Resumos
Objetivo: mastocitose é o termo usado para um grupo de moléstias caracterizadas pelo acúmulo de mastócitos na pele, com ou sem comprometimento de outros órgãos. Os sintomas podem ser apenas cutâneos, associados aos decorrentes dos órgãos envolvidos ou também sistêmicos, pela liberação de mediadores químicos. São descritos três casos de mastocitose sistêmica, cada um com diferentes manifestações próprias da doença.Descrição: no primeiro caso é relatado um paciente com urticária pigmentosa que evoluiu com persistência das lesões até a puberdade, quando iniciaram as manifestações sistêmicas com o comprometimento linforreticular, esplenomegalia e infiltração da medula óssea. No segundo caso, a paciente apresentava mastocitose bolhosa acompanhada de sintomas gastrintestinais, cuja investigação mostrou infiltração de mastócitos na parede intestinal. A terceira paciente ilustra a manifestação cutânea precoce e extensa da mastocitose, com uma evolução dramática para choque e posterior óbito. Comentários: são discutidos aspectos clínicos, laboratoriais, tratamento e diagnóstico.
mastocitose; urticária pigmentosa
Objective: mastocytosis comprises a group of diseases characterized by accumulation of mast cells on the skin, with the possibility of affecting other systems. Symptoms can be exclusively cutaneous, associated with symptoms of the organs involved or even systemic, due to the release of chemical mediators. Three cases of systemic mastocytosis are described, each case presenting different manifestations of the disorder. Description: the first report is about a patient with urticaria pigmentosa that presented persistent lesions until puberty when systemic manifestations initiated with lymphoreticular involvement, splenomegaly and bone marrow infiltration. In the second case, the child had bullous mastocytosis associated with gastrointestinal symptoms, whose investigation showed mast cell infiltration in the intestinal wall. The third patient presented an early and extensive cutaneous manifestation of mastocytosis, with a dramatic evolution to shock and posterior death.Discussion: clinical aspects, treatment and prognosis of such forms of the disease are discussed.
mastocytosis; urticaria pigmentosa
Mastocitose sistêmica na infância: relato de 3 casos
Systemic mastocytosis in childhood: report of 3 cases
Evódie I. Fernandes1 1 Evódie I. Fernandes Especialista em Dermatologia pela Sociedade Brasileira de Dermatologia, mestranda do Programa de Pós-graduação em Medicina: Pediatria da Universidade Federal do Rio Grande do Sul (UFRGS). 2 Beatriz C. de Faria Acadêmica do 10º semestre da Faculdade de Medicina da UFRGS. 3 André Cartell Professor Auxiliar do Dep. de Patologia da UFRGS, dermatopatologista do Hospital de Clínicas de Porto Alegre. 4 Boaventura A. dos Santos Professor Adjunto do Dep. de Pediatria da UFRGS. 5 Tania F. Cestari Professor Adjunto-Doutor do Serviço de Dermatologia da UFRGS. Professora do Curso de Pós-graduação em Medicina Pediatria, UFRGS. , Beatriz C. de Faria2 1 Evódie I. Fernandes Especialista em Dermatologia pela Sociedade Brasileira de Dermatologia, mestranda do Programa de Pós-graduação em Medicina: Pediatria da Universidade Federal do Rio Grande do Sul (UFRGS). 2 Beatriz C. de Faria Acadêmica do 10º semestre da Faculdade de Medicina da UFRGS. 3 André Cartell Professor Auxiliar do Dep. de Patologia da UFRGS, dermatopatologista do Hospital de Clínicas de Porto Alegre. 4 Boaventura A. dos Santos Professor Adjunto do Dep. de Pediatria da UFRGS. 5 Tania F. Cestari Professor Adjunto-Doutor do Serviço de Dermatologia da UFRGS. Professora do Curso de Pós-graduação em Medicina Pediatria, UFRGS. , André Cartell3 1 Evódie I. Fernandes Especialista em Dermatologia pela Sociedade Brasileira de Dermatologia, mestranda do Programa de Pós-graduação em Medicina: Pediatria da Universidade Federal do Rio Grande do Sul (UFRGS). 2 Beatriz C. de Faria Acadêmica do 10º semestre da Faculdade de Medicina da UFRGS. 3 André Cartell Professor Auxiliar do Dep. de Patologia da UFRGS, dermatopatologista do Hospital de Clínicas de Porto Alegre. 4 Boaventura A. dos Santos Professor Adjunto do Dep. de Pediatria da UFRGS. 5 Tania F. Cestari Professor Adjunto-Doutor do Serviço de Dermatologia da UFRGS. Professora do Curso de Pós-graduação em Medicina Pediatria, UFRGS. , Boaventura A. dos Santos4 1 Evódie I. Fernandes Especialista em Dermatologia pela Sociedade Brasileira de Dermatologia, mestranda do Programa de Pós-graduação em Medicina: Pediatria da Universidade Federal do Rio Grande do Sul (UFRGS). 2 Beatriz C. de Faria Acadêmica do 10º semestre da Faculdade de Medicina da UFRGS. 3 André Cartell Professor Auxiliar do Dep. de Patologia da UFRGS, dermatopatologista do Hospital de Clínicas de Porto Alegre. 4 Boaventura A. dos Santos Professor Adjunto do Dep. de Pediatria da UFRGS. 5 Tania F. Cestari Professor Adjunto-Doutor do Serviço de Dermatologia da UFRGS. Professora do Curso de Pós-graduação em Medicina Pediatria, UFRGS. , Tania F. Cestari5 1 Evódie I. Fernandes Especialista em Dermatologia pela Sociedade Brasileira de Dermatologia, mestranda do Programa de Pós-graduação em Medicina: Pediatria da Universidade Federal do Rio Grande do Sul (UFRGS). 2 Beatriz C. de Faria Acadêmica do 10º semestre da Faculdade de Medicina da UFRGS. 3 André Cartell Professor Auxiliar do Dep. de Patologia da UFRGS, dermatopatologista do Hospital de Clínicas de Porto Alegre. 4 Boaventura A. dos Santos Professor Adjunto do Dep. de Pediatria da UFRGS. 5 Tania F. Cestari Professor Adjunto-Doutor do Serviço de Dermatologia da UFRGS. Professora do Curso de Pós-graduação em Medicina Pediatria, UFRGS.
RESUMO
ABSTRACT
Objetivo: mastocitose é o termo usado para um grupo de moléstias caracterizadas pelo acúmulo de mastócitos na pele, com ou sem comprometimento de outros órgãos. Os sintomas podem ser apenas cutâneos, associados aos decorrentes dos órgãos envolvidos ou também sistêmicos, pela liberação de mediadores químicos. São descritos três casos de mastocitose sistêmica, cada um com diferentes manifestações próprias da doença.
Descrição: no primeiro caso é relatado um paciente com urticária pigmentosa que evoluiu com persistência das lesões até a puberdade, quando iniciaram as manifestações sistêmicas com o comprometimento linforreticular, esplenomegalia e infiltração da medula óssea. No segundo caso, a paciente apresentava mastocitose bolhosa acompanhada de sintomas gastrintestinais, cuja investigação mostrou infiltração de mastócitos na parede intestinal. A terceira paciente ilustra a manifestação cutânea precoce e extensa da mastocitose, com uma evolução dramática para choque e posterior óbito.
Comentários: são discutidos aspectos clínicos, laboratoriais, tratamento e diagnóstico.
mastocitose, urticária pigmentosa
Objective: mastocytosis comprises a group of diseases characterized by accumulation of mast cells on the skin, with the possibility of affecting other systems. Symptoms can be exclusively cutaneous, associated with symptoms of the organs involved or even systemic, due to the release of chemical mediators. Three cases of systemic mastocytosis are described, each case presenting different manifestations of the disorder.
Description: the first report is about a patient with urticaria pigmentosa that presented persistent lesions until puberty when systemic manifestations initiated with lymphoreticular involvement, splenomegaly and bone marrow infiltration. In the second case, the child had bullous mastocytosis associated with gastrointestinal symptoms, whose investigation showed mast cell infiltration in the intestinal wall. The third patient presented an early and extensive cutaneous manifestation of mastocytosis, with a dramatic evolution to shock and posterior death.
Discussion: clinical aspects, treatment and prognosis of such forms of the disease are discussed.
mastocytosis, urticaria pigmentosa
-
Introdução
-
Descrição dos Casos
-
Discussão
-
Referências
Introdução
A mastocitose é uma doença caracterizada pelo acúmulo desordenado de mastócitos em vários órgãos, manifestando-se através de um amplo espectro de alterações clínicas. A sua prevalência na população geral é difícil de determinar, pois muitos casos são autolimitados e/ou não diagnosticados, mas estima-se que na infância esteja próxima de 5,4 casos em cada 1.000 crianças atendidas em clínicas de dermatologia pediátrica(1).
A mastocitose é definida como sistêmica quando ocorre um aumento anormal dos mastócitos em diferentes locais, além da pele. Na infância, a doença é considerada, na maioria das vezes, uma condição benigna(2-5), cuja apresentação característica é a cutânea: mastocitoma, urticária pigmentosa e mastocitose cutânea difusa(6). O atrito ou escoriação das lesões causam urticação e eritema perilesional, caracterizando o sinal de Darier. Embora os sintomas sistêmicos sejam raros, evidências de comprometimentos múltiplos podem ser demonstradas em até 50% dos casos, dependendo também do esforço feito para detectá-los(2). A mastocitose pode envolver órgãos internos, incluindo a medula óssea, trato gastrintestinal, sistema esquelético e linforreticular (baço, fígado e linfonodos)(6). Os sintomas podem ser restritos ao órgão envolvido ou sistêmicos, devido à liberação local ou generalizada de histamina ou outros mediadores.
São descritos três casos de mastocitose sistêmica, com apresentação ou evolução diferentes do esperado, servindo de ilustração da complexidade das manifestações e prognósticos dessa doença.
Descrição dos Casos
Caso 1
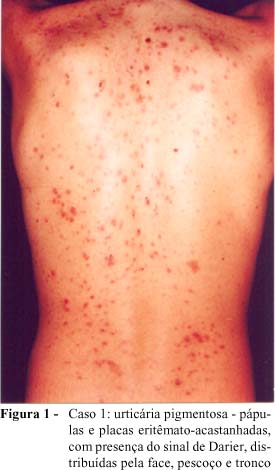
Paciente masculino, branco, 14 anos, em acompanhamento no Serviço de Dermatologia do HCPA, desde os 12 anos, com diagnóstico de urticária pigmentosa, iniciada por lesões cutâneas desde os 6 meses de vida. Ao exame dermatológico, apresentava várias pápulas e placas eritêmato-acastanhadas, com presença do sinal de Darier, distribuídas pela face, pescoço e tronco (Figura 1). O exame anátomo-patológico de biópsia de pele mostrou infiltrado mastocitário perivascular, com contagem média acima de 30 células por campo de grande aumento, compatível com mastocitose. Na avaliação da medula óssea, há dois anos, tanto o medulograma quanto a biópsia não mostraram comprometimento, e as avaliações laboratoriais hematológicas e de função hepática foram sempre normais. O paciente fez uso de corticosteróides tópicos, por períodos intermitentes, e de anti-histamínicos de forma continuada. Nos últimos meses, devido à falta de regressão do quadro cutâneo, permanecendo com lesões papulosas e infiltradas, foi realizada nova avaliação sistêmica, na qual a ecografia abdominal mostrou esplenomegalia, e o aspirado de medula óssea apresentava discreta eosinofilia e a presença de 5% de mastócitos. A biópsia da medula óssea foi normocelular. Foi mantido o uso de anti-histamínicos, com diminuição do prurido, e o paciente segue em acompanhamento hematológico, com reavaliações clínica e laboratorial a cada 3 meses.
Caso 2
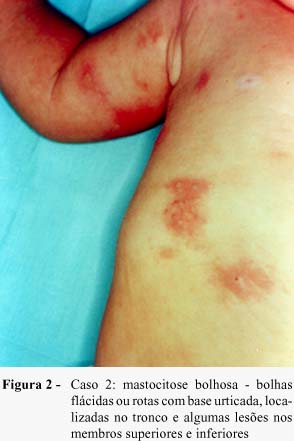
Paciente feminina, branca, 1 ano e 2 meses, residente em área rural do interior do estado, foi encaminhada para o Hospital de Clínicas de Porto Alegre para internação e investigação de lesões cutâneas bolhosas desde os 4 meses de idade, acompanhadas de diarréia e febre intermitentes. Havia feito diversos tratamentos para parasitoses intestinais e piodermites. A criança apresentava bolhas flácidas ou rotas com base urticada, localizadas principalmente no tronco, com a presença de algumas lesões nos membros superiores e inferiores (Figura 2). O hemograma, VHS e provas de função hepática foram normais, bem como as radiografias ósseas realizadas; a ecografia de abdômen mostrou fígado e baço de tamanhos usuais. As biópsias de pele do abdômen, tórax posterior e coxa confirmaram a hipótese de mastocitose bolhosa, com contagem de mastócitos acima de 30 células por campo de grande aumento. Tanto o medulograma quanto a biópsia de medula óssea não mostraram comprometimento, não tendo sido identificados mastócitos nas colorações por azul de toluidina e Giemsa. A colonoscopia e as biópsias de intestino (ceco transverso e descendente) evidenciaram mucosa com aumento da contagem de mastócitos na lâmina própria, com 15 a 20 células por campo de grande aumento. Foi iniciado tratamento com bloqueadores H1 e H2; após alguns dias, foi associado cromoglicato dissódico, mantendo-se por cerca de 10 meses. A resposta ao tratamento foi lenta, porém satisfatória, sem o surgimento de bolhas, nem sintomas digestivos nos últimos 6 meses. A criança mantém-se com hidroxizina (1 mg/kg/dia) e em acompanhamento dermatológico, com revisões a cada 4 meses.
Caso 3
Paciente feminina, branca, 6 meses de idade, foi trazida à emergência pediátrica do Hospital de Clínicas de Porto Alegre devido à sibilância e dificuldade respiratória, acompanhadas de manchas eritematosas e pruriginosas, recorrentes, na face e extremidades, desde os 15 dias de vida, sendo que, inicialmente, as lesões eram vesiculosas. Ao exame, apresentava pápulas eritematosas, com sinal de Darier positivo, distribuídas pela face, membros superiores e inferiores, linfadenopatia cervical bilateral e hepatoesplenomegalia (Figura 3). A paciente foi internada, iniciou tratamento para o quadro respiratório com antibióticos e broncodilatadores, sendo submetida a extensa investigação. Os exames mostraram anemia, plaquetopenia, VHS elevado e alteração das enzimas hepáticas. A ecografia abdominal confirmou a hepatoesplenomegalia. O medulograma evidenciou hiperplasia eritróide e megaloblastose, e a biópsia de medula óssea, o predomínio da série vermelha. A biópsia e exame anátomo-patológico de uma das lesões cutâneas apontou infiltrado mononuclear, com predomínio de linfócitos e mastócitos perivasculares (10 a 15 células por campo de grande aumento). As radiografias e cintilografias ósseas não apresentaram alterações. A criança recebeu, durante a internação, tratamento com anti-histamínicos H1 e H2, cromoglicato, corticóides sistêmicos e ciclosporina. Apesar dos esforços, ela evoluiu desfavoravelmente, com agravamento do quadro respiratório e posterior insuficiência respiratória e óbito, por choque irreversível.
Discussão
Os mastócitos têm a capacidade de gerar uma grande quantidade de mediadores que são responsáveis por efeitos nos órgãos alvos, a curto e a longo prazo3. O conjunto de sintomas clínicos associados à mastocitose é o resultado do aumento da produção ou liberação desses mediadores, os quais agem tanto nos seus locais de produção quanto em zonas distantes da sua origem, independentemente da etiologia do aumento dos mastócitos ou da categoria da doença. Esses mediadores podem circular através dos vasos sangüíneos ou linfáticos para produzir os efeitos biológicos observados nos pacientes com mastocitose(3).
O início das manifestações da mastocitose pode ocorrer em qualquer idade, porém, em mais da metade dos pacientes é visto entre o nascimento e os 2 anos(3,7), como nos casos aqui descritos. Embora muitos órgãos possam estar envolvidos, a apresentação mais comum é a cutânea(8-11). As expressões clínicas da mastocitose cutânea são telangiectasia macularis eruptiva perstans, mastocitomas, mastocitose cutânea difusa e urticária pigmentosa (UP)(8). As quatro categorias têm características clínicas que são devidas ao excesso de produção de mediadores dependentes dos mastócitos, embora alguns aspectos da doença possam predominar em um tipo específico.
O diagnóstico da doença é suspeitado clinicamente e confirmado pela histologia(3). Atualmente, a demonstração de um aumento de mastócitos em lesões cutâneas características se constitui no teste padrão-ouro para o diagnóstico de mastocitose. Em certos pacientes suspeitos da forma sistêmica, mas nos quais faltam os sintomas e os sinais característicos da doença, testes como níveis séricos ou urinários de mediadores de mastócitos ou seus metabólitos podem ser úteis (histamina, prostaglandina D2, triptase e N-metil-histamina, entre outros)(8,12). Os estudos adicionais são determinados pelos sintomas ou pelos achados durante a avaliação rotineira(13).
A mastocitose cutânea recebe a denominação de telangiectasia macularis eruptiva perstans quando as telangiectasias são a característica predominante, com pouca pigmentação e edema. Praticamente não ocorre na infância, mas nos adultos ela é persistente e pouco responsiva ao tratamento.
Lesões solitárias chamadas de mastocitomas são umas das apresentações características da mastocitose em pediatria6. Representam 15% dos casos da moléstia e, na maioria das vezes, as lesões involuem espontaneamente. Apresentam-se como nódulos vermelhos, rosas ou amarelados, com 3 a 4 cm de diâmetro, podendo ser múltiplos. Na infância, são assintomáticos ou com edema quando friccionados, podendo desencadear flushing e hipotensão(6).
A mastocitose cutânea difusa é uma variedade rara e consiste de uma infiltração difusa da pele com mastócitos, podendo ter lesões isoladas. Usualmente todo o tegumento está envolvido, manifestando-se através de um espessamento difuso, de consistência empastada, semelhante ao couro, com coloração normal ou amarelo-acastanhada e prurido intenso. Os locais mais afetados são axilas e região inguinal, porém a apresentação pode ser sistêmica com hepatoesplenomegalia(2,3).
Na infância, entre as expressões cutâneas da mastocitose, a que acorre com maior freqüência é a urticária pigmentosa. Ela é vista em mais de 90% dos pacientes com doença leve, e em menos de 50% daqueles com mastocitose associada à doença hematológica ou com linfadenopatia e eosinofilia2. As lesões de UP caracterizam-se por máculas eritêmato-acastanhadas ou pápulas levemente elevadas e esparsas, com as bordas pouco definidas. As palmas, plantas, face e couro cabeludo podem estar livres de lesões. Traumas leves, incluindo escoriação ou atrito, normalmente causam urticação e eritema perilesional, conhecidos como sinal de Darier. Urticária pigmentosa está associada com uma intensidade variável de prurido, que pode ser exacerbado por alterações climáticas, fricção da pele, ingestão de bebidas quentes ou comidas condimentadas, álcool e certos medicamentos, todos mecanismos que favorecem a degranulação de mastócitos com a liberação de mediadores químicos(8,9,11).
A UP, na maioria dos casos, evolui com regressão espontânea da doença, antes ou durante a puberdade. Porém, quando isso não ocorre, como no caso 1 aqui descrito ou nos pacientes que apresentam alterações sangüíneas periféricas, linfadenopatia, hepato ou esplenomegalia, deve ser realizada uma investigação sistêmica mais detalhada, pois manifestações ou sinais clínicos só são evidentes quando o comprometimento já é extenso. Amostras de tecidos linfáticos, baço, fígado e mucosa gastrintestinal ajudam a definir a extensão da doença(3).
Lesões cutâneas bolhosas podem manifestar-se e acompanhar todas as formas de mastocitose, porém, quando essa apresentação é a predominante, é denominada de mastocitose bolhosa (MB)(14). Esta variedade, mais rara, ocorre em crianças e está associada com envolvimento sistêmico(4) e prognóstico mais reservado(9), inclusive com o relato de casos que evoluíram a óbito(14). De acordo com Murphy, os pacientes com um alto risco de choque ou morte súbita seriam aqueles com lesões bolhosas extensas, sintomas de vasodilatação, flushing ou hipotensão e aqueles com início muito precoce da doença, ainda no período neonatal(14). Para essas crianças, estaria recomendado um acompanhamento mais cuidadoso e orientação dos pais com relação aos potenciais degranuladores de mastócitos. O achado sistêmico da paciente com MB aqui relatada, orientado pelos sintomas e após extensa investigação, foi o comprometimento intestinal. O envolvimento do trato gastrintestinal pela mastocitose é relacionado na literatura como um evento não tão comum(9). As principais manifestações em crianças seriam os sintomas gastrintestinais por ulceração péptica e mal-absorção com atrofia de vilos. Hipersecreção gástrica, devida ao aumento plasmático de histamina resultando em gastrite e úlcera péptica, seriam os problemas mais comuns, bem como diarréia e dor abdominal, relacionadas com mal-absorção em até um terço dos casos(3,6). É interessante destacar que no presente caso os sintomas intestinais eram repetidamente interpretados como secundários à infecção intestinal, vista freqüentemente na infância, principalmente em crianças residentes em meio rural e, ainda mais, quando associada às lesões cutâneas muito semelhantes a piodermites.
Além do comprometimento do trato gastrintestinal, a mastocitose pode atingir outros órgãos, como o sistema linforreticular, sistema esquelético e a medula óssea. Hepatoesplenomegalia pode ser detectada em 40%, envolvimento ósseo visto em radiografias em 57%(15), e alterações hematológicas, como anemia, leucocitose e eosinoflia em mais de 50%(3).
Manifestações hepáticas e esplênicas, inclusive hipertensão porta e ascite, devida à fibrose hepática, são mais comuns em pacientes com mastocitose associada com doenças hematológicas ou naqueles com mastocitose agressiva.
A infiltração da medula óssea com mastócitos pode incluir alterações ósseas, que causam lesões detectáveis em radiografias na maioria dos casos, através de lesões líticas ou condensadas(5). Os ossos longos proximais são os mais comumente afetados, seguidos pela pelve, costelas e crânio. A cintilografia óssea é o exame mais sensível para detectar e localizar lesões ativas. Dores ósseas acometem de 19 a 28% dos pacientes e, naqueles com doença mais grave ou avançada, podem ocorrer fraturas patológicas(3).
As alterações de medula óssea consistem de agregados paratrabeculares de mastócitos fusiformes, freqüentemente misturados com eosinófilos, linfócitos e ocasionalmente plasmócitos, histiócitos e fibroblastos. Elas raramente são vistas em crianças. Anemia, leucopenia, leucocitose, trombocitopenia ou eosinofilia podem, entretanto, ocorrer em associação com doença sistêmica(5). Sangramento prolongado na pele ou no trato gastrintestinal tem sido observado nos pacientes pediátricos com mastocitose, principalmente naqueles com mastocitose cutânea difusa(6). Além dessas manifestações hematológicas, já foi relatada a ocorrência de displasia ou neoplasia de células mielóides com mastocitose sistêmica, principalmente em pacientes adultos(10,16). Há somente dois casos relatados de doença maligna hematológica em crianças, ambos com mastocitose sistêmica, e não há nenhum relato da coexistência de urticária pigmentosa isolada com doença maligna linfoproliferativa em crianças(17).
Pacientes com mastocitose muitas vezes apresentam manifestações cardiovasculares, como flushing, hipotensão, taquicardia, síncope e choque(2,4,8). Tais reações são secundárias aos efeitos dos mediadores, liberados pelos mastócitos por agentes que estimulam a sua degranulação. Entre os degranuladores mais relevantes estão as toxinas bacterianas, os estímulos físicos (calor, frio, luz solar, fricção), venenos (cobras, Hymenoptera), polipeptídeos biológicos (de áscaris, água-viva, lagosta, veneno de vespa e abelha), polímeros (dextrano) e medicamentos (ácido acetilsalicílico, codeína, morfina, polimixina B, quinina, contrastes radiográficos, escopolamina, galamina, decametônio, reserpina)(2,4,8,18).
Uma das causas prováveis de morte na paciente do caso 3 pode ter sido a hipotensão secundária à degranulação maciça dos mastócitos, precipitada por algum agente. Entre esses agentes, poderiam ser relacionados as toxinas bacterianas, uma vez que encontrava-se internada numa unidade de terapia intensiva e em ventilação mecânica. Outra hipótese para o óbito, associada ou não à degranulação dos mastócitos, seria a sepse, com falência de múltiplos órgãos.
O principal objetivo do tratamento de todas as categorias de mastocitose é o controle dos sinais e sintomas determinados ou provocados pela liberação dos mediadores dos mastócitos. Os pacientes devem sempre evitar e prevenir o uso de agentes degranuladores dos mastócitos. Tanto em adultos quanto em crianças, os antagonistas dos receptores H1 são úteis em reduzir prurido, flushing, o sinal de Darier e a taquicardia. Se forem insuficientes, a adição de um antagonista H2 pode ser benéfica. Apesar disso, muitas vezes os sintomas persistem, em parte pela incapacidade de bloquear os efeitos dos altos níveis de histamina e a presença de outros mediadores(3).
O cromoglicato dissódico inibe a degranulação dos mastócitos e pode ter alguma eficácia no tratamento, particularmente no alívio dos sintomas gastrintestinais, apesar de não diminuir os níveis séricos ou urinários de histamina(3).
Entre outros tratamentos citados na literatura, na maioria relatos ou série de casos, estão: cetotifeno, como estabilizador da membrana celular dos mastócitos e com propriedades anti-histamínicas3,5; adrenalina, usada nos episódios de colapso vascular2; tratamento fotoquimioterápico com psoraleno e ultravioleta A, que induz uma diminuição temporária dos mastócitos dérmicos5; interferons associados ou não com corticóides sistêmicos, para pacientes com mastocitose sistêmica19. Glicocorticóides tópicos podem ser usados na UP extensa ou na mastocitose cutânea difusa. O número de mastócitos diminui à medida que as lesões se tornam mais claras; elas eventualmente podem recorrer após o término do tratamento, mas a melhora pode durar mais de 1 ano. Nas crianças, os corticóides tópicos devem ser usados em pequenas áreas e com muita cautela, pelo risco de supressão adrenal devida ao uso crônico(6).
Os pacientes com UP ou com mastocitoma têm, em geral, um excelente prognóstico. O número de lesões na UP pode continuar a aumentar após o seu início, primeiro de uma forma rápida e após gradualmente até atingir um platô. Cerca de metade dos casos de UP na infância apresentarão resolução das lesões e sintomas durante a adolescência, e os casos restantes exibirão uma acentuada redução nos sintomas das lesões cutâneas. Contudo, a progressão para comprometimento sistêmico ocorre em 10% das crianças com UP, nas quais a primeira manifestação aparece após os 5 anos de idade(7). Crianças que apresentam lesões bolhosas como primeiro sinal de mastocitose parecem ter um prognóstico pior em relação àquelas crianças com mastocitose cutânea difusa, com posterior surgimento de bolhas(9,14). É importante salientar que a associação ou o desenvolvimento de doenças malignas, como leucemia de mastócitos, é rara em crianças com mastocitose, ao contrário do que ocorre em adultos(8,11).
Apesar da relativa raridade da mastocitose, seu diagnóstico é importante, tanto pela multiplicidade de manifestações cutâneas quanto pelo risco associado aos sintomas dela decorrentes, ilustrando a complexidade clínica que pode acompanhar a doença e a necessidade da colaboração próxima do pediatra e do dermatologista no tratamento e orientação desses pacientes.
Sobe
Endereço para correspondência:
Dra. Tania F. Cestari
Fax: 51 3346.2773
E-mail: tcestari@terra.com.br
Sobe
Artigo submetido em 03.09.01, aceito em 30.01.02.
- 1. Fernández AT, Campoamor LN, Mora LE, Zambrano AZ. Diagnóstico, tratamiento y clasificación de la mastocitosis pediátrica. Estudio de 172 casos. Actas Dermosifiliogr 1998;89:461-76.
- 2. Greaves MW. Mastocytosis. In: Champion RH, Burton JL, Burns DA, Breathnach SM, eds. Textbook of Dermatology. 6th ed. Oxford: Blackwell Science; 1998. p.2337-46.
- 3. Metcalfe DD. The mastocytosis syndrome. In: Frredberg IM, Eisen AZ, Wolff K, Austen KF, Goldsmith LA, Katz SI, et al., eds. Dermatology in general medicine. 5th ed. Austen: Mc Graw Hill;1999. p.1902-08.
- 4. Longley J, Duffy TP, Kohn S. The mast cell and mast cell disease. J Am Acad Dermatol 1995;32:546-61.
- 5. Katsambas AD, Karpouziz AJ, Koumantaki-Mathioudaki E, Jorizzo JL. Mastocytosis with skin manifestations: current status. J Eur Acad Dermatol Venereol 1999;13:155-65.
- 6. Kettelhut BV, Metcalfe DD. Pediatric mastocytosis. J Invest Dermatol 1991;96:15S-18S.
- 7. Metcalfe DD. Classification and diagnosis of mastocytosis: current status. J Invest Dermatol 1991;96:2S-4S.
- 8. Van Gysel D, Oranje AP. Mastocytosis. In: Harper J, Oranje A, Prose N, eds. Textbook of Pediatric Dermatology. 1st ed. Oxford: Blackwell Science; 2000. p.600-9.
- 9. DiBacco RS, De Leo VA. Mastocytosis and the mast cell. J Am Acad Dermatol 1982;7:709-92.
- 10. Travis WD, Li CH, Bergstralh EJ, Yam LT, Swee RG. Systemic mast cell disease: analysis of 58 cases and literature review. Medicine 1988;67:345-68.
- 11. Stein DH. Mastocytosis: a review. Pediatric Dermatol 1986;3:365-75.
- 12. Tharp MD. Mast cell disease and its diagnosis. J Invest Dermatol 1995;104(6):885-6.
- 13. Setcalfe DD. Conclusions. J Invest Dermatol 1991;96:64S-65S.
- 14. Murphy M, Walsh D, Drumm B, Watson R. Bullous mastocytosis: a fatal outcome. Pediatric Dermatol 1999;16:452-5.
- 15. Horan RF, Austen KF. Systemic mastocytosis: retrospective review of a decade's clinical experience at the Brigham and Women's Hospital. J Invest Dermatol 1991;96:5S-14S.
- 16. Horny HP, Ruck M, Wehrmann M, Kaiserling E. Blood findings in generalized mastocytosis: evidence of frequent simultaneous occurrence of myeloproliferative disorders. Br J Hematol 1990;76:186-93.
- 17. Güler E, Emir S, Kutluk T, Varan A, Büyükpamukçu. Urticaria pigmentosa associated with Wilms tumor. Pediatr Dermatol 2001;18:313-5.
- 18. Leaf FA, Jaecks EP, Rodriguez DR. Bullous urticaria pigmentosa. Cutis 1996;58:358-60.
- 19. Delaporte E, Piérard E, Wolthers BG, Desreumaux P, Janin A, Cartot A, et al. Interferon-a in combination with corticosteroids improves systemic mast cell disease. Br J Dermatol 1995;132:479-94.
Datas de Publicação
-
Publicação nesta coleção
26 Nov 2002 -
Data do Fascículo
Abr 2002
Histórico
-
Recebido
03 Set 2001 -
Aceito
30 Jan 2002