclerosis. Front Neurol. 2017 Sep;8:433. https://doi.org/10.3389/fneur.2017.00433
https://doi.org/10.3389/fneur.2017.00433...
, also have direct correlation with long-term disability, meaning that inflammation leads to irreversible nervous damage.
All these data together strongly suggest the existence of a therapeutic window, a time in the early stages of the inflammatory phase of MS during which therapeutic intervention can substantially influence the timing and condition in which the patient will reach the progressive stage. The era of monoclonal antibodies came in to consolidate the existence of a therapeutic window. A retrospective analysis of controlled clinical trials of alemtuzumab revealed that patients treated earlier in the evolution of the disease with a drug considered to have high efficacy showed better results for the parameters of sustained long-term disability, when compared to the group receiving treatment later after disease onset6464. Coles AJ, Cox A, Le Page E, Jones J, Trip SA, Deans J et al. The window of therapeutic opportunity in multiple sclerosis: evidence from monoclonal antibody therapy. J Neurol. 2006 Jan;253(1):98-108. https://doi.org/10.1007/s00415-005-0934-5
https://doi.org/10.1007/s00415-005-0934-...
. The finding that disease-modifying drugs can delay the conversion from CIS to clinically definite MS6565. Jacobs LD, Beck RW, Simon JH, Kinkel RP, Brownscheidle CM, Murray TJ et al. Intramuscular interferon beta-1a therapy initiated during a first demyelinating event in multiple sclerosis. N Engl J Med. 2000 Sep;343(13):898-904. https://doi.org/10.1056/NEJM200009283431301
https://doi.org/10.1056/NEJM200009283431...
,6666. Comi G, Filippi M, Barkhof F, Durelli L, Edan G, Fernández O, et al. Effect of early interferon treatment on conversion to definite multiple sclerosis: a randomised study. Lancet. 2001 May;357(9268):1576-82. https://doi.org/10.1016/S0140-6736(00)04725-5
https://doi.org/10.1016/S0140-6736(00)04...
,6767. Kappos L, Polman CH, Freedman MS, Edan G, Hartung HP, Miller DH et al. Treatment with interferon beta-1b delays conversion to clinically definite and McDonald MS in patients with clinically isolated syndromes. Neurology. 2006 Oct;67(7):1242-9. https://doi.org/10.1212/01.wnl.0000237641.33768.8d
https://doi.org/10.1212/01.wnl.000023764...
,6868. Comi G, Martinelli V, Rodegher M, Moiola L, Bajenaru O, Carra A et al. Effect of glatiramer acetate on conversion to clinically definite multiple sclerosis in patients with clinically isolated syndrome (PreCISe study): a randomised, double-blind, placebo-controlled trial. Lancet. 2009 Oct;374(9700):1503-11. https://doi.org/10.1016/S0140-6736(09)61259-9
https://doi.org/10.1016/S0140-6736(09)61...
,6969. Comi G, De Stefano N, Freedman MS, Barkhof F, Polman CH, Uitdehaag BM et al. Comparison of two dosing frequencies of subcutaneous interferon beta-1a in patients with a first clinical demyelinating event suggestive of multiple sclerosis (REFLEX): a phase 3 randomised controlled trial. Lancet Neurol. 2012 Jan;11(1):33-41. https://doi.org/10.1016/S1474-4422(11)70262-9
https://doi.org/10.1016/S1474-4422(11)70...
,7070. Miller AE, Wolinsky JS, Kappos L, Comi G, Freedman MS, Olsson TP. Oral teriflunomide for patients with a first clinical episode suggestive of multiple sclerosis (TOPIC): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2014 Oct;13(10):977-86. https://doi.org/10.1016/S1474-4422(14)70191-7
https://doi.org/10.1016/S1474-4422(14)70...
, together with results indicating increased responsiveness to therapies in CIS relative to the relapsing-remitting phase, probably due to the increasing complexity in pathogenic mechanisms as the disease evolves22. Comi G, Radaelli M, Soelberg Sørensen P. Evolving concepts in the treatment of relapsing multiple sclerosis. Lancet. 2017 Apr;389(10076):1347-56. https://doi.org/10.1016/S0140-6736(16)32388-1
https://doi.org/10.1016/S0140-6736(16)32...
,3232. Quintana FJ, Patel B, Yeste A, Nyirenda M, Kenison J, Rahbari R et al. Epitope spreading as an early pathogenic event in pediatric multiple sclerosis. Neurology. 2014 Dec;83(24):2219-26. https://doi.org/10.1212/WNL.0000000000001066
https://doi.org/10.1212/WNL.000000000000...
,7171. Freedman MS, Comi G, De Stefano N, Barkhof F, Polman CH, Uitdehaag BM et al. Moving toward earlier treatment of multiple sclerosis: findings from a decade of clinical trials and implications for clinical practice. Mult Scler Relat Disord. 2014 Mar;3(2):147-55. https://doi.org/10.1016/j.msard.2013.07.001
https://doi.org/10.1016/j.msard.2013.07....
, also helps to strengthen the concept of a therapeutic window.
The therapeutic window is, therefore, the variable and sometimes short time period in which treatment should be optimized, linking the need for early treatment with that of an effective treatment. Within this context, in addition to the regular monitoring of radiological and clinical disease activity for timely recognition of therapeutic failure, early definition of the clinical phenotype is of extreme importance. Relapses involving the pyramidal, sphincter and cerebellar pathways are known to have a higher correlation with the accumulation of disability, when compared to relapses involving other neurological fields, such as visual and sensory, for instance4343. Amato MP, Ponziani G, Bartolozzi ML, Siracusa G. A prospective study on the natural history of multiple sclerosis: clues to the conduct and interpretation of clinical trials. J Neurol Sci. 1999 Oct;168(2):96-106. https://doi.org/10.1016/S0022-510X(99)00143-4
https://doi.org/10.1016/S0022-510X(99)00...
,4747. Bergamaschi R, Berzuini C, Romani A, Cosi V. Predicting secondary progression in relapsing-remitting multiple sclerosis: a Bayesian analysis. J Neurol Sci. 2001 Aug;189(1-2):13-21. https://doi.org/10.1016/S0022-510X(01)00572-X
https://doi.org/10.1016/S0022-510X(01)00...
,4949. Riise T, Grønning M, Fernández O, Lauer K, Midgard R, Minderhoud JM et al. Early prognostic factors for disability in multiple sclerosis, a European multicenter study. Acta Neurol Scand. 1992 Mar;85(3):212-8. https://doi.org/10.1111/j.1600-0404.1992.tb04031.x
https://doi.org/10.1111/j.1600-0404.1992...
,5252. Gholipour T, Healy B, Baruch NF, Weiner HL, Chitnis T. Demographic and clinical characteristics of malignant multiple sclerosis. Neurology. 2011 Jun;76(23):1996-2001. https://doi.org/10.1212/WNL.0b013e31821e559d
https://doi.org/10.1212/WNL.0b013e31821e...
,7272. Stewart T, Spelman T, Havrdova E, Horakova D, Trojano M, Izquierdo G, et al. Contribution of different relapse phenotypes to disability in multiple sclerosis. Mult Scler. 2017 Feb;23(2):266-76. https://doi.org/10.1177/1352458516643392
https://doi.org/10.1177/1352458516643392...
,7373. Damasceno A, Von Glehn F, Brandão CO, Damasceno BP, Cendes F. Prognostic indicators for long-term disability in multiple sclerosis patients. J Neurol Sci. 2013 Jan;324(1-2):29-33. https://doi.org/10.1016/j.jns.2012.09.020
https://doi.org/10.1016/j.jns.2012.09.02...
,7474. Langer-Gould A, Popat RA, Huang SM, Cobb K, Fontoura P, Gould MK et al. Clinical and demographic predictors of long-term disability in patients with relapsing-remitting multiple sclerosis: a systematic review. Arch Neurol. 2006 Dec;63(12):1686-91. https://doi.org/10.1001/archneur.63.12.1686
https://doi.org/10.1001/archneur.63.12.1...
. Knowing this does not occur randomly, as further relapse phenotypes are predicted by the phenotype of previous relapses7575. Mowry EM, Deen S, Malikova I, Pelletier J, Bacchetti P, Waubant E. The onset location of multiple sclerosis predicts the location of subsequent relapses. J Neurol Neurosurg Psychiatry. 2009 Apr;80(4):400-3. https://doi.org/10.1136/jnnp.2008.157305
https://doi.org/10.1136/jnnp.2008.157305...
,7676. Kalincik T, Buzzard K, Jokubaitis V, Trojano M, Duquette P, Izquierdo G et al. Risk of relapse phenotype recurrence in multiple sclerosis. Mult Scler. 2014 Oct;20(11):1511-22. https://doi.org/10.1177/1352458514528762
https://doi.org/10.1177/1352458514528762...
,7777. Deen S, Bacchetti P, High A, Waubant E. Predictors of the location of multiple sclerosis relapse. J Neurol Neurosurg Psychiatry. 2008 Oct;79(10):1190-3. https://doi.org/10.1136/jnnp.2007.136440
https://doi.org/10.1136/jnnp.2007.136440...
, in the vast majority of cases there are elements that are predictors of prognosis in a patient's clinical presentation (Table 1). The suggestion of there being a differential impact of phenotypes on long-term disability can justify, wherever possible, an individualized therapeutic management of different and specific phenotypes. Given the different profiles of effectiveness and risk for the various drugs currently available, the choice of treatment should be carefully considered and based on an individual benefit/risk assessment for the patient, where drug efficacy should be appropriate for the severity of phenotype and thereby justify or offset the risks.
Changes in the diagnostic criteria for MS, essentially aimed at accelerating the diagnostic process, progress in neuroimaging techniques in the daily clinical setting, and the approval of high-efficacy drugs, has led to expectations of greater therapy effectiveness, alongside a trend to tolerate less and less disease activity, increasing the possibility of enhancing MS treatment remarkably7878. Capra R, Cordioli C, Rasia S, Gallo F, Signori A, Sormani MP. Assessing long-term prognosis improvement as a consequence of treatment pattern changes in MS. Mult Scler. 2017 Nov;23(13):1757-61. https://doi.org/10.1177/1352458516687402
https://doi.org/10.1177/1352458516687402...
. The current therapeutic approaches for MS are the so-called escalation or induction strategies7979. Comi G. Induction vs. escalating therapy in multiple sclerosis: practical implications. Neurol Sci. 2008 Sep;29(S2 Suppl 2):S253-5. https://doi.org/10.1007/s10072-008-0954-x
https://doi.org/10.1007/s10072-008-0954-...
.
The rationale for an escalation therapy approach is based on initiating treatment as early as possible using the safest but also less effective DMT, chosen based on the degree of inflammatory activity. This strategy is more often applied to mild or moderate clinical phenotypes, and close monitoring of clinical and MRI activity is advised. Continuing treatment while the disease is stable is recommended, as is promptly switching to a more effective drug when the ongoing treatment becomes ineffective or only partially effective, escalating the efficacy of treatment (but also increasing the risk profile as a consequence)2121. Wingerchuk DM, Carter JL. Multiple sclerosis: current and emerging disease-modifying therapies and treatment strategies. Mayo Clin Proc. 2014 Feb;89(2):225-40. https://doi.org/10.1016/j.mayocp.2013.11.002
https://doi.org/10.1016/j.mayocp.2013.11...
,8080. Fenu G, Lorefice L, Frau F, Coghe GC, Marrosu MG, Cocco E. Induction and escalation therapies in multiple sclerosis. Antiinflamm Antiallergy Agents Med Chem. 2015;14(1):26-34. https://doi.org/10.2174/1871523014666150504122220
https://doi.org/10.2174/1871523014666150...
.
The rationale for an induction approach arises from the observation that a proportion of MS patients with a more aggressive phenotype require more aggressive treatment from disease onset; very potent drugs are used earlier in this type of approach, with more important safety issues regarding autoimmunity and infectious complications than other drugs. These drugs have mechanisms of action that usually not only provoke long-term immunological remission, but also a re-initiation or “reset” of immunological pathways, or immune reconstitution8181. Wiendl H. Cladribine - an old newcomer for pulsed immune reconstitution in MS. Nat Rev Neurol. 2017 Oct;13(10):573-4. https://doi.org/10.1038/nrneurol.2017.119
https://doi.org/10.1038/nrneurol.2017.11...
. This could potentially reduce the epitope spreading, which occurs earlier in the disease course, and the shift in the main site of immune responses from the periphery to the CNS compartment, which seems to occur in later phases of MS8080. Fenu G, Lorefice L, Frau F, Coghe GC, Marrosu MG, Cocco E. Induction and escalation therapies in multiple sclerosis. Antiinflamm Antiallergy Agents Med Chem. 2015;14(1):26-34. https://doi.org/10.2174/1871523014666150504122220
https://doi.org/10.2174/1871523014666150...
.
Evaluation of prognostic factors must be performed every time disease activity is assessed or treatment failure is declared, and treatment decisions regarding drug efficacy or treatment approach must be adjusted accordingly, to follow either a strategy of escalation or induction therapy.
CONSENSUS FOR THE TREATMENT OF MS
Disease-modifying therapy
The choice of a DMT (and the decision to keep it or change it throughout follow-up) depends on several factors, including phenotype, prognostic factors, activity, progression status, severity, comorbidities, safety profile, tolerability, patient preference, convenience, cost, and availability. To date, there are no established biomarkers to predict the response of individual patients to most DMTs.
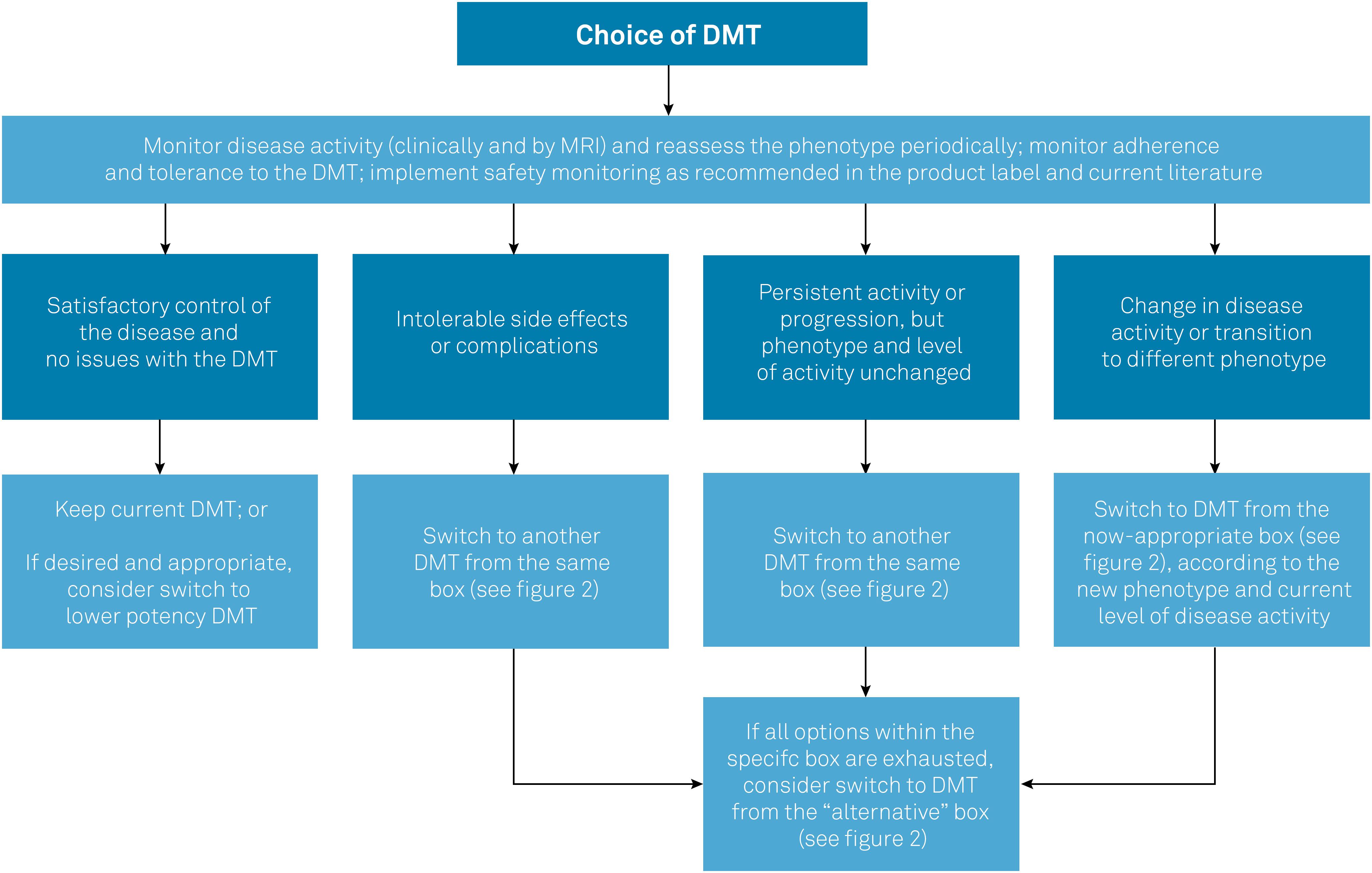
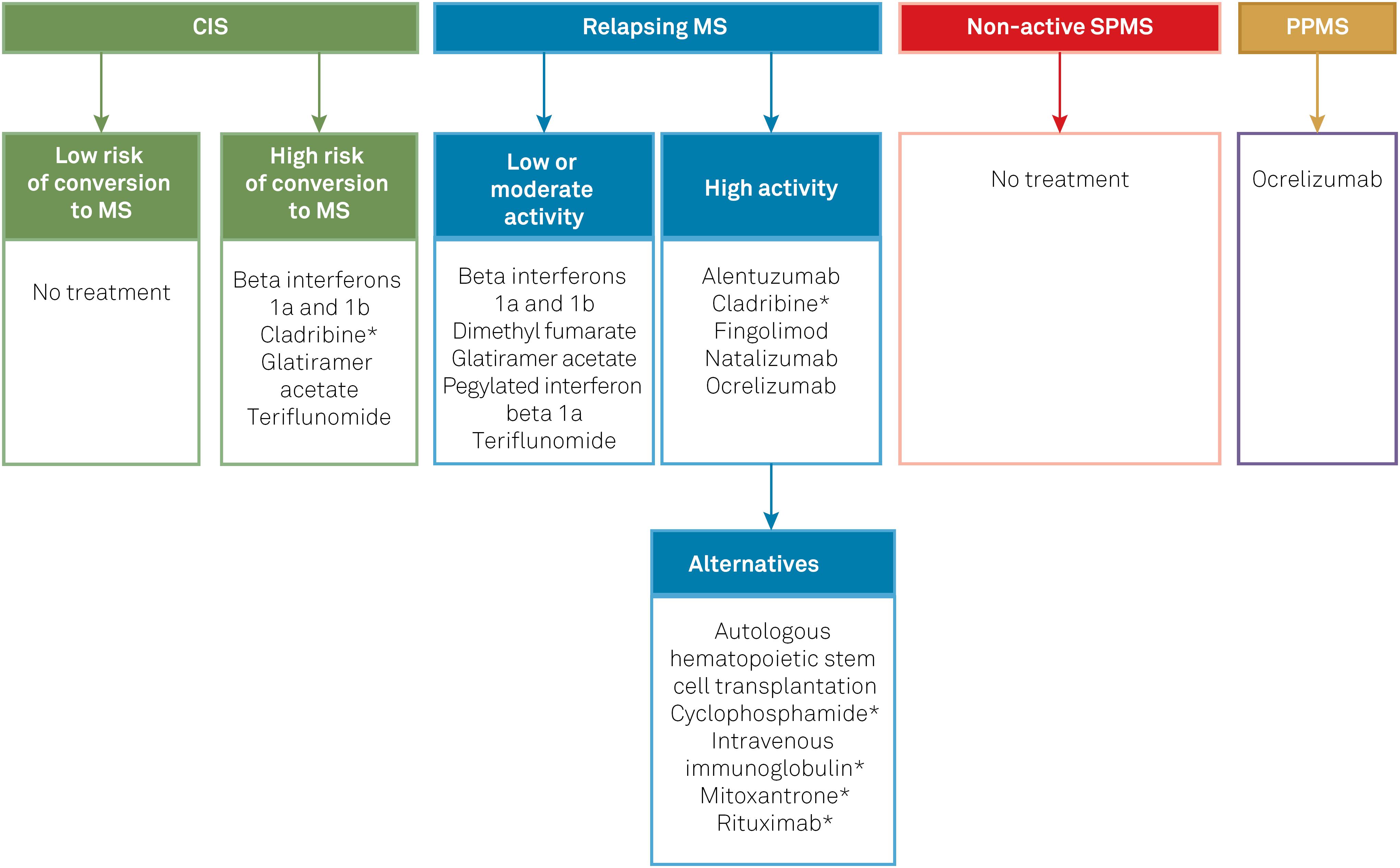
Table 2 compiles the DMTs included in the protocol and summarizes their posology, clinical efficacy parameters in each phenotype (based on the results of key clinical trials that support their use in MS), and approval status. Figure 1 summarizes the general recommendations of the Brazilian Consensus for the Treatment of MS with regard to when and how to switch DMTs in different contexts, such as lack of efficacy or safety concerns. Figure 2 lists the DMTs that have been included in the Consensus for different MS phenotypes, according to the risk of conversion from CIS to MS, as well as the level of disease activity in RRMS and SPMS.
General principles guiding the management of DMTs in MS.
DMT: disease-modifying therapy; MS: multiple sclerosis.
DMTs included in the Brazilian Consensus for the Treatment of MS. The classification of MS guiding the choice of DMT in this flowchart is based on the combined assessment of MS phenotype, level of disease activity, and factors of poorer prognosis (if present).
*DMTs currently not approved for MS by the Brazilian Health Regulatory Agency (Agência Nacional de Vigilância Sanitária, ANVISA). CIS: clinically isolated syndrome; DMT: disease-modifying therapy; MS: multiple sclerosis; PPMS: primary progressive multiple sclerosis; SPMS: secondary progressive multiple sclerosis.
The first set of DMTs presented for each group in Figure 2 represents the most appropriate initial choices. In the event of intolerance, poor adhesion and/or serious adverse effects, DMTs could be switched within the same groups. On the other hand, in the event of therapeutic failure, patients should be reassessed in relation to phenotype and disease activity, and DMTs should be managed accordingly, which may involve switching to higher efficacy drugs. For example, upon therapeutic failure, a patient with CIS will nearly always have met the criteria for RRMS, and a patient with low disease activity may have evolved to high disease activity. Conversely, if the management of highly active disease with potent DMTs has achieved a satisfactory response and stability for several years, it would be acceptable (though not mandatory) to consider switching to a lower potency DMT. Finally, if a patient with relapsing MS fails to achieve satisfactory responses (or presents intolerance or safety concerns) with multiple DMTs from the first group, a switch to DMTs of the second (alternative) group shown in Figure 2 should be considered.
CIS
In patients with CIS, the main therapeutic goal is to prevent (or at least delay) the conversion to MS, i.e., the development of further clinical relapses or new MRI lesions that may lead to fulfillment of the McDonald criteria for RRMS. It is likely that all MS DMTs may be effective in doing so; however, in many CIS cases there remains some uncertainty regarding the likelihood of evolving to MS, thus it seems reasonable to start DMTs only in patients with high-risk CIS, as well as to choose safer drugs.
Up to the present time, efficacy in CIS has been demonstrated with the beta interferons, cladribine, glatiramer acetate, and teriflunomide6565. Jacobs LD, Beck RW, Simon JH, Kinkel RP, Brownscheidle CM, Murray TJ et al. Intramuscular interferon beta-1a therapy initiated during a first demyelinating event in multiple sclerosis. N Engl J Med. 2000 Sep;343(13):898-904. https://doi.org/10.1056/NEJM200009283431301
https://doi.org/10.1056/NEJM200009283431...
66. Comi G, Filippi M, Barkhof F, Durelli L, Edan G, Fernández O, et al. Effect of early interferon treatment on conversion to definite multiple sclerosis: a randomised study. Lancet. 2001 May;357(9268):1576-82. https://doi.org/10.1016/S0140-6736(00)04725-5
https://doi.org/10.1016/S0140-6736(00)04...
67. Kappos L, Polman CH, Freedman MS, Edan G, Hartung HP, Miller DH et al. Treatment with interferon beta-1b delays conversion to clinically definite and McDonald MS in patients with clinically isolated syndromes. Neurology. 2006 Oct;67(7):1242-9. https://doi.org/10.1212/01.wnl.0000237641.33768.8d
https://doi.org/10.1212/01.wnl.000023764...
68. Comi G, Martinelli V, Rodegher M, Moiola L, Bajenaru O, Carra A et al. Effect of glatiramer acetate on conversion to clinically definite multiple sclerosis in patients with clinically isolated syndrome (PreCISe study): a randomised, double-blind, placebo-controlled trial. Lancet. 2009 Oct;374(9700):1503-11. https://doi.org/10.1016/S0140-6736(09)61259-9
https://doi.org/10.1016/S0140-6736(09)61...
69. Comi G, De Stefano N, Freedman MS, Barkhof F, Polman CH, Uitdehaag BM et al. Comparison of two dosing frequencies of subcutaneous interferon beta-1a in patients with a first clinical demyelinating event suggestive of multiple sclerosis (REFLEX): a phase 3 randomised controlled trial. Lancet Neurol. 2012 Jan;11(1):33-41. https://doi.org/10.1016/S1474-4422(11)70262-9
https://doi.org/10.1016/S1474-4422(11)70...
-7070. Miller AE, Wolinsky JS, Kappos L, Comi G, Freedman MS, Olsson TP. Oral teriflunomide for patients with a first clinical episode suggestive of multiple sclerosis (TOPIC): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2014 Oct;13(10):977-86. https://doi.org/10.1016/S1474-4422(14)70191-7
https://doi.org/10.1016/S1474-4422(14)70...
,8282. Leist TP, Comi G, Cree BA, Coyle PK, Freedman MS, Hartung HP et al. Effect of oral cladribine on time to conversion to clinically definite multiple sclerosis in patients with a first demyelinating event (ORACLE MS): a phase 3 randomised trial. Lancet Neurol. 2014 Mar;13(3):257-67. https://doi.org/10.1016/S1474-4422(14)70005-5
https://doi.org/10.1016/S1474-4422(14)70...
. Since no direct comparison between these is available, any of these drugs are deemed appropriate for the treatment of high-risk CIS.
Relapsing MS
For the purpose of this guideline, we define relapsing MS as any case of RRMS, irrespective of disease activity, as well as SPMS with persistence of superimposed relapses or radiologic signs of activity. This approach reflects the panel's view that, in patients transitioning from the relapsing-remitting form to the secondary progressive form, the presence of relapses may suggest a residual “relapsing” pathophysiological component, potentially treatable with DMTs, which may persist for years. This view is further supported by recent clinical trials including both RRMS and SPMS with relapses, grouped under the term “active relapsing MS”8383. Montalban X, Cohen B, Leist T, et al. Efficacy of Cladribine Tablets as add-on to IFN-beta therapy in patients with active relapsing MS: final results from the Phase II ONWARD Study (P3.029). Neurology. 2016;86(16 Supplement)., as well as the recent approval by health authorities of some DMTs for “relapsing forms of MS”. The panel recognizes that many of the pivotal trials for DMTs have included only RRMS patients, with the efficacy of such drugs for SPMS being less clear. On the other hand, it also acknowledges that the distinction between RRMS and SPMS in clinical practice is often challenging and may take months or years until the presence of ongoing disease progression (and thus SPMS) is clearly established, particularly if there remain superimposed relapses.
The main goals in patients with relapsing MS are to reduce the annualized relapse rate and appearance of new or enlarging MRI lesions, and consequently the accumulation of disability. Identifying those with high disease activity (or at higher risk of evolving to a more aggressive form of the disease) is of paramount importance to guide the choice of DMT.
If there are no particular concerns regarding high levels of disease activity, it would seem reasonable to start treatment with interferon beta-1a8484. Jacobs LD, Cookfair DL, Rudick RA, Herndon RM, Richert JR, Salazar AM, et al.; The Multiple Sclerosis Collaborative Research Group (MSCRG). Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. Ann Neurol. 1996 Mar;39(3):285-94. https://doi.org/10.1002/ana.410390304
https://doi.org/10.1002/ana.410390304...
,8585. PRISMS (Prevention of Relapses and Disability by Interferon β-1a Subcutaneously in Multiple Sclerosis) Study Group. Randomised double-blind placebo-controlled study of interferon β-1a in relapsing/remitting multiple sclerosis. Lancet. 1998 Nov;352(9139):1498-504. https://doi.org/10.1016/S0140-6736(98)03334-0
https://doi.org/10.1016/S0140-6736(98)03...
, interferon beta-1b8686. The IFNB Multiple Sclerosis Study Group. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. Neurology. 1993 Apr;43(4):655-61. https://doi.org/10.1212/WNL.43.4.655
https://doi.org/10.1212/WNL.43.4.655...
, glatiramer acetate8787. Johnson KP, Brooks BR, Cohen JA, Ford CC, Goldstein J, Lisak RP et al; The Copolymer 1 Multiple Sclerosis Study Group. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. Neurology. 1995 Jul;45(7):1268-76. https://doi.org/10.1212/WNL.45.7.1268
https://doi.org/10.1212/WNL.45.7.1268...
,8888. Khan O, Rieckmann P, Boyko A, Selmaj K, Zivadinov R. Three times weekly glatiramer acetate in relapsing-remitting multiple sclerosis. Ann Neurol. 2013 Jun;73(6):705-13. https://doi.org/10.1002/ana.23938
https://doi.org/10.1002/ana.23938...
, pegylated interferon beta-1a8989. Calabresi PA, Kieseier BC, Arnold DL, Balcer LJ, Boyko A, Pelletier J et al. Pegylated interferon β-1a for relapsing-remitting multiple sclerosis (ADVANCE): a randomised, phase 3, double-blind study. Lancet Neurol. 2014 Jul;13(7):657-65. https://doi.org/10.1016/S1474-4422(14)70068-7
https://doi.org/10.1016/S1474-4422(14)70...
, dimethyl fumarate9090. Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012 Sep;367(12):1098-107. https://doi.org/10.1056/NEJMoa1114287
https://doi.org/10.1056/NEJMoa1114287...
,9191. Fox RJ, Miller DH, Phillips JT, Hutchinson M, Havrdova E, Kita M et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012 Sep;367(12):1087-97. https://doi.org/10.1056/NEJMoa1206328
https://doi.org/10.1056/NEJMoa1206328...
, or teriflunomide9292. O’Connor P, Wolinsky JS, Confavreux C, Comi G, Kappos L, Olsson TP et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011 Oct;365(14):1293-303. https://doi.org/10.1056/NEJMoa1014656
https://doi.org/10.1056/NEJMoa1014656...
,9393. Confavreux C, O’Connor P, Comi G, Freedman MS, Miller AE, Olsson T, et al. Oral teriflunomide for patients with relapsing multiple sclerosis (TOWER): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2014 Mar;13(3):247-56. https://doi.org/10.1016/S1474-4422(13)70308-9
https://doi.org/10.1016/S1474-4422(13)70...
, which usually have a good safety profile and are often more easily available (including in the Brazilian public health system). Overall, these drugs are associated with a moderate reduction in the annualized relapse rate in comparison to placebo (around 30%; except for dimethyl fumarate with an efficacy of around 50%).
For patients meeting the criteria for highly active relapsing MS, or presenting factors associated with poorer prognosis, the treating neurologist should consider drugs of higher potency (associated with a reduction greater than 50% in the annualized relapse rate, usually compared with placebo), whose efficacy has been well established by phase 3 trials. This group comprises alemtuzumab9494. Cohen JA, Coles AJ, Arnold DL, Confavreux C, Fox EJ, Hartung HP et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. 2012 Nov;380(9856):1819-28. https://doi.org/10.1016/S0140-6736(12)61769-3
https://doi.org/10.1016/S0140-6736(12)61...
,9595. Coles AJ, Twyman CL, Arnold DL, Cohen JA, Confavreux C, Fox EJ et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet. 2012 Nov;380(9856):1829-39. https://doi.org/10.1016/S0140-6736(12)61768-1
https://doi.org/10.1016/S0140-6736(12)61...
, cladribine9696. Giovannoni G, Comi G, Cook S, Rammohan K, Rieckmann P, Soelberg Sørensen P et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. 2010 Feb;362(5):416-26. https://doi.org/10.1056/NEJMoa0902533
https://doi.org/10.1056/NEJMoa0902533...
, fingolimod9797. Kappos L, Radue EW, O’Connor P, Polman C, Hohlfeld R, Calabresi P et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010 Feb;362(5):387-401. https://doi.org/10.1056/NEJMoa0909494
https://doi.org/10.1056/NEJMoa0909494...
,9898. Calabresi PA, Radue EW, Goodin D, Jeffery D, Rammohan KW, Reder AT et al. Safety and efficacy of fingolimod in patients with relapsing-remitting multiple sclerosis (FREEDOMS II): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2014 Jun;13(6):545-56. https://doi.org/10.1016/S1474-4422(14)70049-3
https://doi.org/10.1016/S1474-4422(14)70...
, natalizumab9999. Polman CH, O’Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006 Mar;354(9):899-910. https://doi.org/10.1056/NEJMoa044397
https://doi.org/10.1056/NEJMoa044397...
, and ocrelizumab100100. Hauser SL, Bar-Or A, Comi G, Giovannoni G, Hartung HP, Hemmer B et al. Ocrelizumab versus Interferon Beta-1a in relapsing multiple sclerosis. N Engl J Med. 2017 Jan;376(3):221-34. https://doi.org/10.1056/NEJMoa1601277
https://doi.org/10.1056/NEJMoa1601277...
. These drugs are usually associated with more serious safety concerns, including opportunistic infections or secondary autoimmune diseases, to cite a few. Nevertheless, the need to control aggressive (or potentially aggressive) MS before irreversible disability accumulates often justifies the use of such drugs. At the time of the consensus panel meeting, this group of drugs also included daclizumab101101. Gold R, Giovannoni G, Selmaj K, Havrdova E, Montalban X, Radue EW et al. Daclizumab high-yield process in relapsing-remitting multiple sclerosis (SELECT): a randomised, double-blind, placebo-controlled trial. Lancet. 2013 Jun;381(9884):2167-75. https://doi.org/10.1016/S0140-6736(12)62190-4
https://doi.org/10.1016/S0140-6736(12)62...
,102102. Kappos L, Wiendl H, Selmaj K, Arnold DL, Havrdova E, Boyko A et al. Daclizumab HYP versus Interferon Beta-1a in Relapsing Multiple Sclerosis. N Engl J Med. 2015 Oct;373(15):1418-28. https://doi.org/10.1056/NEJMoa1501481
https://doi.org/10.1056/NEJMoa1501481...
, but it has since been withdrawn from the market (and removed from this guideline) due to recently detected safety issues103103. European Medicines Agency. EMA urgently reviewing multiple sclerosis medicine Zinbryta following cases of inflammatory brain disorders. 2018 [accessed on 2018 Mar 14] Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Press_release/2018/03/WC500244890.pdf
http://www.ema.europa.eu/docs/en_GB/docu...
,104104. Biogen. Biogen and AbbVie Announce the Voluntary Worldwide Withdrawal of Marketing Authorizations for ZINBRYTA® (daclizumab) for Relapsing Multiple Sclerosis. 2018 [accessed on 2018 Mar 14]. Available from: http://media.biogen.com/press-release/autoimmune-diseases/biogenand-abbvie-announce-voluntaryworldwide-withdrawal-marketi
http://media.biogen.com/press-release/au...
.
A subset of highly active relapsing MS patients may persist with breakthrough disease despite treatment with several drugs from the high potency group, characterizing refractory MS. In such cases, subsequent therapy is largely empiric and may include DMTs like cyclophosphamide105105. Killian JM, Bressler RB, Armstrong RM, Huston DP. Controlled pilot trial of monthly intravenous cyclophosphamide in multiple sclerosis. Arch Neurol. 1988 Jan;45(1):27-30. https://doi.org/10.1001/archneur.1988.00520250033014
https://doi.org/10.1001/archneur.1988.00...
, intravenous immunoglobulin106106. Fazekas F, Deisenhammer F, Strasser-Fuchs S, Nahler G, Mamoli B. Randomised placebo-controlled trial of monthly intravenous immunoglobulin therapy in relapsing-remitting multiple sclerosis. Lancet. 1997 Mar;349(9052):589-93. https://doi.org/10.1016/S0140-6736(96)09377-4
https://doi.org/10.1016/S0140-6736(96)09...
, mitoxantrone107107. Hartung HP, Gonsette R, König N, Kwiecinski H, Guseo A, Morrissey SP et al. Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet. 2002 Dec;360(9350):2018-25. https://doi.org/10.1016/S0140-6736(02)12023-X
https://doi.org/10.1016/S0140-6736(02)12...
, rituximab108108. Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008 Feb;358(7):676-88. https://doi.org/10.1056/NEJMoa0706383
https://doi.org/10.1056/NEJMoa0706383...
, and autologous hematopoietic stem cell transplantation109109. Mancardi GL, Sormani MP, Gualandi F, Saiz A, Carreras E, Merelli E et al. Autologous hematopoietic stem cell transplantation in multiple sclerosis: a phase II trial. Neurology. 2015 Mar;84(10):981-8. https://doi.org/10.1212/WNL.0000000000001329
https://doi.org/10.1212/WNL.000000000000...
. These DMTs have had their efficacy suggested by phase 2 trials only (except for mitoxantrone, which was investigated in a phase 3 trial) and are often associated with serious adverse effects, including infertility, neoplasms, or cardiotoxicity). In this group, as an alternative to the aforementioned drugs, experimental DMTs may also be considered, as long as they are offered in a research setting, under appropriate regulatory and ethical approval.
Non-active SPMS
There remains a lack of substantial evidence supporting the benefit of any particular DMT for patients with clearly-established SPMS that no longer presents relapses. Not prescribing a DMT is an acceptable choice in this group. If a DMT is considered (e.g., if there is uncertainty regarding the secondary progressive phenotype in a particular case), the off-label nature of this approach should be clearly discussed with the patient. In this context, interferon beta-1b could be preferred, based on one positive phase 3 trial in SPMS110110. European Study Group on Interferon β-1b in Secondary Progressive MS. Placebo-controlled multicentre randomised trial of interferon beta-1b in treatment of secondary progressive multiple sclerosis. Lancet. 1998;352(9139):1491-7. https://doi.org/10.1016/S0140-6736(98)10039-9
https://doi.org/10.1016/S0140-6736(98)10...
. In cases of rapidly-progressive disease, there is some evidence supporting the use of DMTs such as cyclophosphamide, mitoxantrone, and autologous hematopoietic stem cell transplantation, but these would also represent off-label treatments.
A phase 3 trial of siponimod in patients with SPMS has recently been published, showing modest efficacy in reducing the progression of disability111111. Kappos L, Bar-Or A, Cree BA, Fox RJ, Giovannoni G, Gold R et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study. Lancet. 2018 Mar;391(10127):1263-73. https://doi.org/10.1016/S0140-6736(18)30475-6
https://doi.org/10.1016/S0140-6736(18)30...
. However, evaluation and approval by health authorities are still pending; therefore, siponimod has not been included in this guideline.
PPMS
The current main therapeutic goal in PPMS is to delay the progression of disability. The only drug approved for this purpose is ocrelizumab, with a modest benefit112112. Montalban X, Hauser SL, Kappos L, Arnold DL, Bar-Or A, Comi G et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N Engl J Med. 2017 Jan;376(3):209-20. https://doi.org/10.1056/NEJMoa1606468
https://doi.org/10.1056/NEJMoa1606468...
. Hence, the consensus panel understands that ocrelizumab should be the treatment of choice for patients with PPMS, after consideration of the expected benefits and potential risks on a case-by-case basis.
Special situations
Pediatric MS: To date, the Food and Drug Administration, European Medicines Agency, and Brazilian Health Regulatory Agency have not approved most DMTs for patients younger than 18 years. This reflects the lack of phase 3 randomized clinical trials assessing MS drugs in this age range. Meanwhile, the treatment of pediatric MS remains largely off-label, based on evidence from observational studies. Beta interferons and glatiramer acetate are seemingly safe and effective in this population and should be preferred as first-line therapy, at least until growing evidence and regulatory clearance becomes available for the newer DMTs113113. Ghezzi A, Amato MP, Makhani N, Shreiner T, Gärtner J, Tenembaum S. Pediatric multiple sclerosis: conventional first-line treatment and general management. Neurology. 2016 Aug;87(9 Suppl 2):S97-102. https://doi.org/10.1212/WNL.0000000000002823
https://doi.org/10.1212/WNL.000000000000...
. Attention should be paid to the specific dose initiation and titration schemes recommended for younger children. Upon therapeutic failure, escalation to drugs other than beta interferons and glatiramer acetate may be considered with caution. Positive results of fingolimod in a phase 3 trial in pediatric MS were recently reported in a conference114114. Chitnis T. PARADIGMS: a randomised double-blind study of fingolimod versus interferon β-1a in paediatric multiple sclerosis. ECTRIMS Online Library. 2017 Oct 28;202640., but publication of the full study was still pending during the preparation of this guideline and, therefore, the panel could not make a specific recommendation on whether fingolimod should be preferred over other DMTs in this age group.
Pregnancy and breastfeeding: As for pediatric MS, there is only scarce evidence guiding the decision on whether or not to use DMTs (and how to choose them) during pregnancy and breastfeeding. Treating neurologists should discuss this on a case-by-case basis and, when the use of a DMT is judged necessary, glatiramer acetate should usually be preferred over others DMTs.
Radiologically isolated syndrome: At this point, the consensus panel understands there is insufficient evidence to recommend the use of DMTs for patients with radiologically isolated syndrome, i.e., those with the incidental finding of MS-typical lesions on MRI who lack evidence of symptoms or signs compatible with MS.
Tumefactive demyelination syndromes: This is a group of heterogeneous conditions whose relationship to MS is still to be fully elucidated. The consensus panel recommends that only patients fulfilling the diagnostic criteria for MS should be eligible for DMTs. At this point, there is insufficient evidence to guide the choice of specific DMTs. Beta interferons and glatiramer acetate are the most commonly used drugs in most case series, thus could be regarded as preferred115115. Hardy TA, Reddel SW, Barnett MH, Palace J, Lucchinetti CF, Weinshenker BG. Atypical inflammatory demyelinating syndromes of the CNS. Lancet Neurol. 2016 Aug;15(9):967-81. https://doi.org/10.1016/S1474-4422(16)30043-6
https://doi.org/10.1016/S1474-4422(16)30...
,116116. Algahtani H, Shirah B, Alassiri A. Tumefactive demyelinating lesions: A comprehensive review. Mult Scler Relat Disord. 2017 May;14:72-9. https://doi.org/10.1016/j.msard.2017.04.003
https://doi.org/10.1016/j.msard.2017.04....
. On the other hand, there are reports of a controversial association between fingolimod and the occurrence of tumefactive lesions (as well as unfavorable outcomes in this group of patients); thus the panel recommends particular caution when prescribing this drug to patients with tumefactive demyelinating syndromes116116. Algahtani H, Shirah B, Alassiri A. Tumefactive demyelinating lesions: A comprehensive review. Mult Scler Relat Disord. 2017 May;14:72-9. https://doi.org/10.1016/j.msard.2017.04.003
https://doi.org/10.1016/j.msard.2017.04....
,117117. Faissner S, Hoepner R, Lukas C, Chan A, Gold R, Ellrichmann G. Tumefactive multiple sclerosis lesions in two patients after cessation of fingolimod treatment. Ther Adv Neurol Disorder. 2015 Sep;8(5):233-8. https://doi.org/10.1177/1756285615594575
https://doi.org/10.1177/1756285615594575...
.
Treatment interruption: There is no clear evidence to support a consensus recommendation on whether or when to consider interruption of DMTs in patients with advanced MS or in those with the disputable entity known as “benign MS”118118. Correale J, Ysrraelit MC, Fiol MP. Benign multiple sclerosis: does it exist? Curr Neurol Neurosci Rep. 2012 Oct;12(5):601-9. https://doi.org/10.1007/s11910-012-0292-5
https://doi.org/10.1007/s11910-012-0292-...
. The treating neurologist should discuss this with patients and their family on an individual basis.
For further guidance on the management of special situations, we refer the reader to the recommendations of the Brazilian Academy of Neurology.
Management of relapses
Figure 3 summarizes the recommendations for the management of relapses. The initial choice is usually intravenous methylprednisolone, 1 g/day, infused over at least 30 minutes, for three to five consecutive days119119. Berkovich R. Treatment of acute relapses in multiple sclerosis. Neurotherapeutics. 2013 Jan;10(1):97-105. https://doi.org/10.1007/s13311-012-0160-7
https://doi.org/10.1007/s13311-012-0160-...
. If intravenous methylprednisolone fails to promote satisfactory recovery, an additional course may be given, with the same or higher dose (1–2 g/day), again for three to five days. High-dose oral methylprednisolone (1 g/day) or dexamethasone (150 mg/day), for three to five consecutive days, may be considered in the outpatient setting120120. Le Page E, Veillard D, Laplaud DA, Hamonic S, Wardi R, Lebrun C, et al; COPOUSEP investigators; West Network for Excellence in Neuroscience. Oral versus intravenous high-dose methylprednisolone for treatment of relapses in patients with multiple sclerosis (COPOUSEP): a randomised, controlled, double-blind, non-inferiority trial. Lancet. 2015 Sep;386(9997):974-81. https://doi.org/10.1016/S0140-6736(15)61137-0
https://doi.org/10.1016/S0140-6736(15)61...
,121121. National Clinical Advisory Board, National Multiple Sclerosis Society. Recommendations Regarding Corticosteroids in the Management of Multiple Sclerosis. US Neurol. 2008;4(1):22. https://doi.org/10.17925/USN.2008.04.01.22.
https://doi.org/10.17925/USN.2008.04.01....
.
Patients who fail to respond (or have contraindications) to corticosteroids may be eligible for therapeutic plasma exchange (five to seven sessions scheduled every other day) or intravenous human immunoglobulin G (usually 1 g/kg/day for two days, or 0.4 g/kg/day for five days)119119. Berkovich R. Treatment of acute relapses in multiple sclerosis. Neurotherapeutics. 2013 Jan;10(1):97-105. https://doi.org/10.1007/s13311-012-0160-7
https://doi.org/10.1007/s13311-012-0160-...
. It is noted that plasma exchange is likely more effective than intravenous immunoglobulin; however, it is not easily available in most centers in Brazil.
Symptomatic treatment and rehabilitation
Although a comprehensive review of symptomatic treatment and rehabilitation is beyond the scope of this guideline, the panel acknowledges the paramount importance of such aspects in the treatment of MS and highlights the need to proactively consider these resources and offer them to patients in need, whenever possible. We recommend accessing the Brazilian Academy of Neurology recommendations to review these treatments.
Symptoms and disability in the context of MS, such as neurogenic bladder and bowel dysfunction, neuropsychological conditions, sleep disorders, neuropathic pain and spasticity, are usually managed similarly as in the context of other neurological conditions. Nonetheless, it is worth bearing in mind that some MS-specific symptomatic drugs are available, namely tetrahydrocannabinol + cannabidiol for refractory spasticity and fampridine for gait dysfunction122122. Toosy A, Ciccarelli O, Thompson A. Symptomatic treatment and management of multiple sclerosis. In: Goodin DS, editor. Handbook of Clinical Neurology. [S. l.}: Elsevier; 2014. p. 513-62..
Vitamin D3 supplementation
Vitamin D3 (cholecalciferol) is a fat-soluble hormone with numerous physiologic responses, including immune regulation123123. Shoemaker TJ, Mowry EM. A review of vitamin D supplementation as disease-modifying therapy. Mult Scler. 2018 Jan;24(1):6-11. https://doi.org/10.1177/1352458517738131
https://doi.org/10.1177/1352458517738131...
. Preliminary studies of vitamin D3 as an add-on therapy in MS have shown promising results, but none have yet provided significant evidence of a clinically meaningful effect on disease activity. Until the results of ongoing larger randomized controlled trials help elucidate the role of vitamin D supplementation in MS, the panel recommends to check the serum level of 25-OH-vitamin D3 in all patients with MS, and to consider supplementation on a case-by-case basis, in physiologic doses, i.e., sufficient to achieve serum levels between 40 and 100 ng/mL (100–150 nmol/L)124124. Brum DG, Comini-Frota ER, Vasconcelos CC, Dias-Tosta E. Supplementation and therapeutic use of vitamin D in patients with multiple sclerosis: consensus of the Scientific Department of Neuroimmunology of the Brazilian Academy of Neurology. Arq Neuropsiquiatr. 2014 Feb;72(2):152-6. https://doi.org/10.1590/0004-282X20130252
https://doi.org/10.1590/0004-282X2013025...
. After starting supplementation, serum levels should be re-checked after three months, to further tailor the dose, as MS patients may have a less robust response to supplementation compared to those without comorbidities123123. Shoemaker TJ, Mowry EM. A review of vitamin D supplementation as disease-modifying therapy. Mult Scler. 2018 Jan;24(1):6-11. https://doi.org/10.1177/1352458517738131
https://doi.org/10.1177/1352458517738131...
. The panel herein emphasizes that supraphysiological doses of vitamin D3, i.e., those leading to a serum level higher than 100 ng/mL, are not indicated, neither as an add-on nor as monotherapy in MS, due to potential systemic toxicity124124. Brum DG, Comini-Frota ER, Vasconcelos CC, Dias-Tosta E. Supplementation and therapeutic use of vitamin D in patients with multiple sclerosis: consensus of the Scientific Department of Neuroimmunology of the Brazilian Academy of Neurology. Arq Neuropsiquiatr. 2014 Feb;72(2):152-6. https://doi.org/10.1590/0004-282X20130252
https://doi.org/10.1590/0004-282X2013025...
.
FINAL REMARKS
This guideline provides an overview of the principles and general directions that should guide therapeutic decisions in MS. Overall, we highlight the need to start DMTs early in the course of MS, always conforming properly to the clinical phenotype, and changing them promptly in the case of treatment failure to prevent further relapses and accumulation of disability. We advocate for flexibility in the management of DMTs, so it can be tailored to individual circumstances (especially disease activity), rather than adhering to a rigid sequence of treatment lines.
We acknowledge that these recommendations are unable to cover all the situations that neurologists may face in the management of MS. Furthermore, the need for periodic revisions is anticipated, as further understanding of the disease and new medications become more rapidly available. Therefore, we encourage treating physicians to always review additional information (especially from each drug's label), and to examine all the relevant factors on a case-by-case basis.
References
-
1Multiple Sclerosis International Federation. Atlas of MS 2013: mapping multiple sclerosis around the world. London: Multiple Sclerosis International Federation; 2013.
-
2Comi G, Radaelli M, Soelberg Sørensen P. Evolving concepts in the treatment of relapsing multiple sclerosis. Lancet. 2017 Apr;389(10076):1347-56. https://doi.org/10.1016/S0140-6736(16)32388-1
» https://doi.org/10.1016/S0140-6736(16)32388-1 -
3Ministério da Saúde (Brasil). Secretaria de Atenção à Saúde. Secretaria de Ciência, Tecnologia e Insumos Estratégicos. Portaria Conjunta Nº 10, de 2 de abril de 2018. Aprova o protocolo clínico e diretrizes trapêuticas da esclerose múltipla. Brasília, DF, 2018 [accessed 2018 May 8] Available from: http://portalarquivos2.saude.gov.br/images/pdf/2018/abril/09/PORTARIA-CONJUNTA-N-10-ESCLEROSE-MULTIPLA.09.04.2018.pdf
» http://portalarquivos2.saude.gov.br/images/pdf/2018/abril/09/PORTARIA-CONJUNTA-N-10-ESCLEROSE-MULTIPLA.09.04.2018.pdf -
4Tilbery CP, Moreira MA, Mendes MF, Lana-Peixoto MA. [Recommendations for the use of immunomodulatory drugs in multiple sclerosis: the BCTRIMS consensus]. Arq Neuropsiquiatr. 2000 Sep;58 3A:769-76. Portuguese. https://doi.org/10.1590/S0004-282X2000000400030
» https://doi.org/10.1590/S0004-282X2000000400030 -
5Callegaro D, Lana-Peixoto MA, Moreira MA, Marchiori PE, Bacheschi LA, Arruda WO et al. [The BCTRIMS Expanded Consensus on treatment of multiple sclerosis: I. The evidences for the use of immunosuppressive agents, plasma exchange and autologous hematopoietic stem cell transplantation]. Arq Neuropsiquiatr. 2002 Sep;60 3-B:869-74. Portuguese. https://doi.org/10.1590/S0004-282X2002000500035
» https://doi.org/10.1590/S0004-282X2002000500035 -
6Moreira MA, Lana-Peixoto MA, Callegaro D, Haussen SR, Gama PD, Gabbai AA et al. [The BCTRIMS expanded consensus on treatment of multiple sclerosis: II. The evidences for the use of glucocorticoids and immunomodulatory treatments]. Arq Neuropsiquiatr. 2002 Sep;60 3-B:875-80. Portuguese. https://doi.org/10.1590/S0004-282X2002000500036
» https://doi.org/10.1590/S0004-282X2002000500036 -
7Lana-Peixoto MA, Callegaro D, Moreira MA, Campos GB, Marchiori PE, Gabbai AA et al. [The BCTRIMS Expanded Consensus on treatment of multiple sclerosis: III. Evidence and recommendation-based guidelines]. Arq Neuropsiquiatr. 2002 Sep;60 3-B:881-6. Portuguese. https://doi.org/10.1590/S0004-282X2002000500037
» https://doi.org/10.1590/S0004-282X2002000500037 -
8Machado S. Recomendações Esclerose Múltipla - Academia Brasileira de Neurologia. São Paulo: Omnifarma; 2012.
-
9Comini-Frota ER, Vasconcelos CCF, Mendes MF. Guideline for multiple sclerosis treatment in Brazil: Consensus from the Neuroimmunology Scientific Department of the Brazilian Academy of Neurology. Arq Neuropsiquiatr. 2017 Jan;75(1):57-65. https://doi.org/10.1590/0004-282X20160185
» https://doi.org/10.1590/0004-282X20160185 -
10Freedman MS, Selchen D, Arnold DL, Prat A, Banwell B, Yeung M et al. Treatment optimization in MS: Canadian MS Working Group updated recommendations. Can J Neurol Sci. 2013 May;40(3):307-23. https://doi.org/10.1017/S0317167100014244
» https://doi.org/10.1017/S0317167100014244 -
11Scolding N, Barnes D, Cader S, Chataway J, Chaudhuri A, Coles A et al. Association of British Neurologists: revised (2015) guidelines for prescribing disease-modifying treatments in multiple sclerosis. Pract Neurol. 2015 Aug;15(4):273-9. https://doi.org/10.1136/practneurol-2015-001139
» https://doi.org/10.1136/practneurol-2015-001139 -
12Montalban X, Gold R, Thompson AJ, Otero-Romero S, Amato MP, Chandraratna D et al. ECTRIMS/EAN guideline on the pharmacological treatment of people with multiple sclerosis. Eur J Neurol. 2018 Feb;25(2):215-37. https://doi.org/10.1111/ene.13536
» https://doi.org/10.1111/ene.13536 -
13Thompson AJ, Banwell BL, Barkhof F, Carrol WM, Coetzee T, Comi G et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018 Feb;17(2):162-73. https://doi.org/10.1016/S1474-4422(17)30470-2
» https://doi.org/10.1016/S1474-4422(17)30470-2 -
14Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sørensen PS, Thompson AJ et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014 Jul;83(3):278-86. https://doi.org/10.1212/WNL.0000000000000560
» https://doi.org/10.1212/WNL.0000000000000560 -
15Tintore M, Rovira À, Río J, Otero-Romero S, Arrambide G, Tur C et al. Defining high, medium and low impact prognostic factors for developing multiple sclerosis. Brain. 2015 Jul;138(Pt 7):1863-74. https://doi.org/10.1093/brain/awv105
» https://doi.org/10.1093/brain/awv105 -
16Fazekas F, Bajenaru O, Berger T, Fabjan TH, Ledinek AH, Jakab G et al. How does fingolimod (Gilenya®) fit in the treatment algorithm for highly active relapsing-remitting multiple sclerosis? Front Neurol. 2013 May;4:10. https://doi.org/10.3389/fneur.2013.00010
» https://doi.org/10.3389/fneur.2013.00010 -
17Devonshire V, Havrdova E, Radue EW, O’Connor P, Zhang-Auberson L, Agoropoulou C et al. Relapse and disability outcomes in patients with multiple sclerosis treated with fingolimod: subgroup analyses of the double-blind, randomised, placebo-controlled FREEDOMS study. Lancet Neurol. 2012 May;11(5):420-8. https://doi.org/10.1016/S1474-4422(12)70056-X
» https://doi.org/10.1016/S1474-4422(12)70056-X -
18Rush CA, MacLean HJ, Freedman MS. Aggressive multiple sclerosis: proposed definition and treatment algorithm. Nat Rev Neurol. 2015 Jul;11(7):379-89. https://doi.org/10.1038/nrneurol.2015.85
» https://doi.org/10.1038/nrneurol.2015.85 -
19Menon S, Shirani A, Zhao Y, Oger J, Traboulsee A, Freedman MS et al. Characterising aggressive multiple sclerosis. J Neurol Neurosurg Psychiatry. 2013 Nov;84(11):1192-8. https://doi.org/10.1136/jnnp-2013-304951
» https://doi.org/10.1136/jnnp-2013-304951 -
20Ingwersen J, Aktas O, Hartung HP. Advances in and algorithms for the treatment of relapsing-remitting multiple sclerosis. Neurotherapeutics. 2016 Jan;13(1):47-57. https://doi.org/10.1007/s13311-015-0412-4
» https://doi.org/10.1007/s13311-015-0412-4 -
21Wingerchuk DM, Carter JL. Multiple sclerosis: current and emerging disease-modifying therapies and treatment strategies. Mayo Clin Proc. 2014 Feb;89(2):225-40. https://doi.org/10.1016/j.mayocp.2013.11.002
» https://doi.org/10.1016/j.mayocp.2013.11.002 -
22Vosoughi R, Freedman MS. Therapy of MS. Clin Neurol Neurosurg. 2010 Jun;112(5):365-85. https://doi.org/10.1016/j.clineuro.2010.03.010
» https://doi.org/10.1016/j.clineuro.2010.03.010 -
23Balabanov P, Haas M, Elferink A, Bakchine S, Broich K. Addressing the regulatory and scientific challenges in multiple sclerosis: a statement from the EU regulators. Mult Scler. 2014 Sep;20(10):1282-7. https://doi.org/10.1177/1352458514546876
» https://doi.org/10.1177/1352458514546876 -
24Sorensen PS. New management algorithms in multiple sclerosis. Curr Opin Neurol. 2014 Jun;27(3):246-59. https://doi.org/10.1097/WCO.0000000000000096
» https://doi.org/10.1097/WCO.0000000000000096 -
25Torkildsen Ø, Myhr KM, Bø L. Disease-modifying treatments for multiple sclerosis - a review of approved medications. Eur J Neurol. 2016 Jan;23 Suppl 1:18-27. https://doi.org/10.1111/ene.12883
» https://doi.org/10.1111/ene.12883 -
26Cross AH, Naismith RT. Established and novel disease-modifying treatments in multiple sclerosis. J Intern Med. 2014 Apr;275(4):350-63. https://doi.org/10.1111/joim.12203
» https://doi.org/10.1111/joim.12203 -
27Carrithers MD. Update on disease-modifying treatments for multiple sclerosis. Clin Ther. 2014 Dec;36(12):1938-45. https://doi.org/10.1016/j.clinthera.2014.08.006
» https://doi.org/10.1016/j.clinthera.2014.08.006 -
28Michel L, Larochelle C, Prat A. Update on treatments in multiple sclerosis. Presse Med. 2015 Apr;44(4 Pt 2):e137-51. https://doi.org/10.1016/j.lpm.2015.02.008
» https://doi.org/10.1016/j.lpm.2015.02.008 -
29Broadley SA, Barnett MH, Boggild M, Brew BJ, Butzkueven H, Heard R et al. Therapeutic approaches to disease modifying therapy for multiple sclerosis in adults: an Australian and New Zealand perspective: part 3 treatment practicalities and recommendations. J Clin Neurosci. 2014 Nov;21(11):1857-65. https://doi.org/10.1016/j.jocn.2014.01.017
» https://doi.org/10.1016/j.jocn.2014.01.017 -
30García-Merino A, Fernández O, Montalbán X, Andrés C, Oreja-Guevar C, Rodríguez-Antigüedad A et al. Documento del Grupo de Consenso de la Sociedad Española de Neurología sobre el uso de medicamentos en esclerosis múltiple. Neurología. 2013;28(6):375-8. https://doi.org/10.1016/j.nrl.2013.01.009
» https://doi.org/10.1016/j.nrl.2013.01.009 -
31Limmroth V. Treatment of relapsing-remitting multiple sclerosis: current and future algorithms. Eur Neurol. 2014;72(s1 Suppl 1):35-8. https://doi.org/10.1159/000367624
» https://doi.org/10.1159/000367624 -
32Quintana FJ, Patel B, Yeste A, Nyirenda M, Kenison J, Rahbari R et al. Epitope spreading as an early pathogenic event in pediatric multiple sclerosis. Neurology. 2014 Dec;83(24):2219-26. https://doi.org/10.1212/WNL.0000000000001066
» https://doi.org/10.1212/WNL.0000000000001066 -
33Hohlfeld R, Dornmair K, Meinl E, Wekerle H. The search for the target antigens of multiple sclerosis, part 2: CD8+ T cells, B cells, and antibodies in the focus of reverse-translational research. Lancet Neurol. 2016 Mar;15(3):317-31. https://doi.org/10.1016/S1474-4422(15)00313-0
» https://doi.org/10.1016/S1474-4422(15)00313-0 -
34Hohlfeld R, Dornmair K, Meinl E, Wekerle H. The search for the target antigens of multiple sclerosis, part 1: autoreactive CD4+ T lymphocytes as pathogenic effectors and therapeutic targets. Lancet Neurol. 2016 Feb;15(2):198-209. https://doi.org/10.1016/S1474-4422(15)00334-8
» https://doi.org/10.1016/S1474-4422(15)00334-8 -
35Leray E, Yaouanq J, Le Page E, Coustans M, Laplaud D, Oger J et al. Evidence for a two-stage disability progression in multiple sclerosis. Brain. 2010 Jul;133(Pt 7):1900-13. https://doi.org/10.1093/brain/awq076
» https://doi.org/10.1093/brain/awq076 -
36Michel L, Touil H, Pikor NB, Gommerman JL, Prat A, Bar-Or A. B Cells in the multiple sclerosis central nervous system: trafficking and contribution to CNS-compartmentalized inflammation. Front Immunol. 2015 Dec;6:636. https://doi.org/10.3389/fimmu.2015.00636
» https://doi.org/10.3389/fimmu.2015.00636 -
37Pikor NB, Prat A, Bar-Or A, Gommerman JL. Meningeal tertiary lymphoid tissues and multiple sclerosis: a gathering place for diverse types of immune cells during CNS autoimmunity. Front Immunol. 2016 Jan;6:657. https://doi.org/10.3389/fimmu.2015.00657
» https://doi.org/10.3389/fimmu.2015.00657 -
38Meinl E, Krumbholz M, Derfuss T, Junker A, Hohlfeld R. Compartmentalization of inflammation in the CNS: a major mechanism driving progressive multiple sclerosis. J Neurol Sci. 2008 Nov;274(1-2):42-4. https://doi.org/10.1016/j.jns.2008.06.032
» https://doi.org/10.1016/j.jns.2008.06.032 -
39Jokubaitis VG, Spelman T, Kalincik T, Lorscheider J, Havrdova E, Horakova D et al. Predictors of long-term disability accrual in relapse-onset multiple sclerosis. Ann Neurol. 2016 Jul;80(1):89-100. https://doi.org/10.1002/ana.24682
» https://doi.org/10.1002/ana.24682 -
40Scalfari A, Neuhaus A, Degenhardt A, Rice GP, Muraro PA, Daumer M et al. The natural history of multiple sclerosis: a geographically based study 10: relapses and long-term disability. Brain. 2010 Jul;133(Pt 7):1914-29. https://doi.org/10.1093/brain/awq118
» https://doi.org/10.1093/brain/awq118 -
41Scalfari A, Neuhaus A, Daumer M, Muraro PA, Ebers GC. Onset of secondary progressive phase and long-term evolution of multiple sclerosis. J Neurol Neurosurg Psychiatry. 2014 Jan;85(1):67-75. https://doi.org/10.1136/jnnp-2012-304333
» https://doi.org/10.1136/jnnp-2012-304333 -
42Myhr KM, Riise T, Vedeler C, Nortvedt MW, Grønning R, Midgard R et al. Disability and prognosis in multiple sclerosis: demographic and clinical variables important for the ability to walk and awarding of disability pension. Mult Scler. 2001 Feb;7(1):59-65. https://doi.org/10.1177/135245850100700110
» https://doi.org/10.1177/135245850100700110 -
43Amato MP, Ponziani G, Bartolozzi ML, Siracusa G. A prospective study on the natural history of multiple sclerosis: clues to the conduct and interpretation of clinical trials. J Neurol Sci. 1999 Oct;168(2):96-106. https://doi.org/10.1016/S0022-510X(99)00143-4
» https://doi.org/10.1016/S0022-510X(99)00143-4 -
44Lublin FD, Baier M, Cutter G. Effect of relapses on development of residual deficit in multiple sclerosis. Neurology. 2003 Dec;61(11):1528-32. https://doi.org/10.1212/01.WNL.0000096175.39831.21
» https://doi.org/10.1212/01.WNL.0000096175.39831.21 -
45Hirst C, Ingram G, Pearson O, Pickersgill T, Scolding N, Robertson N. Contribution of relapses to disability in multiple sclerosis. J Neurol. 2008 Feb;255(2):280-7. https://doi.org/10.1007/s00415-008-0743-8
» https://doi.org/10.1007/s00415-008-0743-8 -
46Vercellino M, Romagnolo A, Mattioda A, Masera S, Piacentino C, Merola A et al. Multiple sclerosis relapses: a multivariable analysis of residual disability determinants. Acta Neurol Scand. 2009 Feb;119(2):126-30. https://doi.org/10.1111/j.1600-0404.2008.01076.x
» https://doi.org/10.1111/j.1600-0404.2008.01076.x -
47Bergamaschi R, Berzuini C, Romani A, Cosi V. Predicting secondary progression in relapsing-remitting multiple sclerosis: a Bayesian analysis. J Neurol Sci. 2001 Aug;189(1-2):13-21. https://doi.org/10.1016/S0022-510X(01)00572-X
» https://doi.org/10.1016/S0022-510X(01)00572-X -
48Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998 Jan;338(5):278-85. https://doi.org/10.1056/NEJM199801293380502
» https://doi.org/10.1056/NEJM199801293380502 -
49Riise T, Grønning M, Fernández O, Lauer K, Midgard R, Minderhoud JM et al. Early prognostic factors for disability in multiple sclerosis, a European multicenter study. Acta Neurol Scand. 1992 Mar;85(3):212-8. https://doi.org/10.1111/j.1600-0404.1992.tb04031.x
» https://doi.org/10.1111/j.1600-0404.1992.tb04031.x -
50Tremlett H, Yinshan Zhao, Devonshire V. Natural history of secondary-progressive multiple sclerosis. Mult Scler. 2008 Apr;14(3):314-24. https://doi.org/10.1177/1352458507084264
» https://doi.org/10.1177/1352458507084264 -
51Scalfari A, Neuhaus A, Daumer M, Ebers GC, Muraro PA. Age and disability accumulation in multiple sclerosis. Neurology. 2011 Sep;77(13):1246-52. https://doi.org/10.1212/WNL.0b013e318230a17d
» https://doi.org/10.1212/WNL.0b013e318230a17d -
52Gholipour T, Healy B, Baruch NF, Weiner HL, Chitnis T. Demographic and clinical characteristics of malignant multiple sclerosis. Neurology. 2011 Jun;76(23):1996-2001. https://doi.org/10.1212/WNL.0b013e31821e559d
» https://doi.org/10.1212/WNL.0b013e31821e559d -
53Cossburn M, Ingram G, Hirst C, Ben-Shlomo Y, Pickersgill TP, Robertson NP. Age at onset as a determinant of presenting phenotype and initial relapse recovery in multiple sclerosis. Mult Scler. 2012 Jan;18(1):45-54. https://doi.org/10.1177/1352458511417479
» https://doi.org/10.1177/1352458511417479 -
54Bermel RA, You X, Foulds P, Hyde R, Simon JH, Fisher E et al. Predictors of long-term outcome in multiple sclerosis patients treated with interferon β. Ann Neurol. 2013 Jan;73(1):95-103. https://doi.org/10.1002/ana.23758
» https://doi.org/10.1002/ana.23758 -
55Río J, Rovira A, Tintoré M, Huerga E, Nos C, Tellez N et al. Relationship between MRI lesion activity and response to IFN-β in relapsing–remitting multiple sclerosis patients. Mult. Scler. J. 2008;14(4):479-84. https://doi.org/10.1177/1352458507085555
» https://doi.org/10.1177/1352458507085555 -
56Losseff NA, Miller DH, Kidd D, Thompson AJ. The predictive value of gadolinium enhancement for long term disability in relapsing-remitting multiple sclerosis: preliminary results. Mult Scler. 2001 Feb;7(1):23-5.
-
57Fisniku LK, Brex PA, Altmann DR, Miszkiel KA, Benton CE, Lanyon R et al. Disability and T2 MRI lesions: a 20-year follow-up of patients with relapse onset of multiple sclerosis. Brain. 2008 Mar;131(Pt 3):808-17. https://doi.org/10.1093/brain/awm329
» https://doi.org/10.1093/brain/awm329 -
58Maghzi AH, Revirajan N, Julian LJ, Spain R, Mowry EM, Liu S et al. Magnetic resonance imaging correlates of clinical outcomes in early multiple sclerosis. Mult Scler Relat Disord. 2014 Nov;3(6):720-7. https://doi.org/10.1016/j.msard.2014.07.003
» https://doi.org/10.1016/j.msard.2014.07.003 -
59Sormani MP, Arnold DL, De Stefano N. Treatment effect on brain atrophy correlates with treatment effect on disability in multiple sclerosis. Ann Neurol. 2014 Jan;75(1):43-9. https://doi.org/10.1002/ana.24018
» https://doi.org/10.1002/ana.24018 -
60De Stefano N, Narayanan S, Francis GS, Arnaoutelis R, Tartaglia MC, Antel JP et al. Evidence of axonal damage in the early stages of multiple sclerosis and its relevance to disability. Arch Neurol. 2001 Jan;58(1):65-70. https://doi.org/10.1001/archneur.58.1.65 PMID:11176938
» https://doi.org/10.1001/archneur.58.1.65 -
61von Gumberz J, Mahmoudi M, Young K, Schippling S, Martin R, Heesen C et al. Short-term MRI measurements as predictors of EDSS progression in relapsing-remitting multiple sclerosis: grey matter atrophy but not lesions are predictive in a real-life setting. PeerJ. 2016 Sep;4:e2442. https://doi.org/10.7717/peerj.2442
» https://doi.org/10.7717/peerj.2442 -
62Kalincik T, Vaneckova M, Tyblova M, Krasensky J, Seidl Z, Havrdova E, et al. Volumetric MRI markers and predictors of disease activity in early multiple sclerosis: a longitudinal cohort study. PLoS One. 2012;7(11):e50101. https://doi.org/10.1371/journal.pone.0050101
» https://doi.org/10.1371/journal.pone.0050101 -
63Rocca MA, Comi G, Filippi M. The role of T1-weighted derived measures of neurodegeneration for assessing disability progression in multiple sclerosis. Front Neurol. 2017 Sep;8:433. https://doi.org/10.3389/fneur.2017.00433
» https://doi.org/10.3389/fneur.2017.00433 -
64Coles AJ, Cox A, Le Page E, Jones J, Trip SA, Deans J et al. The window of therapeutic opportunity in multiple sclerosis: evidence from monoclonal antibody therapy. J Neurol. 2006 Jan;253(1):98-108. https://doi.org/10.1007/s00415-005-0934-5
» https://doi.org/10.1007/s00415-005-0934-5 -
65Jacobs LD, Beck RW, Simon JH, Kinkel RP, Brownscheidle CM, Murray TJ et al. Intramuscular interferon beta-1a therapy initiated during a first demyelinating event in multiple sclerosis. N Engl J Med. 2000 Sep;343(13):898-904. https://doi.org/10.1056/NEJM200009283431301
» https://doi.org/10.1056/NEJM200009283431301 -
66Comi G, Filippi M, Barkhof F, Durelli L, Edan G, Fernández O, et al. Effect of early interferon treatment on conversion to definite multiple sclerosis: a randomised study. Lancet. 2001 May;357(9268):1576-82. https://doi.org/10.1016/S0140-6736(00)04725-5
» https://doi.org/10.1016/S0140-6736(00)04725-5 -
67Kappos L, Polman CH, Freedman MS, Edan G, Hartung HP, Miller DH et al. Treatment with interferon beta-1b delays conversion to clinically definite and McDonald MS in patients with clinically isolated syndromes. Neurology. 2006 Oct;67(7):1242-9. https://doi.org/10.1212/01.wnl.0000237641.33768.8d
» https://doi.org/10.1212/01.wnl.0000237641.33768.8d -
68Comi G, Martinelli V, Rodegher M, Moiola L, Bajenaru O, Carra A et al. Effect of glatiramer acetate on conversion to clinically definite multiple sclerosis in patients with clinically isolated syndrome (PreCISe study): a randomised, double-blind, placebo-controlled trial. Lancet. 2009 Oct;374(9700):1503-11. https://doi.org/10.1016/S0140-6736(09)61259-9
» https://doi.org/10.1016/S0140-6736(09)61259-9 -
69Comi G, De Stefano N, Freedman MS, Barkhof F, Polman CH, Uitdehaag BM et al. Comparison of two dosing frequencies of subcutaneous interferon beta-1a in patients with a first clinical demyelinating event suggestive of multiple sclerosis (REFLEX): a phase 3 randomised controlled trial. Lancet Neurol. 2012 Jan;11(1):33-41. https://doi.org/10.1016/S1474-4422(11)70262-9
» https://doi.org/10.1016/S1474-4422(11)70262-9 -
70Miller AE, Wolinsky JS, Kappos L, Comi G, Freedman MS, Olsson TP. Oral teriflunomide for patients with a first clinical episode suggestive of multiple sclerosis (TOPIC): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2014 Oct;13(10):977-86. https://doi.org/10.1016/S1474-4422(14)70191-7
» https://doi.org/10.1016/S1474-4422(14)70191-7 -
71Freedman MS, Comi G, De Stefano N, Barkhof F, Polman CH, Uitdehaag BM et al. Moving toward earlier treatment of multiple sclerosis: findings from a decade of clinical trials and implications for clinical practice. Mult Scler Relat Disord. 2014 Mar;3(2):147-55. https://doi.org/10.1016/j.msard.2013.07.001
» https://doi.org/10.1016/j.msard.2013.07.001 -
72Stewart T, Spelman T, Havrdova E, Horakova D, Trojano M, Izquierdo G, et al. Contribution of different relapse phenotypes to disability in multiple sclerosis. Mult Scler. 2017 Feb;23(2):266-76. https://doi.org/10.1177/1352458516643392
» https://doi.org/10.1177/1352458516643392 -
73Damasceno A, Von Glehn F, Brandão CO, Damasceno BP, Cendes F. Prognostic indicators for long-term disability in multiple sclerosis patients. J Neurol Sci. 2013 Jan;324(1-2):29-33. https://doi.org/10.1016/j.jns.2012.09.020
» https://doi.org/10.1016/j.jns.2012.09.020 -
74Langer-Gould A, Popat RA, Huang SM, Cobb K, Fontoura P, Gould MK et al. Clinical and demographic predictors of long-term disability in patients with relapsing-remitting multiple sclerosis: a systematic review. Arch Neurol. 2006 Dec;63(12):1686-91. https://doi.org/10.1001/archneur.63.12.1686
» https://doi.org/10.1001/archneur.63.12.1686 -
75Mowry EM, Deen S, Malikova I, Pelletier J, Bacchetti P, Waubant E. The onset location of multiple sclerosis predicts the location of subsequent relapses. J Neurol Neurosurg Psychiatry. 2009 Apr;80(4):400-3. https://doi.org/10.1136/jnnp.2008.157305
» https://doi.org/10.1136/jnnp.2008.157305 -
76Kalincik T, Buzzard K, Jokubaitis V, Trojano M, Duquette P, Izquierdo G et al. Risk of relapse phenotype recurrence in multiple sclerosis. Mult Scler. 2014 Oct;20(11):1511-22. https://doi.org/10.1177/1352458514528762
» https://doi.org/10.1177/1352458514528762 -
77Deen S, Bacchetti P, High A, Waubant E. Predictors of the location of multiple sclerosis relapse. J Neurol Neurosurg Psychiatry. 2008 Oct;79(10):1190-3. https://doi.org/10.1136/jnnp.2007.136440
» https://doi.org/10.1136/jnnp.2007.136440 -
78Capra R, Cordioli C, Rasia S, Gallo F, Signori A, Sormani MP. Assessing long-term prognosis improvement as a consequence of treatment pattern changes in MS. Mult Scler. 2017 Nov;23(13):1757-61. https://doi.org/10.1177/1352458516687402
» https://doi.org/10.1177/1352458516687402 -
79Comi G. Induction vs. escalating therapy in multiple sclerosis: practical implications. Neurol Sci. 2008 Sep;29(S2 Suppl 2):S253-5. https://doi.org/10.1007/s10072-008-0954-x
» https://doi.org/10.1007/s10072-008-0954-x -
80Fenu G, Lorefice L, Frau F, Coghe GC, Marrosu MG, Cocco E. Induction and escalation therapies in multiple sclerosis. Antiinflamm Antiallergy Agents Med Chem. 2015;14(1):26-34. https://doi.org/10.2174/1871523014666150504122220
» https://doi.org/10.2174/1871523014666150504122220 -
81Wiendl H. Cladribine - an old newcomer for pulsed immune reconstitution in MS. Nat Rev Neurol. 2017 Oct;13(10):573-4. https://doi.org/10.1038/nrneurol.2017.119
» https://doi.org/10.1038/nrneurol.2017.119 -
82Leist TP, Comi G, Cree BA, Coyle PK, Freedman MS, Hartung HP et al. Effect of oral cladribine on time to conversion to clinically definite multiple sclerosis in patients with a first demyelinating event (ORACLE MS): a phase 3 randomised trial. Lancet Neurol. 2014 Mar;13(3):257-67. https://doi.org/10.1016/S1474-4422(14)70005-5
» https://doi.org/10.1016/S1474-4422(14)70005-5 -
83Montalban X, Cohen B, Leist T, et al. Efficacy of Cladribine Tablets as add-on to IFN-beta therapy in patients with active relapsing MS: final results from the Phase II ONWARD Study (P3.029). Neurology. 2016;86(16 Supplement).
-
84Jacobs LD, Cookfair DL, Rudick RA, Herndon RM, Richert JR, Salazar AM, et al.; The Multiple Sclerosis Collaborative Research Group (MSCRG). Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. Ann Neurol. 1996 Mar;39(3):285-94. https://doi.org/10.1002/ana.410390304
» https://doi.org/10.1002/ana.410390304 -
85PRISMS (Prevention of Relapses and Disability by Interferon β-1a Subcutaneously in Multiple Sclerosis) Study Group. Randomised double-blind placebo-controlled study of interferon β-1a in relapsing/remitting multiple sclerosis. Lancet. 1998 Nov;352(9139):1498-504. https://doi.org/10.1016/S0140-6736(98)03334-0
» https://doi.org/10.1016/S0140-6736(98)03334-0 -
86The IFNB Multiple Sclerosis Study Group. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. Neurology. 1993 Apr;43(4):655-61. https://doi.org/10.1212/WNL.43.4.655
» https://doi.org/10.1212/WNL.43.4.655 -
87Johnson KP, Brooks BR, Cohen JA, Ford CC, Goldstein J, Lisak RP et al; The Copolymer 1 Multiple Sclerosis Study Group. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. Neurology. 1995 Jul;45(7):1268-76. https://doi.org/10.1212/WNL.45.7.1268
» https://doi.org/10.1212/WNL.45.7.1268 -
88Khan O, Rieckmann P, Boyko A, Selmaj K, Zivadinov R. Three times weekly glatiramer acetate in relapsing-remitting multiple sclerosis. Ann Neurol. 2013 Jun;73(6):705-13. https://doi.org/10.1002/ana.23938
» https://doi.org/10.1002/ana.23938 -
89Calabresi PA, Kieseier BC, Arnold DL, Balcer LJ, Boyko A, Pelletier J et al. Pegylated interferon β-1a for relapsing-remitting multiple sclerosis (ADVANCE): a randomised, phase 3, double-blind study. Lancet Neurol. 2014 Jul;13(7):657-65. https://doi.org/10.1016/S1474-4422(14)70068-7
» https://doi.org/10.1016/S1474-4422(14)70068-7 -
90Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012 Sep;367(12):1098-107. https://doi.org/10.1056/NEJMoa1114287
» https://doi.org/10.1056/NEJMoa1114287 -
91Fox RJ, Miller DH, Phillips JT, Hutchinson M, Havrdova E, Kita M et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012 Sep;367(12):1087-97. https://doi.org/10.1056/NEJMoa1206328
» https://doi.org/10.1056/NEJMoa1206328 -
92O’Connor P, Wolinsky JS, Confavreux C, Comi G, Kappos L, Olsson TP et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011 Oct;365(14):1293-303. https://doi.org/10.1056/NEJMoa1014656
» https://doi.org/10.1056/NEJMoa1014656 -
93Confavreux C, O’Connor P, Comi G, Freedman MS, Miller AE, Olsson T, et al. Oral teriflunomide for patients with relapsing multiple sclerosis (TOWER): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2014 Mar;13(3):247-56. https://doi.org/10.1016/S1474-4422(13)70308-9
» https://doi.org/10.1016/S1474-4422(13)70308-9 -
94Cohen JA, Coles AJ, Arnold DL, Confavreux C, Fox EJ, Hartung HP et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. 2012 Nov;380(9856):1819-28. https://doi.org/10.1016/S0140-6736(12)61769-3
» https://doi.org/10.1016/S0140-6736(12)61769-3 -
95Coles AJ, Twyman CL, Arnold DL, Cohen JA, Confavreux C, Fox EJ et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet. 2012 Nov;380(9856):1829-39. https://doi.org/10.1016/S0140-6736(12)61768-1
» https://doi.org/10.1016/S0140-6736(12)61768-1 -
96Giovannoni G, Comi G, Cook S, Rammohan K, Rieckmann P, Soelberg Sørensen P et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. 2010 Feb;362(5):416-26. https://doi.org/10.1056/NEJMoa0902533
» https://doi.org/10.1056/NEJMoa0902533 -
97Kappos L, Radue EW, O’Connor P, Polman C, Hohlfeld R, Calabresi P et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010 Feb;362(5):387-401. https://doi.org/10.1056/NEJMoa0909494
» https://doi.org/10.1056/NEJMoa0909494 -
98Calabresi PA, Radue EW, Goodin D, Jeffery D, Rammohan KW, Reder AT et al. Safety and efficacy of fingolimod in patients with relapsing-remitting multiple sclerosis (FREEDOMS II): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2014 Jun;13(6):545-56. https://doi.org/10.1016/S1474-4422(14)70049-3
» https://doi.org/10.1016/S1474-4422(14)70049-3 -
99Polman CH, O’Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006 Mar;354(9):899-910. https://doi.org/10.1056/NEJMoa044397
» https://doi.org/10.1056/NEJMoa044397 -
100Hauser SL, Bar-Or A, Comi G, Giovannoni G, Hartung HP, Hemmer B et al. Ocrelizumab versus Interferon Beta-1a in relapsing multiple sclerosis. N Engl J Med. 2017 Jan;376(3):221-34. https://doi.org/10.1056/NEJMoa1601277
» https://doi.org/10.1056/NEJMoa1601277 -
101Gold R, Giovannoni G, Selmaj K, Havrdova E, Montalban X, Radue EW et al. Daclizumab high-yield process in relapsing-remitting multiple sclerosis (SELECT): a randomised, double-blind, placebo-controlled trial. Lancet. 2013 Jun;381(9884):2167-75. https://doi.org/10.1016/S0140-6736(12)62190-4
» https://doi.org/10.1016/S0140-6736(12)62190-4 -
102Kappos L, Wiendl H, Selmaj K, Arnold DL, Havrdova E, Boyko A et al. Daclizumab HYP versus Interferon Beta-1a in Relapsing Multiple Sclerosis. N Engl J Med. 2015 Oct;373(15):1418-28. https://doi.org/10.1056/NEJMoa1501481
» https://doi.org/10.1056/NEJMoa1501481 -
103European Medicines Agency. EMA urgently reviewing multiple sclerosis medicine Zinbryta following cases of inflammatory brain disorders. 2018 [accessed on 2018 Mar 14] Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Press_release/2018/03/WC500244890.pdf
» http://www.ema.europa.eu/docs/en_GB/document_library/Press_release/2018/03/WC500244890.pdf -
104Biogen. Biogen and AbbVie Announce the Voluntary Worldwide Withdrawal of Marketing Authorizations for ZINBRYTA® (daclizumab) for Relapsing Multiple Sclerosis. 2018 [accessed on 2018 Mar 14]. Available from: http://media.biogen.com/press-release/autoimmune-diseases/biogenand-abbvie-announce-voluntaryworldwide-withdrawal-marketi
» http://media.biogen.com/press-release/autoimmune-diseases/biogenand-abbvie-announce-voluntaryworldwide-withdrawal-marketi -
105Killian JM, Bressler RB, Armstrong RM, Huston DP. Controlled pilot trial of monthly intravenous cyclophosphamide in multiple sclerosis. Arch Neurol. 1988 Jan;45(1):27-30. https://doi.org/10.1001/archneur.1988.00520250033014
» https://doi.org/10.1001/archneur.1988.00520250033014 -
106Fazekas F, Deisenhammer F, Strasser-Fuchs S, Nahler G, Mamoli B. Randomised placebo-controlled trial of monthly intravenous immunoglobulin therapy in relapsing-remitting multiple sclerosis. Lancet. 1997 Mar;349(9052):589-93. https://doi.org/10.1016/S0140-6736(96)09377-4
» https://doi.org/10.1016/S0140-6736(96)09377-4 -
107Hartung HP, Gonsette R, König N, Kwiecinski H, Guseo A, Morrissey SP et al. Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet. 2002 Dec;360(9350):2018-25. https://doi.org/10.1016/S0140-6736(02)12023-X
» https://doi.org/10.1016/S0140-6736(02)12023-X -
108Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008 Feb;358(7):676-88. https://doi.org/10.1056/NEJMoa0706383
» https://doi.org/10.1056/NEJMoa0706383 -
109Mancardi GL, Sormani MP, Gualandi F, Saiz A, Carreras E, Merelli E et al. Autologous hematopoietic stem cell transplantation in multiple sclerosis: a phase II trial. Neurology. 2015 Mar;84(10):981-8. https://doi.org/10.1212/WNL.0000000000001329
» https://doi.org/10.1212/WNL.0000000000001329 -
110European Study Group on Interferon β-1b in Secondary Progressive MS. Placebo-controlled multicentre randomised trial of interferon beta-1b in treatment of secondary progressive multiple sclerosis. Lancet. 1998;352(9139):1491-7. https://doi.org/10.1016/S0140-6736(98)10039-9
» https://doi.org/10.1016/S0140-6736(98)10039-9 -
111Kappos L, Bar-Or A, Cree BA, Fox RJ, Giovannoni G, Gold R et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study. Lancet. 2018 Mar;391(10127):1263-73. https://doi.org/10.1016/S0140-6736(18)30475-6
» https://doi.org/10.1016/S0140-6736(18)30475-6 -
112Montalban X, Hauser SL, Kappos L, Arnold DL, Bar-Or A, Comi G et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N Engl J Med. 2017 Jan;376(3):209-20. https://doi.org/10.1056/NEJMoa1606468
» https://doi.org/10.1056/NEJMoa1606468 -
113Ghezzi A, Amato MP, Makhani N, Shreiner T, Gärtner J, Tenembaum S. Pediatric multiple sclerosis: conventional first-line treatment and general management. Neurology. 2016 Aug;87(9 Suppl 2):S97-102. https://doi.org/10.1212/WNL.0000000000002823
» https://doi.org/10.1212/WNL.0000000000002823 -
114Chitnis T. PARADIGMS: a randomised double-blind study of fingolimod versus interferon β-1a in paediatric multiple sclerosis. ECTRIMS Online Library. 2017 Oct 28;202640.
-
115Hardy TA, Reddel SW, Barnett MH, Palace J, Lucchinetti CF, Weinshenker BG. Atypical inflammatory demyelinating syndromes of the CNS. Lancet Neurol. 2016 Aug;15(9):967-81. https://doi.org/10.1016/S1474-4422(16)30043-6
» https://doi.org/10.1016/S1474-4422(16)30043-6 -
116Algahtani H, Shirah B, Alassiri A. Tumefactive demyelinating lesions: A comprehensive review. Mult Scler Relat Disord. 2017 May;14:72-9. https://doi.org/10.1016/j.msard.2017.04.003
» https://doi.org/10.1016/j.msard.2017.04.003 -
117Faissner S, Hoepner R, Lukas C, Chan A, Gold R, Ellrichmann G. Tumefactive multiple sclerosis lesions in two patients after cessation of fingolimod treatment. Ther Adv Neurol Disorder. 2015 Sep;8(5):233-8. https://doi.org/10.1177/1756285615594575
» https://doi.org/10.1177/1756285615594575 -
118Correale J, Ysrraelit MC, Fiol MP. Benign multiple sclerosis: does it exist? Curr Neurol Neurosci Rep. 2012 Oct;12(5):601-9. https://doi.org/10.1007/s11910-012-0292-5
» https://doi.org/10.1007/s11910-012-0292-5 -
119Berkovich R. Treatment of acute relapses in multiple sclerosis. Neurotherapeutics. 2013 Jan;10(1):97-105. https://doi.org/10.1007/s13311-012-0160-7
» https://doi.org/10.1007/s13311-012-0160-7 -
120Le Page E, Veillard D, Laplaud DA, Hamonic S, Wardi R, Lebrun C, et al; COPOUSEP investigators; West Network for Excellence in Neuroscience. Oral versus intravenous high-dose methylprednisolone for treatment of relapses in patients with multiple sclerosis (COPOUSEP): a randomised, controlled, double-blind, non-inferiority trial. Lancet. 2015 Sep;386(9997):974-81. https://doi.org/10.1016/S0140-6736(15)61137-0
» https://doi.org/10.1016/S0140-6736(15)61137-0 -
121National Clinical Advisory Board, National Multiple Sclerosis Society. Recommendations Regarding Corticosteroids in the Management of Multiple Sclerosis. US Neurol. 2008;4(1):22. https://doi.org/10.17925/USN.2008.04.01.22
» https://doi.org/10.17925/USN.2008.04.01.22 -
122Toosy A, Ciccarelli O, Thompson A. Symptomatic treatment and management of multiple sclerosis. In: Goodin DS, editor. Handbook of Clinical Neurology. [S. l.}: Elsevier; 2014. p. 513-62.
-
123Shoemaker TJ, Mowry EM. A review of vitamin D supplementation as disease-modifying therapy. Mult Scler. 2018 Jan;24(1):6-11. https://doi.org/10.1177/1352458517738131
» https://doi.org/10.1177/1352458517738131 -
124Brum DG, Comini-Frota ER, Vasconcelos CC, Dias-Tosta E. Supplementation and therapeutic use of vitamin D in patients with multiple sclerosis: consensus of the Scientific Department of Neuroimmunology of the Brazilian Academy of Neurology. Arq Neuropsiquiatr. 2014 Feb;72(2):152-6. https://doi.org/10.1590/0004-282X20130252
» https://doi.org/10.1590/0004-282X20130252 -
125Brochet B, Deloire MSA, Perez P, Loock T, Baschet L, Debouverie M et al; PROMESS study investigators. Double-Blind Controlled Randomized Trial of Cyclophosphamide versus Methylprednisolone in Secondary Progressive Multiple Sclerosis. PLOS One. 2017 Jan; 12(1):e0168834. https://doi.org/10.1371/journal.pone.0168834
» https://doi.org/10.1371/journal.pone.0168834
APPENDIX
Conflict of interest: Marques VD has received financial support for giving lectures, for participating in advisory boards and for congresses from Biogen Idec, Genzyme, Merck, Novartis, Rocha, and Teva.
Passos GR has received funding for research from Novartis and Roche; has been employed as an intern at Novartis for a 3-month internship in partnership with the University of Basel; has received sponsorship for attendance at scientific meetings from Roche, Sanofi-Genzyme, and Teva; has received fees for preparation of editorial content from Bayer, Merck Serono, and Roche; and has received scholarships from the World Federation of Neurology and the European Committee on Treatment and Research in Multiple Sclerosis.
Mendes MF has received fees for giving a lecture/organizing teaching activities from Biogen, Merck, Novartis, and Roche; has received funding for conducting research from Biogen and Schering; and has received financial support for congresses or scientific or promotional events from Biogen, Merck, Novartis, Roche, and Schering.
Callegaro D is the chief of a reference center for research and treatment of multiple sclerosis and neuromyelitis optica spectrum disorders and has received honoraria for participating in medical courses and meetings from Roche and Biogen.
Lana-Peixoto MA has received sponsorship for travel to the ECTRIMS Meeting in 2016 (from Genzyme) and in 2017 (from Roche).
Comini-Frota ER has received fees for making presentations for Genzyme and Teva Pharmaceuticals.
Vasconcelos CCF has received honoraria as consultant for advisory boards/lectures at meetings and sponsorship for participation in congresses from Genzyme, Biogen Indec, Merck-Serono, Teva, and Roche.
Sato DK has received research support from Teva (EMOCEMP Study – NCT no. 61080516.4.1001.5336), Euroimmun AG; speaking honoraria from Biogen, Novartis, Genzyme, Teva, Merck-Sorono, Roche, and Bayer; and fees for participation in advisory boards from Shire, Roche, Teva, Merck-Serono, and Quest/Athena Diagnostics.
Ferreira MLB has received reimbursement for attendance at a symposium, fees for making a presentation or giving a lecture, and funding for conducting research from Merck, Genzyme, Biogen, and Teva.
Parolin MKF has received support for attendance at scientific events from Roche, Teva, Biogen, and Merck.
Damasceno A has received a post-doctoral grant from São Paulo Research Foundation (FAPESP) and reimbursement for attendance at a symposium and/or fees for consultancy from Sanofi-Genzyme, Biogen, and Roche.
Grzesiuk AK has received honoraria as consultant from Biogen and Teva, and sponsorship for participation in congresses from Merck-Serono, Biogen, Teva, Novartis, Genzyme, Roche and Bayer.
Muniz A has received financial support for giving lectures and for congresses from Biogen Idec, Genzyme, and Roche.
Matta APC is currently employed by Biogen, but was not at the time this manuscript was prepared; he has received speaking honoraria from Biogen, Teva, Bayer, Merck, Genzyme, and Novartis and sponsorship for attending the AAN Meeting 2017 from the Partners MS Center, Harvard Medical School.
Oliveira BES has received support for attendance at scientific events from Sanofi-Genzyme, Biogen, Merck-Serono, and Roche and speaking honoraria from Merck-Serono and Sanofi-Genzyme.
Tauil CB has received speaking honoraria and research or travel grants from Biogen, São Paulo Research Foundation (FAPESP), Distrito Federal Research Foundation (FAP-DF), Libbs, Novartis, Roche, Sanofi, Serono, Shire, and Teva Pharmaceuticals.
Maciel DRK reports no conflicts of interest.
Diniz DS reports no conflicts of interest.
Correa EC has received sponsorship for congresses and meetings from Merck, Biogen, Genzyme, and Roche.
Rocha FCG reports no conflicts of interest.
Jorge FMH reports no conflicts of interest.
Sato HK has received financial support for giving lectures, for participating in advisory boards and for congresses from Biogen Idec, Genzyme, Merck, Novartis, Roche, Bayer, and Teva.
Gonçalves MVM reports no conflicts of interest.
Sousa NAC has received sponsorship for travel, accommodation and enrolment in congresses from Teva, Biogen, Roche, and Merck.
Nascimento OJM reports no conflicts of interest.
Gama PD has received sponsorship for attendance at scientific meetings from Novartis, Merck, Teva, Sonofi-Genzyme, Biogen, Roche, and Bayer.
Domingues RB reports no conflicts of interest.
Simm RF has received funds to attend conferences from Teva and Sanofi and to participate in advisory board from Sanofi and Biogen.
Thomaz RB has received funds or fees for research, consultancy, presentation, lectures, speech lectures and membership in advisory boards from Roche, Sanofi, Biogen, and Novartis-PPD.
Morales RR has received support for attendance at conferences and medical education events from the Brazilian National Council for Scientific and Technological Development (CNPq), Teva, Bayer, Merck, Biogen, Genzyme, and Shire; and honoraria for lectures from Teva, Merck, and Shire.
Dias RM has received fees for participation in advisory boards from Teva and Roche and for lectures from Biogen.
Pereira SLA reports that, as a member of BCTRIMS and ABN, she and some members of her team have participated as attendants or speakers in national and international meetings with support from Biogen Idec, Genzyme, and Merck Serono.
Machado SCN reports no conflicts of interest.
Junqueira TF reports no conflicts of interest.
Becker J has received speaking honoraria and research or travel grants from Bayer Health Care, Biogen, Ipsen, Merck-Serono, Novartis, Roche, Sanofi Genzyme, and Teva Pharmaceuticals.
Publication Dates
-
Publication in this collection
Aug 2018
History
-
Received
17 Mar 2018 -
Reviewed
09 May 2018 -
Accepted
16 May 2018