ABSTRACT
Transthyretin amyloidosis (ATTR) is characterized by the deposit of mutant or wild-type transthyretin that forms amyloid fibrils, which are extracellularly deposited within tissues and organs. Clinical manifestations of familial amyloid polyneuropathy vary according to the mutation, age at onset and geographical location. This study aimed to describe baseline disease characteristics of Brazilian patients with transthyretin familial amyloid polyneuropathy (ATTR-FAP) enrolled in the Transthyretin Amyloidosis Outcome Survey (THAOS). Methods: The THAOS is an international, noninterventional, longitudinal, observational, web-based registry designed to characterize ATTR. The outcome measures included demographics (age at symptom onset, gender, time from onset of symptoms to diagnosis, family history), genotype, and clinical characteristics (presence of amyloid deposit, frequency of misdiagnosis, presenting symptomatology). The analysis was conducted in a dataset from Brazilian patients (from November 2008 to January 2016). Results: One hundred and sixty participants (52.5% male) were included in the analysis. The majority of participants (90.6%) reported a positive family history of ATTR-FAP Median age at symptom onset was 32.5 years. Val30Met mutation was found in 91.9%. Misdiagnosis was observed in 26.6% of symptomatic patients. Over one-third (35.3%) of the misdiagnosed patients experienced a delay of more than one year before receiving a correct diagnosis. At presentation, 79.7% of the patients had motor, 87.5% sensory and 93.8% autonomic symptoms. Conclusion: ATTR-FAP in Brazil starts early, has a strong family history and the majority has Val30Met mutation. Misdiagnosis is common and the most common presentation is of a sensorimotor and autonomic neuropathy.
Keywords:
Amyloidosis; amyloid neuropathies, familial; polyneuropathy
RESUMO
Amiloidose ligada à transtirretina (ATTR) é caracterizada por depósito de transtirretina que forma fibrilas amiloides, que são depositadas extracelularmente dentro de tecidos e órgãos. As manifestações clínicas de polineuropatia amiloidótica familiar (ATTR-PAF) variam de acordo com a mutação, idade de início e localização geográfica. Este estudo tem como objetivo descrever as características dos pacientes com ATTR no Brasil, com base nos dados coletados no THAOS. Métodos: THAOS é um registro internacional longitudinal observacional desenhado para caracterizar ATTR. As medidas de desfecho incluíram dados demográficos (idade do início dos sintomas, gênero, tempo do início dos sintomas até diagnóstico, histórico familiar), genotipagem e características clínicas (presença de depósito amiloide, frequências de diagnósticos errôneos, sintomatologia presente). Esta analise foi conduzida com dados de pacientes brasileiros registrados no THAOS de Novembro 2008 a Janeiro de 2016. Resultado: Cento e sessenta pacientes (52,5% homens) foram incluídos na análise. Na maioria dos casos (90,6%) observou-se história familiar positiva de ATTR-FAP A idade média de inicio dos sintomas foi 32,5 anos. A mutação Val30Met foi encontrada em 91,9%. Erros diagnósticos foram observados em 26,6% dos casos sintomáticos. Aproximadamente um terço dos pacientes diagnosticados erroneamente tiveram atraso de mais de um ano para receber um diagnostico correto. No momento do diagnóstico 79,7% dos pacientes possuíam sintomas motores, 87,5% sintomas sensitivos e 93,8% sintomas autonômicos. Conclusão: No brasil a ATTR-FAP tem início precoce, historia familiar fortemente positiva e em sua maioria são portadores da mutação Val30Met. Erros diagnósticos são comuns e a apresentação mais comum é polineuropatia sensitivo-motora com disautonomia.
Palavras-chave:
Amiloidose; neuropatias amiloides familiares; polineuropatias
Transthyretin amyloidosis (ATTR) belongs to a group of severe systemic conditions characterized by deposit of wild-type or mutant proteins in tissues and organs11. Planté-Bordeneuve V, Kerschen P. Transthyretin familial amyloid polyneuropathy. Handb Clin Neurol. 2013;115:643-58. https://doi.org/10.1016/B978-0-444-52902-2.00038-2
https://doi.org/10.1016/B978-0-444-52902...
,22. Maurer MS, Hanna M, Grogan M, Dispenzieri A, Witteles R, Drachman B et al.. Genotype and Phenotype of Transthyretin Cardiac Amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J Am Coll Cardiol. 2016 Jul;68(2):161-72. https://doi.org/10.1016/j.jacc.2016.03.596
https://doi.org/10.1016/j.jacc.2016.03.5...
.
More specifically, hereditary ATTR is caused by mutations in the gene encoding transthyretin (TTR), a plasma protein, mainly synthesized and excreted by the liver, which circulates in soluble form in the peripheral blood and cerebrospinal fluid and is involved in the transport of thyroxin and retinol. Pathogenic mutations decrease the stability of TTR tetramers and enhance their dissociation into monomers. These monomers are susceptible to misfolding and self-aggregate into insoluble amyloid fibrils capable of systemic extracellular deposition11. Planté-Bordeneuve V, Kerschen P. Transthyretin familial amyloid polyneuropathy. Handb Clin Neurol. 2013;115:643-58. https://doi.org/10.1016/B978-0-444-52902-2.00038-2
https://doi.org/10.1016/B978-0-444-52902...
,33. Carvalho A, Rocha A, Lobato L. Liver transplantation in transthyretin amyloidosis: issues and challenges. Liver Transpl. 2015 Mar;21(3):282-92. https://doi.org/10.1002/lt.24058
https://doi.org/10.1002/lt.24058...
.
Clinical manifestations vary according to the mutation, age of onset and geographical location, and can be predominantly neuropathic (known as familial amyloid polyneuropathy [ATTR-FAP]), predominantly cardiac (or TTR cardiac amyloidosis), or mixed11. Planté-Bordeneuve V, Kerschen P. Transthyretin familial amyloid polyneuropathy. Handb Clin Neurol. 2013;115:643-58. https://doi.org/10.1016/B978-0-444-52902-2.00038-2
https://doi.org/10.1016/B978-0-444-52902...
,22. Maurer MS, Hanna M, Grogan M, Dispenzieri A, Witteles R, Drachman B et al.. Genotype and Phenotype of Transthyretin Cardiac Amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J Am Coll Cardiol. 2016 Jul;68(2):161-72. https://doi.org/10.1016/j.jacc.2016.03.596
https://doi.org/10.1016/j.jacc.2016.03.5...
. In the peripheral nervous system, amyloid deposits are in the interstitium, scattered in the endoneurium and/or around blood vessels11. Planté-Bordeneuve V, Kerschen P. Transthyretin familial amyloid polyneuropathy. Handb Clin Neurol. 2013;115:643-58. https://doi.org/10.1016/B978-0-444-52902-2.00038-2
https://doi.org/10.1016/B978-0-444-52902...
. Familial amyloid polyneuropathy is a slowly-progressive disease in most patients and has been divided into three stages following the length-dependent progression of sensorimotor polyneuropathy. Sensory symptoms are the most frequent initial complaint. They include distal paresthesias, numbness sometimes associated with pain, and burning or tingling sensations, without walking impairment (stage 1 of Coutinho). At a later stage, patients experience more generalized sensorimotor neuropathy, affecting the upper limbs, and require assistance for walking (stage 2 of Coutinho). Ultimately, there is a flaccid paralysis in all four limbs and the patient is confined to a wheelchair or is bedridden (stage 3 of Coutinho)44. Coutinho PD, Lima JL, Barbosa AR. Forty years of experience with type I amyloid neuropathy: review of 483 cases. In: Glenner GG, de Freitas AF, editors. Amyloid and amyloidosis. Amsterdam: Excerpta Medica; 1980. pp. 88-98.).
In an average of 10 years, the disease is fatal, usually due to infections, cachexia or bed sores11. Planté-Bordeneuve V, Kerschen P. Transthyretin familial amyloid polyneuropathy. Handb Clin Neurol. 2013;115:643-58. https://doi.org/10.1016/B978-0-444-52902-2.00038-2
https://doi.org/10.1016/B978-0-444-52902...
,55. Adams D, Théaudin M, Cauquil C, Algalarrondo V, Slama M. FAP neuropathy and emerging treatments. Curr Neurol Neurosci Rep. 2014 Mar;14(3):435. https://doi.org/10.1007/s11910-013-0435-3
https://doi.org/10.1007/s11910-013-0435-...
. In addition, autonomic neuropathy is almost constant during the course of disease, causing a major impact on patient disability, with organ dysfunction directly related to amyloid deposits; cardiovascular (orthostatic hypotension that can cause fatigue, blurred vision, dizziness, faintness or repeated postural syncope); gastrointestinal (early satiety, slow digestion, nausea, anorexia, recurrent postprandial vomiting [later stage], dehydration and progressive weight loss); and genitourinary (erectile dysfunction, dysuria, urinary retention). The disease also directly affects other organs such as: ocular (vitreous opacity, keratoconjunctivitis, glaucoma), heart (conduction abnormalities and cardiac failure), and kidney (early proteinuria progressing to kidney failure)11. Planté-Bordeneuve V, Kerschen P. Transthyretin familial amyloid polyneuropathy. Handb Clin Neurol. 2013;115:643-58. https://doi.org/10.1016/B978-0-444-52902-2.00038-2
https://doi.org/10.1016/B978-0-444-52902...
,66. Ando Y, Nakamura M, Araki S. Transthyretin-related familial amyloidotic polyneuropathy. Arch Neurol. 2005 Jul;62(7):1057-62. https://doi.org/10.1001/archneur.62.71057
https://doi.org/10.1001/archneur.62.7105...
.
Transthyretin familial amyloid polyneuropathy is distributed worldwide, but precise epidemiological data are scarce. Its low incidence (< 1/200) makes it a rare disease. Portugal, Sweden and Japan remain the largest focus of the disease, but it is likely that the disease remains largely underdiagnosed in some areas11. Planté-Bordeneuve V, Kerschen P. Transthyretin familial amyloid polyneuropathy. Handb Clin Neurol. 2013;115:643-58. https://doi.org/10.1016/B978-0-444-52902-2.00038-2
https://doi.org/10.1016/B978-0-444-52902...
,66. Ando Y, Nakamura M, Araki S. Transthyretin-related familial amyloidotic polyneuropathy. Arch Neurol. 2005 Jul;62(7):1057-62. https://doi.org/10.1001/archneur.62.71057
https://doi.org/10.1001/archneur.62.7105...
. In Brazil, the population of ATTR-FAP cases is estimated to be around 5,000 patients77. Saporta MA, Zaros C, Cruz MW, André C, Misrahi M, Bonaiti-Pellié C et al. Penetrance estimation of TTR familial amyloid polyneuropathy (type I) in Brazilian families. Eur J Neurol. 2009 Mar;16(3):337-41. https://doi.org/10.1111/j.1468-1331.2008.02429.x
https://doi.org/10.1111/j.1468-1331.2008...
,88. Schmidt H, Cruz MW, Botteman MF, Carter JA, Chopra A, Stewart M et al. Global epidemiology of transthyretin hereditary amyloid polyneuropathy: a systematic review. Amyloid. 2017 Mar 16;24(sup1):111-2. https://doi.org/10.1080/13506129.2017.1292903
https://doi.org/10.1080/13506129.2017.12...
. In order to characterize the natural history of ATTR, an international noninterventional registry has been implemented - the Transthyretin Amyloidosis Outcomes Survey (THAOS)99. Planté-Bordeneuve V, Suhr OB, Maurer MS, White B, Grogan DR, Coelho T. The Transthyretin Amyloidosis Outcomes Survey (THAOS) registry: design and methodology. Curr Med Res Opin. 2013 Jan;29(1):77-84. https://doi.org/10.1185/03007995.2012.754349
https://doi.org/10.1185/03007995.2012.75...
.
This study aimed to describe the characteristics of patients suffering from ATTR in Brazil, based on data collected in THAOS.
METHODS
The THAOS is an ongoing, international, noninterventional, longitudinal, observational, web-based registry designed to characterize ATTR (ClinicalTrials.gov
NCT00628745). It is open to all patients with ATTR (familial and wild-type) and individuals with TTR gene mutations without a diagnosis of hereditary ATTR (asymptomatic)22. Maurer MS, Hanna M, Grogan M, Dispenzieri A, Witteles R, Drachman B et al.. Genotype and Phenotype of Transthyretin Cardiac Amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J Am Coll Cardiol. 2016 Jul;68(2):161-72. https://doi.org/10.1016/j.jacc.2016.03.596
https://doi.org/10.1016/j.jacc.2016.03.5...
,99. Planté-Bordeneuve V, Suhr OB, Maurer MS, White B, Grogan DR, Coelho T. The Transthyretin Amyloidosis Outcomes Survey (THAOS) registry: design and methodology. Curr Med Res Opin. 2013 Jan;29(1):77-84. https://doi.org/10.1185/03007995.2012.754349
https://doi.org/10.1185/03007995.2012.75...
,1010. Kristen AV, Maurer MS, Rapezzi C, Mundayat R, Suhr OB, Damy T. Impact of genotype and phenotype on cardiac biomarkers in patients with transthyretin amyloidosis - Report from the Transthyretin Amyloidosis Outcome Survey (THAOS). PLoS One. 2017 Apr;12(4):e0173086. https://doi.org/10.1371/journal.pone.0173086
https://doi.org/10.1371/journal.pone.017...
. Prior to inclusion, the site was required to obtain all local regulatory approvals, and participating patients had to provide their written informed consent.
The analysis was conducted on data from patients enrolled in the only Brazilian site contributing to the registry (CEPARM - National Brazilian Amyloidosis referral center located at the Federal University of Rio de Janeiro) from November 13, 2008 to January 14, 2016.
The outcome measures included demographics (age at symptom onset, gender, time from onset of symptoms to diagnosis, family history), genotype, and clinical characteristics (presence of amyloid deposit, frequency of misdiagnosis, presenting symptomatology)1111. Cruz MW, Foguel D, Berensztejn AC, Pedrosa RC, Mundayat R, Ong ML. The demographic, genetic, and clinical characteristics of Brazilian subjects enrolled in the Transthyretin Amyloidosis Outcomes Survey. Amyloid. 2017 Mar;24 sup1:103-4. https://doi.org/10.1080/13506129.2017.1291423
https://doi.org/10.1080/13506129.2017.12...
.
Statistical analysis
Descriptive summary statistics are presented with categorical data shown as frequency and distribution, and continuous data as mean, median, minimum, maximum.
RESULTS
In total, 160 participants (52.5% male) were included in the analysis. The majority of participants (90.6%) reported a known family history of symptomatic ATTR. Amyloid deposit was found in 80.8% of the biopsies performed in symptomatic participants (the most common tissue type tested was salivary gland followed by nerve). Genetic evaluation showed Val30Met (n = 147, 91.9%) and non-Val30Met (n = 13, 8.1%) mutations. Among the non-Val30Met mutations identified were: Ile07Val, Val122Ile, Ala19Asp and Glu109Lys. The overall median age at symptom onset was 32.5 years and median time from symptom onset to diagnosis for men and women was 2.6 years and 5.0 years, respectively.
Misdiagnosis was observed in 26.6% of the symptomatic patients, with chronic inflammatory demyelinating polyneuropathy (CIDP) being the most common diagnosis. Over one-third (35.3%) of the patients experienced a delay of more than one year before receiving a correct diagnosis (Table).
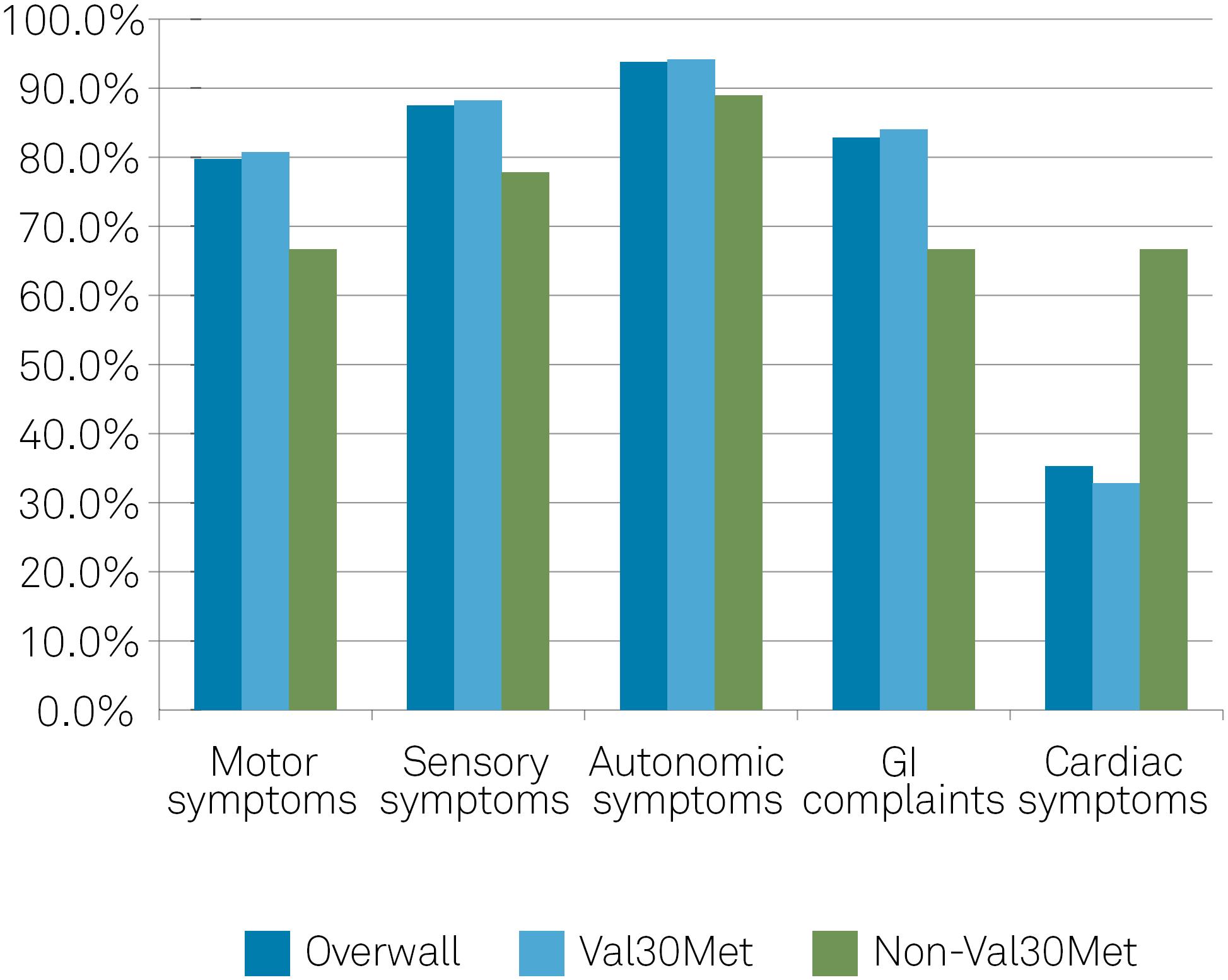
Of the 128 symptomatic patients at enrollment, 79.7% presented with motor symptoms (80.7% Val30Met and 66.7% non-Val30Met); 87.5% presented with sensory symptoms (88.2% Val30Met and 77.8% non-Val30Met); 93.8% presented with autonomic symptoms (94.1% Val30Met and 88.9% non-Val-30Met); and 82.8% presented with gastrointestinal complaints (84.0% Val30Met and 66.7% non-Val30Met). Unintentional weight loss was noted in 50.8% of the patients. Cardiac involvement was present in 35.2% of symptomatic participants (32.8% Val30Met and 66.7% non-Val30Met) (Figure).
When considering the age of onset, 82.0% of early-onset patients (< 50 years old) and 74.1% of late-onset patients (≥ 50 years old) presented with motor symptoms. Corresponding values for sensory symptoms were 90.0% (early-onset) and 81.5% (late-onset), and for autonomic symptoms were 95.0% (early-onset) and 88.9% (late-onset).
DISCUSSION
Transthyretin familial amyloid polyneuropathy was described in 1952 by Corino de Andrade1212. Andrade C. A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain. 1952 Sep;75(3):408-27 https://doi.org/10.1093/brain/75.3.408).
https://doi.org/10.1093/brain/75.3.408...
and was originally considered to be an endemic condition in some areas of Portugal, Japan and Sweden. Over the past decades, about 120 different mutations related to hereditary ATTR have been reported; this genetic variation results in a condition with many different forms, with considerable phenotypic variation across individuals and geographic location1313. Ando Y, Coelho T, Berk JL, Cruz MW, Ericzon BG, Ikeda S et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013 Feb;8(1):31. https://doi.org/10.1186/1750-1172-8-31
https://doi.org/10.1186/1750-1172-8-31...
. Therefore, there are several unanswered questions in terms of natural history and response to therapy. For this purpose, the coordination of an international registry will provide relevant information about the condition and its management.
This is a report based on data collected from Brazilian patients. The majority of patients (> 90%) had Val30Met mutation. Val30Met is the mutation linked to ATTR-FAP, and is endemically reported in Portugal, Japan and Sweden. Of note, the Brazilian population has a strong Portuguese influence but is also a very mixed population with other ethnic influences such as African, Italian, German and Japanese. No study has been performed to confirm the impact of such influences on the profile of the clinical presentation of this disease in Brazil.
Recently, Lavigne-Moreira et al.1414. Lavigne-Moreira C, Marques VD, Gonçalves MV, Oliveira MF, Tomaselli PJ, Nunez JC et al. The genetic heterogeneity of hereditary transthyretin amyloidosis in a sample of the Brazilian population. J Peripher Nerv Syst. 2018 Jun;23(2):134-7. https://doi.org/10.1111/jns.12259
https://doi.org/10.1111/jns.12259...
reported the genetic heterogeneity of 128 Brazilian patients with mutations in the TTR gene. Val30Met was the most common mutation identified (90.6%). The four non-Val30Met mutations were Ile107Val (n = 2), Asp38Tyr (n = 2), Val122Ile (n = 1) and Val71Ala (n = 1). Two nonpathogenic mutations Gly6Ser (n = 5) and Thr119Thr (n = 1) were also identified1414. Lavigne-Moreira C, Marques VD, Gonçalves MV, Oliveira MF, Tomaselli PJ, Nunez JC et al. The genetic heterogeneity of hereditary transthyretin amyloidosis in a sample of the Brazilian population. J Peripher Nerv Syst. 2018 Jun;23(2):134-7. https://doi.org/10.1111/jns.12259
https://doi.org/10.1111/jns.12259...
. This study was conducted in the city of Ribeirao Preto (São Paulo state), which is in the southeast region of Brazil, the same region where our center is located. Taken together, our findings and those of Lavigne-Moreira et al. raise the possibility that the southeast region of Brazil is an endemic region of ATTR-FAP.
Transthyretin familial amyloid polyneuropathy is a progressive fatal disease, with patients presenting with motor disability within five years of diagnosis and generally being fatal within a decade without treatment11. Planté-Bordeneuve V, Kerschen P. Transthyretin familial amyloid polyneuropathy. Handb Clin Neurol. 2013;115:643-58. https://doi.org/10.1016/B978-0-444-52902-2.00038-2
https://doi.org/10.1016/B978-0-444-52902...
. Therefore, the need for early diagnosis is clear given that therapeutic interventions (liver transplantation or anti-amyloidogenic therapy) are designed to reduce further deposition, but do not address the effect of already-deposited amyloid, and appear to be effective in early disease11. Planté-Bordeneuve V, Kerschen P. Transthyretin familial amyloid polyneuropathy. Handb Clin Neurol. 2013;115:643-58. https://doi.org/10.1016/B978-0-444-52902-2.00038-2
https://doi.org/10.1016/B978-0-444-52902...
,22. Maurer MS, Hanna M, Grogan M, Dispenzieri A, Witteles R, Drachman B et al.. Genotype and Phenotype of Transthyretin Cardiac Amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J Am Coll Cardiol. 2016 Jul;68(2):161-72. https://doi.org/10.1016/j.jacc.2016.03.596
https://doi.org/10.1016/j.jacc.2016.03.5...
,55. Adams D, Théaudin M, Cauquil C, Algalarrondo V, Slama M. FAP neuropathy and emerging treatments. Curr Neurol Neurosci Rep. 2014 Mar;14(3):435. https://doi.org/10.1007/s11910-013-0435-3
https://doi.org/10.1007/s11910-013-0435-...
,1515. Cortese A, Vegezzi E, Lozza A, Alfonsi E, Montini A, Moglia A et al. Diagnostic challenges in hereditary transthyretin amyloidosis with polyneuropathy: avoiding misdiagnosis of a treatable hereditary neuropathy. J Neurol Neurosurg Psychiatry. 2017 May;88(5):457-8. https://doi.org/10.1136/jnnp-2016-315262
https://doi.org/10.1136/jnnp-2016-315262...
. The diagnosis of ATTR-FAP remains challenging in the early stages of the disease and requires expertise, adequate diagnostic methods and a multidisciplinary approach55. Adams D, Théaudin M, Cauquil C, Algalarrondo V, Slama M. FAP neuropathy and emerging treatments. Curr Neurol Neurosci Rep. 2014 Mar;14(3):435. https://doi.org/10.1007/s11910-013-0435-3
https://doi.org/10.1007/s11910-013-0435-...
,1313. Ando Y, Coelho T, Berk JL, Cruz MW, Ericzon BG, Ikeda S et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013 Feb;8(1):31. https://doi.org/10.1186/1750-1172-8-31
https://doi.org/10.1186/1750-1172-8-31...
. The median age at onset of disease-related symptoms was 32.5 years old, and the median time from symptom onset to diagnosis for men and women was 2.6 years and 5.0 years, respectively, in the Brazilian database. The early erectile dysfunction encountered in the majority of our male patients probably helped in the shorter time for diagnosis compared to women. Our findings agree with the Portuguese and Japanese population described with early onset (mean age of 33 years) and average time for diagnosis varying from two to six years44. Coutinho PD, Lima JL, Barbosa AR. Forty years of experience with type I amyloid neuropathy: review of 483 cases. In: Glenner GG, de Freitas AF, editors. Amyloid and amyloidosis. Amsterdam: Excerpta Medica; 1980. pp. 88-98.),1313. Ando Y, Coelho T, Berk JL, Cruz MW, Ericzon BG, Ikeda S et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013 Feb;8(1):31. https://doi.org/10.1186/1750-1172-8-31
https://doi.org/10.1186/1750-1172-8-31...
. The main reasons for this long time for diagnosis are: the absence of family history, atypical clinical presentations (varied phenotypes), difficulties in detecting amyloid deposits in a single tissue biopsy, and the rare use of TTR gene analysis in the screening of idiopathic polyneuropathy33. Carvalho A, Rocha A, Lobato L. Liver transplantation in transthyretin amyloidosis: issues and challenges. Liver Transpl. 2015 Mar;21(3):282-92. https://doi.org/10.1002/lt.24058
https://doi.org/10.1002/lt.24058...
.
In addition, overall, approximately 25% of symptomatic patients are misdiagnosed; and correct diagnosis was delayed by more than a year in about one-third of them; CIDP being the most common disease for misdiagnosis. This result is similar to the one reported by Cortese et al.,1515. Cortese A, Vegezzi E, Lozza A, Alfonsi E, Montini A, Moglia A et al. Diagnostic challenges in hereditary transthyretin amyloidosis with polyneuropathy: avoiding misdiagnosis of a treatable hereditary neuropathy. J Neurol Neurosurg Psychiatry. 2017 May;88(5):457-8. https://doi.org/10.1136/jnnp-2016-315262
https://doi.org/10.1136/jnnp-2016-315262...
who conducted a review on the medical records of 150 patients in Pavia, Italy, and found 49 misdiagnosed patients (32%), in which CIDP was the most frequent error. The following typical findings in ATTR-FAP help in the differentiation from CIDP: small fiber predominant sensation loss; absence of nonuniform demyelination on nerve conduction studies (temporal dispersion and/or conduction blocks); weight loss; distal predominant weakness and moderate to severe autonomic dysfunction. The latter seems to be one of the most important. We found autonomic symptoms in approximately 94% of our patients, which are usually seen in less than 25% of patients with CIDP, who have no or minimal autonomic dysfunction1616. Figueroa JJ, Dyck PJ, Laughlin RS, Mercado JA, Massie R, Sandroni P et al. Autonomic dysfunction in chronic inflammatory demyelinating polyradiculoneuropathy. Neurology. 2012 Mar;78(10):702-8. https://doi.org/10.1212/WNL.0b013e3182494d66
https://doi.org/10.1212/WNL.0b013e318249...
. Other differential diagnoses of ATTR-FAP are diabetic neuropathy, inherited sensory and autonomic neuropathies, leprosy, toxic neuropathies, immunoglobulin light-chain amyloidosis and Fabry's disease. The Brazilian consensus on ATTR-FAP recommends that it should be strongly considered in patients with progressive polyneuropathy of unknown etiology who have one or more of the following: family history of neuropathy; orthostatic hypotension; erectile dysfunction; unexplained weight loss; arrhythmias and/or cardiomyopathy; bilateral carpal tunnel syndrome; renal abnormalities; vitreous opacities; gastrointestinal complaints (chronic diarrhea, constipation or diarrhea/constipation); rapid progression and/or prior treatment failure1717. Pinto MV, Barreira AA, Bulle AS, Freitas MR, França MC Jr, Gondim FA et al. Brazilian consensus for diagnosis, management and treatment of transthyretin familial amyloid polyneuropathy. Arq Neuropsiquiatr. 2018 Sep;76(9):609-21. https://doi.org/10.1590/0004-282x20180094
https://doi.org/10.1590/0004-282x2018009...
.
Histological confirmation of the disease requires tissue sampling to demonstrate amyloid deposition. The site of biopsy needs to be guided by clinical features and the expertise of the center. Tissues like salivary glands, subcutaneous abdominal fat or rectal mucosa should be chosen first. However, the severity of polyneuropathy often requires a nerve biopsy11. Planté-Bordeneuve V, Kerschen P. Transthyretin familial amyloid polyneuropathy. Handb Clin Neurol. 2013;115:643-58. https://doi.org/10.1016/B978-0-444-52902-2.00038-2
https://doi.org/10.1016/B978-0-444-52902...
. When the presentation is cardiac, the cardiac biopsy or scintigraphy could help in the differential diagnosis with other forms of amyloidosis and also of another cardiomyopathy. Also, immunohistochemistry studies or mass spectrometry-based proteomics can be done to identify the amyloid protein, and the latter can even differentiate wild- type from mutated transthyretin1313. Ando Y, Coelho T, Berk JL, Cruz MW, Ericzon BG, Ikeda S et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013 Feb;8(1):31. https://doi.org/10.1186/1750-1172-8-31
https://doi.org/10.1186/1750-1172-8-31...
.
Of note is the fact that, at the present moment, the center enrolled 220 patients in the THAOS database, meaning an increase of 37.5% in the number of new patients in the last two years. This reflects an increase in medical knowledge about the disease and interest in new available therapies.
In conclusion, ATTR-FAP in Brazilian patients starts early, has a strong family history, the majority has the Val30Met mutation and the most common presentation is of a sensorimotor and autonomic neuropathy. A delay of more than one year before receiving a correct diagnosis is common and approximately one-quarter of the patients were misdiagnosed, further delaying the introduction of correct management. Awareness should be raised among physicians about the identification and diagnostics work-up of ATTR-FAP.
-
Support: This study was funded by Pfizer Inc. Editorial support was provided by Roberta Trefiglio and was funded by Pfizer Inc.
References
-
1Planté-Bordeneuve V, Kerschen P. Transthyretin familial amyloid polyneuropathy. Handb Clin Neurol. 2013;115:643-58. https://doi.org/10.1016/B978-0-444-52902-2.00038-2
» https://doi.org/10.1016/B978-0-444-52902-2.00038-2 -
2Maurer MS, Hanna M, Grogan M, Dispenzieri A, Witteles R, Drachman B et al.. Genotype and Phenotype of Transthyretin Cardiac Amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J Am Coll Cardiol. 2016 Jul;68(2):161-72. https://doi.org/10.1016/j.jacc.2016.03.596
» https://doi.org/10.1016/j.jacc.2016.03.596 -
3Carvalho A, Rocha A, Lobato L. Liver transplantation in transthyretin amyloidosis: issues and challenges. Liver Transpl. 2015 Mar;21(3):282-92. https://doi.org/10.1002/lt.24058
» https://doi.org/10.1002/lt.24058 -
4Coutinho PD, Lima JL, Barbosa AR. Forty years of experience with type I amyloid neuropathy: review of 483 cases. In: Glenner GG, de Freitas AF, editors. Amyloid and amyloidosis. Amsterdam: Excerpta Medica; 1980. pp. 88-98.)
-
5Adams D, Théaudin M, Cauquil C, Algalarrondo V, Slama M. FAP neuropathy and emerging treatments. Curr Neurol Neurosci Rep. 2014 Mar;14(3):435. https://doi.org/10.1007/s11910-013-0435-3
» https://doi.org/10.1007/s11910-013-0435-3 -
6Ando Y, Nakamura M, Araki S. Transthyretin-related familial amyloidotic polyneuropathy. Arch Neurol. 2005 Jul;62(7):1057-62. https://doi.org/10.1001/archneur.62.71057
» https://doi.org/10.1001/archneur.62.71057 -
7Saporta MA, Zaros C, Cruz MW, André C, Misrahi M, Bonaiti-Pellié C et al. Penetrance estimation of TTR familial amyloid polyneuropathy (type I) in Brazilian families. Eur J Neurol. 2009 Mar;16(3):337-41. https://doi.org/10.1111/j.1468-1331.2008.02429.x
» https://doi.org/10.1111/j.1468-1331.2008.02429.x -
8Schmidt H, Cruz MW, Botteman MF, Carter JA, Chopra A, Stewart M et al. Global epidemiology of transthyretin hereditary amyloid polyneuropathy: a systematic review. Amyloid. 2017 Mar 16;24(sup1):111-2. https://doi.org/10.1080/13506129.2017.1292903
» https://doi.org/10.1080/13506129.2017.1292903 -
9Planté-Bordeneuve V, Suhr OB, Maurer MS, White B, Grogan DR, Coelho T. The Transthyretin Amyloidosis Outcomes Survey (THAOS) registry: design and methodology. Curr Med Res Opin. 2013 Jan;29(1):77-84. https://doi.org/10.1185/03007995.2012.754349
» https://doi.org/10.1185/03007995.2012.754349 -
10Kristen AV, Maurer MS, Rapezzi C, Mundayat R, Suhr OB, Damy T. Impact of genotype and phenotype on cardiac biomarkers in patients with transthyretin amyloidosis - Report from the Transthyretin Amyloidosis Outcome Survey (THAOS). PLoS One. 2017 Apr;12(4):e0173086. https://doi.org/10.1371/journal.pone.0173086
» https://doi.org/10.1371/journal.pone.0173086 -
11Cruz MW, Foguel D, Berensztejn AC, Pedrosa RC, Mundayat R, Ong ML. The demographic, genetic, and clinical characteristics of Brazilian subjects enrolled in the Transthyretin Amyloidosis Outcomes Survey. Amyloid. 2017 Mar;24 sup1:103-4. https://doi.org/10.1080/13506129.2017.1291423
» https://doi.org/10.1080/13506129.2017.1291423 -
12Andrade C. A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain. 1952 Sep;75(3):408-27 https://doi.org/10.1093/brain/75.3.408).
» https://doi.org/10.1093/brain/75.3.408 -
13Ando Y, Coelho T, Berk JL, Cruz MW, Ericzon BG, Ikeda S et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013 Feb;8(1):31. https://doi.org/10.1186/1750-1172-8-31
» https://doi.org/10.1186/1750-1172-8-31 -
14Lavigne-Moreira C, Marques VD, Gonçalves MV, Oliveira MF, Tomaselli PJ, Nunez JC et al. The genetic heterogeneity of hereditary transthyretin amyloidosis in a sample of the Brazilian population. J Peripher Nerv Syst. 2018 Jun;23(2):134-7. https://doi.org/10.1111/jns.12259
» https://doi.org/10.1111/jns.12259 -
15Cortese A, Vegezzi E, Lozza A, Alfonsi E, Montini A, Moglia A et al. Diagnostic challenges in hereditary transthyretin amyloidosis with polyneuropathy: avoiding misdiagnosis of a treatable hereditary neuropathy. J Neurol Neurosurg Psychiatry. 2017 May;88(5):457-8. https://doi.org/10.1136/jnnp-2016-315262
» https://doi.org/10.1136/jnnp-2016-315262 -
16Figueroa JJ, Dyck PJ, Laughlin RS, Mercado JA, Massie R, Sandroni P et al. Autonomic dysfunction in chronic inflammatory demyelinating polyradiculoneuropathy. Neurology. 2012 Mar;78(10):702-8. https://doi.org/10.1212/WNL.0b013e3182494d66
» https://doi.org/10.1212/WNL.0b013e3182494d66 -
17Pinto MV, Barreira AA, Bulle AS, Freitas MR, França MC Jr, Gondim FA et al. Brazilian consensus for diagnosis, management and treatment of transthyretin familial amyloid polyneuropathy. Arq Neuropsiquiatr. 2018 Sep;76(9):609-21. https://doi.org/10.1590/0004-282x20180094
» https://doi.org/10.1590/0004-282x20180094
Publication Dates
-
Publication in this collection
Feb 2019
History
-
Received
09 Aug 2018 -
Reviewed
10 Oct 2018 -
Accepted
23 Oct 2018