Abstracts
OBJECTIVES: Although recognized for decades, little is known about the etiology, physiopathology, and prevention of persistent pulmonary hypertension of the newborn (PPHN). and its treatment remains a major challenge for neonatologists. In this review, the clinical features and physiopathology of the syndrome will be addressed, as well as its general and specific treatments. DATA SOURCE: A review was carried out in PubMed, Cochrane Library, and MRei consult databases, searching for articles related to the syndrome and published between 1995 and 2011. DATA SYNTHESIS: Risk factors and the physiopathological mechanisms of the syndrome are discussed. The clinical picture depends on the different factors involved, which are probably related to the etiology and physiopathological mechanisms. In addition to the measures used to allow the decrease in pulmonary vascular resistance after birth, some cases will require pulmonary vasodilators. Although nitric oxide has proved effective, other vasodilators have been recently used, but clinical evidence is still lacking to demonstrate their benefits in the treatment of PPHN. CONCLUSIONS: Despite recent technological advances and new physiopathological knowledge, mortality associated with PPHN remains at 10%. More clinical research and evidence-based experimental results are needed to prevent, treat, and reduce the morbidity/mortality associated with this neonatal syndrome.
Pulmonary hypertension; Pulmonary vasodilators; Nitric oxide
OBJETIVOS: Embora reconhecida há décadas, ainda pouco se sabe a respeito da etiologia, fisiopatologia e prevenção da hipertensão pulmonar persistente neonatal (HPPN), e seu tratamento continua a ser um grande desafio para os neonatologistas. Nesta revisão, vamos abordar as características clínicas e os mecanismos fisiopatológicos da síndrome, assim como seu tratamento geral e específico. FONTES DE DADOS: Fizemos uma revisão nas bases de dados PubMed, Cochrane Library e MRei Consult , procurando por artigos relacionados à síndrome e publicados entre 1995 e 2011. SÍNTESE DE DADOS: São discutidos os fatores de risco e os mecanismos fisiopatológicos da síndrome. O quadro clínico depende dos diferentes fatores envolvidos, que provavelmente estão relacionados com a etiologia e o mecanismo fisiopatológico. Além das medidas utilizadas para permitir a queda da resistência vascular pulmonar após o nascimento, alguns casos necessitam de vasodilatadores pulmonares. Embora o óxido nítrico tenha se provado efetivo, recentemente, outros vasodilatadores têm sido usados, mas ainda faltam evidências clínicas para comprovar seus benefícios no tratamento da HPPN. CONCLUSÕES: Apesar dos recentes avanços tecnológicos e dos novos conhecimentos fisiopatológicos, a mortalidade associada à HPPN ainda é de 10%. São necessárias mais pesquisas clínicas e resultados experimentais baseados em evidências para prevenir, tratar e reduzir a morbimortalidade associada a esta síndrome neonatal.
Hipertensão pulmonar; Vasodilatadores pulmonares; Óxido nítrico
REVIEW ARTICLE
INeonatologist, Hospital São Luiz, São Paulo, SP, Brazil
IINeonatologist. Professor of Pediatrics and Physiology, University of Toronto, The Hospital for Sick Children, Toronto, Canada
ABSTRACT
OBJECTIVES: Although recognized for decades, little is known about the etiology, physiopathology, and prevention of persistent pulmonary hypertension of the newborn (PPHN), and its treatment remains a major challenge for neonatologists. In this review, the clinical features and physiopathology of the syndrome will be addressed, as well as its general and specific treatments.
DATA SOURCE: A review was carried out in PubMed, Cochrane Library, and MRei consult databases, searching for articles related to the syndrome and published between 1995 and 2011.
DATA SYNTHESIS: Risk factors and the physiopathological mechanisms of the syndrome are discussed. The clinical presentation depends on the different factors involved. These are related to the etiology and physiopathology of the different forms of the disease. In addition to the measures used to allow for the decrease in pulmonary vascular resistance after birth, in some instances pulmonary vasodilators will be required. Although inhaled nitric oxide has proved effective, other vasodilators have been recently used, but clinical evidence is still lacking to demonstrate their benefits in the treatment of PPHN.
CONCLUSIONS: Despite recent technological advances and new physiopathological knowledge of this disease, mortality associated with PPHN remains at 10%. More clinical research and evidence-based experimental results are needed to prevent, treat, and reduce the morbidity/mortality associated with this neonatal syndrome.
Keywords: Pulmonary hypertension; Pulmonary vasodilators; Nitric oxide
Introduction
Persistent pulmonary hypertension of the newborn (PPHN) is a syndrome that, although recognized for over 30 years, continues to challenge physicians, and little is known about its etiology, pathogenesis, and prevention. With the exception of inhaled nitric oxide (NO), treatment is limited and the use of new drugs is based solely on experimental evidence, or in the treatment of adults with primary pulmonary hypertension.
The clinical features of the syndrome and its general and specific treatment were reviewed in the present study. For a better understanding of its pathogenesis and the use of certain pharmacological interventions, an overview of the factors responsible for the control of pulmonary vascular tone and hemodynamic alterations during the transition from fetal to postnatal life is necessary.
Treatment consists of general measures to allow the physiologic decrease in pulmonary vascular resistance after birth and the use of specific therapies, when indicated. Despite the common terminology, pulmonary hypertension of the newborn and primary pulmonary hypertension in adults are quite distinct diseases. The neonatal form is much more frequent than the adult disease, but it has a better prognosis (Table 1).
Pulmonary vascular tone control
The control of vascular resistance to blood flow is exerted by the arterial musculature. Contraction of this muscle, present in the arterial wall middle layer, leads to reduction of its lumen, increased resistance to blood flow, and consequent decreased distal perfusion. Although both arteries and veins contain smooth musculature, the control of blood flow is mainly performed by small-caliber arterioles.
The pulmonary circulation is completely distinct from the systemic one. In the postnatal period, the systemic circulation exhibits a much higher vascular resistance than the pulmonary, and responds to stimuli such as partial oxygen pressure and alterations in blood differently than the pulmonary circulation. Hypoxemia dilates the systemic circulation, whereas the opposite is observed in the pulmonary arteries. In this review only the pulmonary circulation will be discussed, but the reader should keep in mind that the physiology of the two circulations is largely distinct, and this fact has important clinical implications, mainly regarding the use of vasoactive drugs.
The control of the vascular muscle is largely determined by factors produced by endothelial cells. Production of NO, prostacyclin, and vascular endothelial growth factor (VEGF) by endothelial cells leads to relaxation, whereas endothelin, thromboxane, and prostaglandin F2e induce contraction of the pulmonary vascular smooth muscle. Blood flow has a direct effect on dilation/constriction of the vessel through the shear stress phenomenon. NO is produced in proportion to this shear stress. Therefore, the higher the flow, greater is the shear stress and therefore, the tissue perfusion.
Some blood factors also control the pulmonary flow, and oxygen and pH have the greatest clinical importance. The location of the oxygen sensors in the lungs remains unclear, but probably involves precapillary arterioles close to the alveolus. Thus, it is not an increase in arterial oxygen pressure (PaO2) that leads to pulmonary vasodilation, but alveolar oxygen tension. Regarding the pH, the pulmonary circulation responds with vasodilation in the presence of alkalosis and vasoconstriction in response to acidosis.
Fig. 1 shows the complexity of the cascade responsible for pulmonary vasodilation that depends on NO (cyclic GMP) and prostacyclin (cyclic AMP). Both lead to stimulation of the myosin light chain phosphatase, and dephosphorylation of the latter leads to relaxation of the vascular smooth muscle. Conversely, many factors have an inhibitory effect on the vasorelaxation process. Below is a summarized description of some of these factors according to their importance in different clinical presentations of this syndrome and the use of drugs that act through their modulation.
NO is synthesized by the vascular endothelium from Larginine in response to several stimuli.1 The nitric oxide synthase converts L-arginine to L-citrulline, releasing NO in this reaction. There are three nitric oxide synthase isoforms. The endothelial (eNOS), present in endothelial cells, and the neuronal (nNOS), present in muscle cells, are responsible for production of NO under physiological conditions, whereas the inducible form (iNOS) is only activated during inflammatory processes.
Based on evidence from animal models , which have demonstrated that the expression of eNOS is maximal at birth, an important role has been attributed to NO produced by this enzyme in pulmonary vasodilation.2,3 However, it is currently known that an endogenous inhibitor of this enzyme, asymmetric dimethylarginine (ADMA) is at much higher levels in the fetus and newborn, including humans, in comparison to adults.4 This, associated with the fact that the concentration of arginases, which are enzymes that compete with eNOS for the L-arginine substrate, is increased during fetal and immediate postnatal periods,5 calls into question the importance of NO in the pulmonary vasodilation that occurs at birth.
NO stimulates the soluble guanylate cyclase (sGC) enzyme in pulmonary vascular smooth muscle cells, leading to the conversion of guanosine triphosphate (GTP) nucleotide into cyclic guanosine monophosphate (cGMP). The increase in intracellular cGMP leads to a decrease in calcium influx and relaxation of smooth muscle cells by stimulating protein kinase G.6,7 Based on evidence obtained in animal models, it is considered that the contents of pulmonary sGC is higher in the fetus and newborn and decreases with age.8--11 The phosphodiesterase 5 (PG5) enzyme present in the pulmonary vasculature degrades cGMP, and thus controls the degree of vasodilation.12 Factors related to development have a great importance in the generation of cGMP. Pulmonary hypertension is associated with increased PG5 activity,13,14 and inhibition of this enzyme with sildenafil is currently one of the pharmacological interventions used in this disease.15,16
The prostaglandin (PG) system is also involved in the fetal-newborn pulmonary circulation transition. It causes vasodilatation through a parallel pathway that is complementary to NO, and can thus potentiate its action (Fig. 1). Prostaglandins are synthesized from arachidonic acid, and vascular smooth muscle cells activate the adenylate cyclase enzyme, which converts adenosine triphosphate (ATP) into cyclic adenosine monophosphate (cAMP). An intracellular increase of this enzyme also results in relaxation of the vascular smooth musculature by decreasing the calcium influx. Prostacyclin (PGI2) is considered the most potent in this system, although prostin (PGE1), normally used to maintain patency of the ductus arteriosus, has also been shown to be an effective pulmonary vasodilator in newborns with pulmonary hypertension.17
The phosphodiesterase type III (PG3) enzyme degrades cAMP and controls the degree of vasodilation.18 Endothelin 1 (ET-1) is a member of the endothelin family, and its effect on pulmonary vascular resistance is mediated by two receptor subtypes: ETA and ETB. Both are present in the smooth muscle cell and cause vasoconstriction and cell proliferation. ETB receptors are also present in the vascular endothelium, where they contribute to the regulation of pulmonary vascular tone through the release of NO. During fetal life, the maintenance of high pulmonary vascular tone is supported in part by ET-1, and its level is elevated in animal models of PPHN.19
Several other substances involved in the fetal circulation transition are being identified. Experimental studies suggest that superoxide radicals, generated by oxidative stress, decouple eNOS (Fig. 1), and compete with and inhibit the biological action of NO by generating peroxynitrite.20 This molecule, in addition to its deleterious effects on several cell functions, also has an important vasoconstrictor effect in the newborn rat.21 Recent studies have demonstrated that VEGF releases NO, and induces pulmonary vasodilation by increasing cGMP activity.22 Pharmacological inhibition of VEGF induces pulmonary hypertension in newborn and adult rats, showing the importance of this factor in pulmonary vascular angiogenesis.23--25 The intracellular levels of cyclic cGMP are also increased by natriuretic, atrial (ANP), and type B (BNP) peptides, which stimulate particulate guanylate cyclase, an isoform of sGC.7,26,27
Another molecule that plays an important role in the control of pulmonary vascular tone is the rho-kinase. When activated, this molecule has an inhibitory effect on the myosin light chain phosphatase, preventing the relaxation of vascular smooth muscle (Fig. 1). Rho-kinase is increased in animal models of pulmonary hypertension of the newborn, and its inhibition reduces disease severity in several animal models of pulmonary hypertension of the newborn, and its inhibition reduces disease severity in several animal models.28-32
Hemodynamic alterations during birth
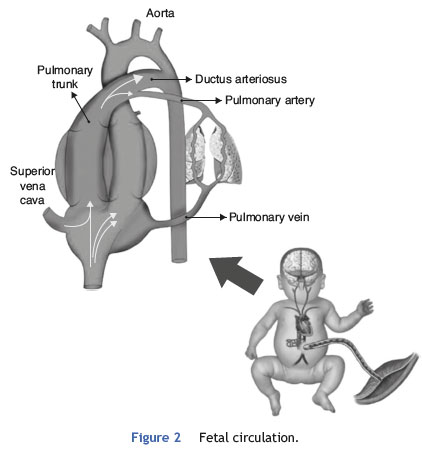
Fetal pulmonary circulation is characterized by elevated pulmonary vascular resistance and the presence of right-toleft shunting via the ductus arteriosus and foramen ovale. These channels allow blood flow from the right atrium to reach the aorta, since only 10% of the cardiac output of the right ventricle reaches the lungs, as a result of high fetal pulmonary vascular resistance.33,34Fig. 2 outlines the characteristics of fetal circulation.
Several mechanisms contribute to the maintenance of high pulmonary vascular resistance in the fetal period. The main mechanisms include low oxygen tension, decreased production of vasodilators (such as NO and prostaglandins with vasodilator characteristic [prostacyclin]), and increased production of vasoconstrictive prostaglandin (thromboxane) and others such as endothelins.
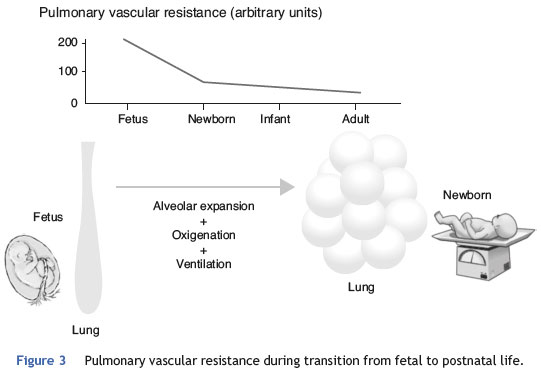
Minutes after birth, the pulmonary arterial pressure rapidly drops to 50% of the systemic pressure, and pulmonary blood flow increases by approximately ten times with the onset of respiration. The factors responsible for this sudden decrease in vascular resistance after birth are related to the expansion and oxygenation of the alveoli, onset of continuous respiration, and clamping of the umbilical cord (Fig. 3). It is believed that pulmonary vascular resistance depends on the association between humoral constricting and dilating factors. During fetal life, constricting factors predominate, whereas dilating factors prevail after birth.
Definition
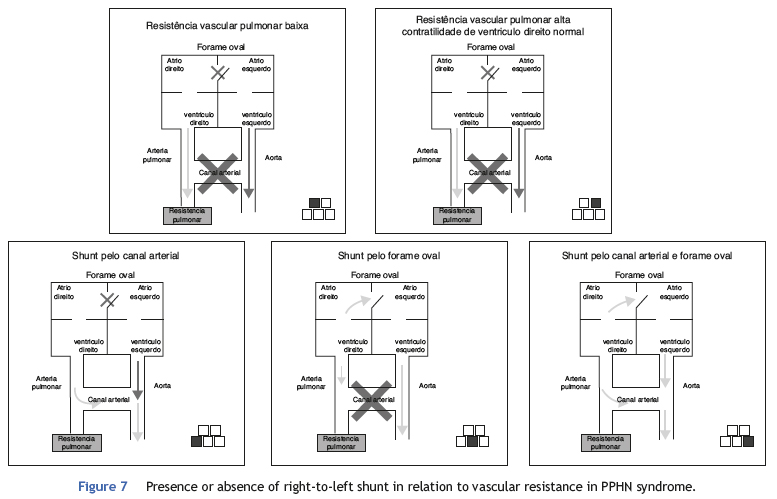
PPHN is a syndrome characterized by the presence of elevated pulmonary vascular resistance and right-left shunt through the ductus arteriosus and/or foramen ovale. Contrary to primary pulmonary hypertension in adults, the newborn syndrome is not defined by a specific pressure of the pulmonary circulation (Table 1). The diagnosis of PPHN is confirmed regardless of the pulmonary arterial pressure, as long as it is accompanied by right-left shunt and absence of congenital heart abnormalities. This concept is very important, as not only the increase in pulmonary vascular resistance, but also the capacity of the right ventricle to overcome this resistance, are determining factors in neonatal pulmonary hypertension.
Incidence and mortality prevalence
PPHN affects mainly at-term or post-term newborns, births.37 In comparison with these international statistics, although also present in premature infants.35 The prevabetween 2000 and 2005, Hospital São Luiz, a service with lence of PPHN in newborns is not well characterized, and 8,000 deliveries/year and a high-complexity neonatalis probably underestimated due to failure in its detection intensive care unit with 54 beds, recorded 2 cases/1,000 when associated with parenchymal pathology. A recent live births, and mortality rates of 11.6%.
Etiology
The etiology of PPHN is considered to be multiple. Certain maternal conditions such as obesity, diabetes, asthma, black or Asian ethnicity, and other neonatal factors such as postmaturity and neonates born large for gestational age are associated with a higher incidence of PPHN.38
The condition most commonly associated with PPHN in the United States is the meconium aspiration syndrome (42%), followed by the idiopathic (27%). Other conditions include respiratory distress syndrome, sepsis, asphyxia, and pulmonary hypoplasia secondary to congenital diaphragmatic hernia.39
In the Hospital São Luiz, the prevalence of the idiopathic form was 70%, much higher when compared to U.S. services.39 This is probably due to the high incidence of cesarean delivery in Brazil. Newborns delivered by cesarean section with no labor have a significantly higher chance of developing PPHN than infants born to mothers with labor or with normal vaginal delivery.40 Labor is associated with the interruption of production and increased resorption of alveolar fluid, thus preventing respiratory distress, characteristic of the transient tachypnea of the newborn.41 Other conditions associated with PPHN at Hospital São Luiz included infection, meconium aspiration syndrome, and respiratory distress syndrome (Fig. 4).
Among the drugs used by the mother, the most commonly associated with PPHN are anti-inflammatory drugs and serotonin-reuptake inhibitors antidepressants. The antiinflammatory drugs lead to premature closing of the ductus arteriosus by decreasing the release of prostaglandins, through the inhibition of cyclooxygenase enzymes.
In animal models, early closure of the ductus arteriosus induces alterations in the pulmonary vasculature very similar to those observed in newborns with PPHN.42,43 Early closure of the ductus arteriosus is probably an uncommon cause of pulmonary hypertension syndrome. Fetal echocardiographic studies have shown that there is indeed a constrictive effect when inhibitors cyclooxygenase inhibitors are used, and that this effect is increased when corticosteroids are also administered.44,45 This constricting effect of cyclooxygenase inhibitors, however, may is transient even in women receiving continuous treatment.46
More data are needed to determine the role of the early closure of the ductus arteriosus during pregnancy in the pathogenesis of PPHN.
Recent studies have demonstrated an association between antidepressants used by mothers in the third rimester of pregnancy and PPHN syndrome.47-49 The prevalence in newborns exposed during fetal life to selective serotonin-reuptake inhibitors is four fold increased. In pregnant rats, administration of fluoxetine induces pulmonary vascular changes in the fetus, comparable to those observed in human infants with PPHN.50 Recently, however, at least one study involving 1,104 infants born to mothers who received antidepressants in the third trimester and an equal number of controls failed to demonstrate this association.51 More research in this area is needed to clarify whether there is an association between PPHN and exposure during fetal life to selective serotonin-reuptake inhibitors.
Physiopathology
Considering that PPHN is a syndrome secondary to increased pulmonary vascular resistance, to the point of inducing right-left shunt, its physiopathology is related to the major factors that lead to these alterations during the perinatal period. The following classification illustrates the different categories of diseases associated with increased pulmonary vascular resistance. A disease classified in one category often has manifestations characteristics of another form. Fig. 5 illustrates some of the forms described below.
Vasoconstriction
This category includes pathologies where the number of vessels, the pulmonary structure, and the pulmonary vascular branching are normal, but the smooth musculature of vessels responsible for the resistance to blood flow is constricted (Fig. 5A). These pathologies show disequilibrium in the balance of vasoactive substances, with a predominance of vasoconstrictor substances over vasodilators. This category is usually associated with diseases or conditions of acute characteristics, such as perinatal asphyxia, sepsis, or metabolic acidosis. As they are associated to vasoconstriction, pathologies related to this form of PPHN are amenable to treatment with vasodilators.
Pulmonary vascular remodeling
Diseases classified in this category exhibit typical histological characteristics, with an increased layer of arterial vascular smooth muscle and its extension to intra-acinar arteries, which are not usually muscularized (Fig. 5B). Evidence obtained from animal models with pulmonary hypertension indicates that excessive musculature of the vascular wall has an important role in vasoconstriction. The increase in pulmonary vascular resistance in these pathologies is associated with the alterations caused by this geometric remodeling that leads to the closing of the lumen, impairing blood flow.
The pathogenesis of these conditions involves stimulation of a number of factors that regulate smooth muscle proliferation and extracellular matrix deposition. These diseases have a chronic characteristic, which develops during fetal life as in cases of placental dysfunction associated with chronic fetal hypoxemia, premature closure of the ductus arteriosus, or exposure to drugs during the fetal period. This pathological manifestation may also be present in infants who develop PPHN only after birth. It is believed that in these cases, the maintenance of pulmonary vasoconstriction with an increase in pulmonary artery pressure for a prolonged period of time leads to vascular remodeling.52 The response to vasodilators in neonates with pulmonary vascular remodeling is limited or absent, and mortality is high.
Pulmonary vascular hypoplasia
The growth and development of the pulmonary vasculature depend on an actual process of generating new blood vessels (vasculogenesis) and their progressive branching (angiogenesis) to allow adequate gas exchange in lung level (Fig. 5C).
Diseases classified in this category present significant alterations in vasculogenesis or angiogenesis, resulting in hypoplasia of the pulmonary vascular bed. The increase in pulmonary vascular resistance in these diseases is related to the incapacity of the pulmonary vasculature to accommodate right ventricular cardiac output. The alterations in PaCO2 and PaO2 observed in these diseases are secondary, not only to the right-left shunt, but also to inadequate perfusion of the lung. Examples include congenital diaphragmatic hernia and pulmonary hypoplasia secondary to early and prolonged oligohydramnios.
Data obtained from animal models of congenital unilateral diaphragmatic hernia demonstrate that not only there is vascular hypoplasia in the lung affected by the hernia, but also that there is evidence of vascular remodeling in the other lung.53,54 Minimal or absent response to vasodilators is usually observed in diseases of this category, and the prognosis is guarded.
Obstruction
The lung in this category presents a normal number and structure of vessels. However, there is pulmonary blood flow restriction caused by alterations in blood viscosity (polycythemia) or by anomalous pulmonary venous drainage.
Pulmonary parenchymal pathology
The hypoxic pulmonary vasoconstriction response ensures an optimum balance between alveolar ventilation and perfusion, by reducing blood flow to unventilated units. This physiological behavior has an important effect on pulmonary vascular resistance. Diseases associated with decreased alveolar ventilation (such as pneumonia, aspiration, and surfactant deficiency) lead to an increase in pulmonary vascular resistance to prevent the perfusion of alveolar units affected by these diseases. To maintain gas exchange, the resistance of the vasculature involved in alveolar ventilation decreases. If the pulmonary process involves a small percentage of the lung, the pulmonary vascular resistance is not altered. However, in the presence of extensive pulmonary disease, vascular resistance increases to the point of inducing right-left shunt.
Diseases in this category are erroneously classified as associated with PPHN syndrome. The primary process, however, is the abnormal alveolar ventilation, with appropriate vasoconstrictor response of the pulmonary circulation.
Congenital deficiency of surfactant protein B is an example of a condition that resembles PPHN, however clearly distinct.55 Lastly, certain congenital vascular conditions, such as capillary alveolar dysplasia, in which the presence of pulmonary hypertension has been reported,56,57 are also best placed into this category. This congenital disease, where there is a misalignment and a drifting apart between the alveolus and its capillaries, is probably associated with hypoxic pulmonary vasoconstriction and vascular hypoplasia.
In theory, pulmonary parenchymal diseases associated with pulmonary hypertension and shunt should not be classified as PPHN. However, from a practical viewpoint, it is difficult to separate this category from the others. The treatment of diseases in this category should be aimed at the primary lung condition and not at pulmonary vasodilation, as vasoconstriction in these cases has a protective effect on the ventilation/perfusion ratio. Data from animal models have shown that the use of a vasodilator (sildenafil) in the presence of pulmonary lobar atelectasis leads to worsening in oxygenation, by interfering with the physiological hypoxic pulmonary vasoconstriction response.58
Iatrogenic factors
A commonly overlooked factor responsible for the increase in pulmonary vascular resistance is the use of high mean pressures during mechanical ventilation. Depending on lung compliance, part of the pressure used in mechanical ventilation can be transmitted to the lung vasculature. Failure to reduce the pressure used to ventilate infants with clinical and lung compliance improvements can lead to pulmonary hypertension. This occurs more often in infants ventilated with high frequency modes, as compared with conventional ventilation. Reducing the ventilaion mean airway pressure results in decreased pulmonary vascular resistance and abolition of the right-to-left shunting in some infants.
Right ventricular myocardial dysfunction
As discussed above, PPHN is characterized by right-to-left shunt and not by a specific pulmonary artery pressure. This leads to the possibility of a shunt at the level of the foramen ovale in clinical conditions where the pulmonary vascular resistance is not very high, but right ventricular contractility is abnormal, resulting in a higher right, as compared with left atrial pressure. This was demonstrated in newborn pigs, where constriction of the main pulmonary artery to the point of inducing right ventricular failure leads to right-to-left shunt at the foramen ovale.59 In these conditions, therapies directed to enhancing the contractility of the right ventricle lead to improved oxygenation (shunt reduction), even though the pulmonary vascular resistance remains high.
Clinical features and diagnosis
The diagnosis of PPHN should be suspected when the level of hypoxemia is disproportionate to the degree of respiratory distress and pulmonary parenchymal radiological findings. Infants with PPHN exhibit oxygenation lability and progressive cyanosis in the first hours of life.
Cardiac auscultation shows increased intensity of the second heart sound due to pulmonary artery hypertension and systolic murmur of the tricuspid regurgitation.
Echocardiography with Doppler flow should always be performed upon suspicion of PPHN. It is a noninvasive method that not only allows the physician to assess the presence of shunt at the ductus arteriosus and foramen ovale level, but also confirms the absence of congenital heart disease and aids in the evaluation of myocardial contractility.
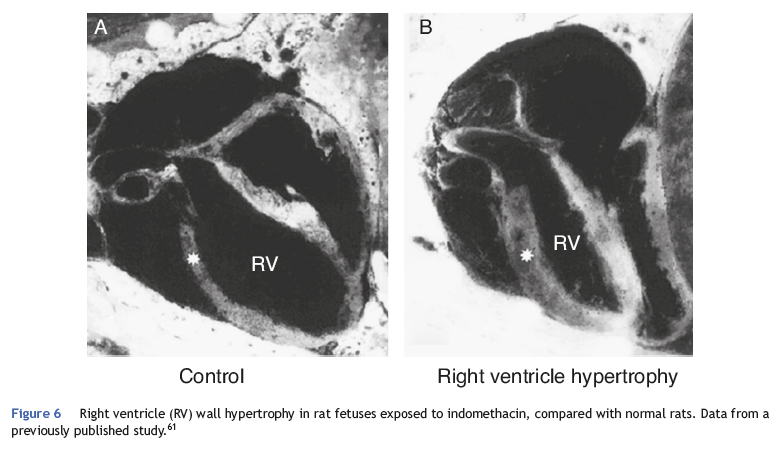
Echocardiography provides an estimate of pulmonary artery pressure by determining the peak velocity of tricuspid regurgitation, and the pulmonary vascular resistance through by measuring the ratio of pulmonary artery acceleration time (ACT), and right ventricular ejection time (RVET). The lower this ratio (ACT/RVET), the higher the pulmonary vascular resistance. In the absence of tricuspid regurgitation, which occurs in 30% of cases, another parameter that can be used to estimate pulmonary artery pressure is the measurement of pulmonary artery flow, estimated by the velocity-time integral of the pulmonary artery (pulmonary VTI).60Fig. 6 illustrates right ventricular hypertrophy in an animal model of PPHN induced in fetal rat after pharmacological closure of the ductus arteriosus.61
The right-to-left shunt, typical of the disease when it occurs exclusively through the ductus arteriosus (50% of cases) can be demonstrated by the difference in preand post-ductal oxygenation. A PaO2 difference > 20 mmHg between the right radial artery (pre-ductal) and the umbilical artery is considered indicative of a right-to-left shunt at the ductus arteriosus level. The same measurement can be obtained noninvasively through the comparative assessment of pulse oximetry between pre-(upper right limb) and postductal areas (lower extremities). A difference > 5% is also indicative of shunting. It is important to remember that the absence of a difference in pre-and post-ductal oxygenation only indicates that there is no right-to-left shunt at the ductus arteriosus. The presence of a shunt at the foramen ovale is only diagnosed by echocardiography. Fig. 7 shows the different scenarios illustrating that not only the presence of the shunt, but also the capacity of the right ventricle to overcome the increased vascular resistance, determines the presence and severity of disease.
Treatment
General care
Maintenance of a normal body temperature and correction of electrolyte and metabolic disturbances are essential. Hypoxemia, hypercapnia, and metabolic acidosis lead to pulmonary vasoconstriction and should be promptly corrected.
In addition to general care, the treatment strategy is to maintain systemic blood pressure at appropriate levels, decrease pulmonary vascular resistance, ensure oxygen release to tissues, and minimize lesions induced by oxygen and ventilation.
In the presence of parenchymal lung disease, ventilatory assistance should have as strategy the improvement of alveolar recruitment, always preventing excessive lung inflation. When indicated (hyaline membrane disease, blood or meconium aspiration), the use of surfactant is of great therapeutic value. Continuous heart, blood pressure, and oxygen saturation monitoring, preferably pre-and post-ductal, are essential. Children with PPHN are extremely labile and unstable. Thus, manipulation should be minimal.
Sedatives have significant side effects, and the use of narcotics such as morphine commonly leads to hypotension. Sedation, although necessary, should be maintain at the lowest possible level and withdrawn as soon as there is clinical improvement. Muscle relaxants should be reserved only for newborns in whom there is great difficulty establishing adequate ventilation, and unresponsive to sedation.
Hemodynamic support
Myocardial activity is commonly compromised in this disease, leading not only to a worsening in the right-to-left shunt at the foramen ovale (right ventricular dysfunction), but also to a decrease in the cardiac output due to left ventricular impairment. The use of inotropic agents is generally indicated.62
It is worth mentioning that the use of quick corrections with colloid or crystalloid solutions, unless there is evidence of intravascular depletion, is contraindicated, since the right atrial pressure is usually high (increased pulmonary vascular resistance and right ventricular dysfunction). Excessive administration of fluids in these circumstances results in further increase in right atrial pressure and exacerbation of right-to-left shunt at the foramen ovale and hypoxemia.
Inotropic and vasopressor agents should be introduced early in an attempt to optimize cardiac function, stabilize systemic blood pressure, and reduce extrapulmonary shunt. Each of these drugs has a distinct effect that should be taken into account when choosing a specific therapy for each newborn (Table 2), and all of them have a significant inotropic effect. Dopamine, through its adrenergic effect, is the most effective in increasing blood pressure and therefore the most frequently used. However, there is some concern regarding a possible pulmonary vasoconstrictor effect when used at high doses. Dobutamine has an effect of reducing left ventricular overload, which together with its inotropic characteristics contribute to an increased cardiac output. Choosing the ideal vasopressor for the treatment of newborns with hypotension remains a subject of great debate.63-65 Attention to the cause of hypotension and myocardial performance is of great help in therapeutic decision-making.
Epinephrine, in spite of being the drug with greatest inotropic effect, also has an adrenergic vasoconstriction effect in systemic and pulmonary circulation, which sometimes leads to a significant reduction in peripheral and pulmonary blood flow. Recently, a study showed that norepinephrine improved oxygenation and decreased pulmonary vascular resistance in newborns with PPHN through an unknown mechanism.66 In sheep, norepinephrine decreases pulmonary vascular tone and increases blood flow by activating alpha-receptors and NO release.67,68
Pulmonary vasodilatation
In response to many vasoactive drugs, the pulmonary circulation has a similar behavior to that of systemic circulation. Thus, the great difficulty in the pharmacological treatment of PPHN is to dilate the pulmonary vessels without causing systemic. As the pulmonary circulation shows vasodilation with pH elevation, hyperventilation and alkalization have been widely used for the treatment of this disease in the past.69,70 Hyperventilation is no longer used due to a significant reduction in cerebral perfusion when PaCO2 is maintained < 25 mmhg, and pulmonary trauma. alkalization, in turn, when appropriately used, has beneficial therapeutic effect with minimal side effects.
Inhaled NO
This method is currently considered the standard treatment.
When administered by inhalation (iNO), it reaches the alveolar space and diffuses into vascular smooth muscle of the adjacent pulmonary arteries, where it causes vasodilation by increasing cGMP levels. iNO continues to disseminate, and in the pulmonary artery lumen, it is rapidly bound to hemoglobin, restricting its effect on the pulmonary circulation, without any effect on the systemic circulation (Fig. 8). iNO is preferably distributed to the ventilated segments of the lungs, with increased perfusion in those areas. This results in an improved ventilation/perfusion ratio, decreasing intra-alveolar shunting and improving oxygenation. When the response is positive, improved oxygenation is evident within a few minutes.
Since 1997, several clinical trials have demonstrated a significant improvement in oxygenation, decreased need for extracorporeal membrane oxygenation (ECMO) and/or reduced mortality in neonates older than 34 weeks with severe respiratory failure.71 Clinical improvement in infants with PPHN occurs in approximately 70% of patients, and the best results are observed in the idiopathic type.72
In the past, treatment with i NO was initiated at rather late stages of the disease, usually when the oxygenation index was > 25. Such late intervention resulted the mean incidence of ECMO and/or mortality was still 40%. An earlier introduction of iNO is currently recommended, before prolonged exposure to high concentrations of oxygen and high ventilatory occurs parameters.
Currently, the initial recommended concentration of iNO is 20 ppm. Higher concentrations are not more effective, and are associated with a higher incidence of methemoglobinemia and formation of nitrogen dioxide.73 Some studies have shown that concentrations of up to 5 ppm are effective in improving oxygenation;74,75 lower concentrations (2 ppm), in addition to not being effective, reduce the response to the subsequent increase in concentration to 20 ppm.76 1 ppm/4 h. Such slow weaning is geared at preventing the phenomenon of rebound vasoconstriction, which may be related to the decreased endogenous production of NO. During the use of inhaled NO, continuous monitoring of nitrogen dioxide (generated by the reaction of NO with oxygen) and daily serum levels of methemoglobin should be obtained. The methemoglobin level must be kept below 5%. Inhibition of platelet aggregation has been reported in humans; however, the occurrence of this side effect remains controversial.77,78 Until this fact is clarified, the use of iNO is not recommend in the presence of significant intracranial bleeding.
Pulmonary vasodilators
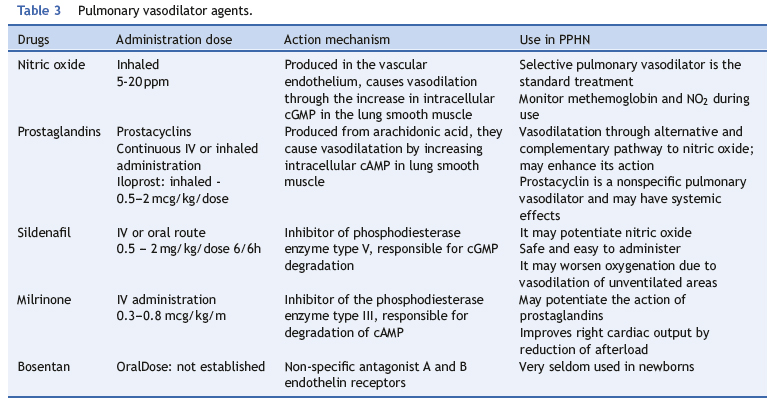
Besides NO, there are no other drugs with specific vasodilator effects for the pulmonary circulation. Several drugs, however, have a predominant vasodilator effect in PPHN; they are discussed below and are summarized in Table 3.
Prostaglandins
Prostacyclin is the most potent vasodilator among prostaglandins. Its intravenous use has been longestablished in the treatment in adults with pulmonary hypertension. Most studies in infants have shown a similar or greater effect when compared to iNO in decreasing pulmonary artery pressure and improving oxygenation.79,80 Its use, however, requires a permanent vascular access and often leads to hypotension, as it is not a selective pulmonary vasodilator. Its administration via inhalation allows for selective pulmonary vasodilation, but with very short half-life, which makes its administration difficult.81 Iloprost is a longer-acting prostacyclin analogue with specific effect in pulmonary circulation.82
Some studies have shown its effectiveness, even in cases refractory to NO.83,84 It can be used in the inhaled form in intubated patients or in the nebulized form in spontaneously breathing patients.85
Phosphodiesterase inhibitors
There are 11 isoforms of phosphodiesterases (PDEs). The most important are the PDE3 and PDE5, which preferentially degrade cAMP and cGMP, respectively.
Sildenafil produces vasodilation by inhibiting PDE5. Dipyridamole, zaprinast, and pentoxifylline are PDE5 inhibitors that are seldom used, as they have important systemic effects.
Studies in neonates with pulmonary hypertension have shown that sildenafil selectively reduces pulmonary vascular resistance with few systemic effects.86-8<9 Its effect on pulmonary vasculature appears to be independent from the underlying cause, and thus effective in idiopathic pulmonary hypertension associated with heart conditions, lung disease, and PPHN.90 In the postoperative period of heart disease, sildenafil decreased pulmonary artery pressure and prevented rebound after withdrawal of NO.91
In a Cochrane meta-analysis with 37 newborns from centers that lacked NO and high frequency ventilation, significant improvement in oxygenation was observed in the group receiving sildenafil.92
Sildenafil is safe, effective, and easily administered. As discussed earlier, its vasodilator effect extends to poorly ventilated areas in the lungs, which changes the ventilation/perfusion ratio, increases the intrapulmonary shunt, and worsens oxygenation. In an animal model of neonatal lobar atelectasis, sildenafil inhibited pulmonary vasoconstriction in response to hypoxia and worsened oxygenation.58 Using tadalafil, another PDE5 inhibitor in the same animal model without lobar atelectasis, a decrease in pulmonary vascular resistance and improved oxygenation were demonstrated.93
Milrinone causes pulmonary vasodilation by inhibiting PDE3. Originally used to reduce the afterload and as an inotropic agent, it also has been shown to be effective in the treatment of neonatal pulmonary hypertension. Recently, Lakshminrusimha et al. demonstrated, in an animal model of PPHN, that pretreatment with milrinone increased the effect of prostaglandin in pulmonary artery relaxation.94 This would explain the findings of Bassller and MacNamara, who, when studying four and nine newborns, respectively, demonstrated a significant improvement in oxygenation with the use of milrinone after unsatisfactory response to iNO.95,96
Endothelin blockers
In adults, the use of bosentan, a nonspecific antagonist of receptors A and B, significantly improves symptoms and exercise capacity in patients with pulmonary hypertension. In newborns, the plasma levels of ET1 are high, with a linear correlation with disease severity.97 These data have stimulated the use and publication of some case reports with the use of bosentan in neonates with PPHN.98,99 The usefulness of these agents in the treatment of PPHN still requires further clinical investigation before it can be recommended.
Other drugs
Other drugs have been tested in animal models with promising effects. Rho-kinase is a protein that prevents vasodilatation by inhibiting the dephosphorylation of myosin in smooth muscle cells. Fasudil and Y-27632, rho-kinase inhibitors, cause potent vasodilation in experimental models of PPHN.29,32 These studies suggest that rho-kinase inhibitors may play an important role in the treatment of PPHN.
Superoxide dismutase removes superoxide radicals from circulation, generated by oxidative stress, leading to pulmonary vasoconstriction by binding and competing with NO. In animals, administration of superoxide dismutase reduces pulmonary artery pressure and improves the response to NO.100
The adenosine nucleotide and its trinucleotide ATP are potent selective pulmonary vasodilators, through intracellular increase of AMP. Intravenous infusion of adenosine in infants with PPHN syndrome has shown a significant improve ment in oxygenation.101-103
Magnesium sulphate has been described in some case reports.104 However, the Cochrane meta-analysis did not show enough evidence to recommend the use of this drug in PPHN.105
Mechanical ventilation and surfactant
Mechanical ventilation facilitates alveolar recruitment and improves ventilation/perfusion and oxygenation.
It is still debatable whether high-frequency ventilation is superior to conventional ventilation in newborns with PPHN. Some studies have shown that high-frequency ventilation improves the efficacy of inhaled NO in infants with parenchymal lung disease.106
Certain causes of PPHN, such as meconium aspiration syndrome and diaphragmatic hernia, are associated with surfactant deficiency or reduced activity,107 and its use in neonates born to term with severe respiratory failure decreases the need for ECMO.108
Prevention
Despite recent advances, mortality associated with this syndrome remains at 10%. Due to the limited knowledge of its etiology and pathogenesis, little is progress has been made regarding prevention. Based on data from Hospital São Luiz, 70% of cases of PPHN are idiopathic. Considering that elective C-sections without labor are associated with most of these cases, attention regarding this practice and proper monitoring of these infants may play an important preventive role. Much has been achieved in the diagnosis and treatment of PPHN in the last 30 years, but more studies are necessary to adequately prevent or avoid this syndrome.
Funding
Canadian Institutes of Health and Research (CIHR) Grant # MOP-93710.
Conflicts of interest
The authors declare no conflicts of interest.
References
- 1. Klinger JR. The nitric oxide/cGMP signaling pathway in pulmonary hypertension. Clin Chest Med. 2007;28:143-67.
- 2. Nowicki MJ, Shi D, Cai Z, Bishop PR, May WL. Developmental expression of endothelial nitric oxide synthase (eNOS) in the rat liver. Pediatr Res. 2003;54:732-8.
- 3. Parker TA, le Cras TD, Kinsella JP, Abman SH. Developmental changes in endothelial nitric oxide synthase expression and activity in ovine fetal lung. Am J Physiol Lung Cell Mol Physiol. 2000;278:L202-8.
- 4. Lucke T, Kanzelmeyer N, Kemper MJ, Tsikas D, Das AM. Developmental changes in the L-arginine/nitric oxide pathway from infancy to adulthood: plasma asymmetric dimethylarginine levels decrease with age. Clin Chem Lab Med. 2007;45:1525-30.
- 5. Belik J, Shehnaz D, Pan J, Grasemann H. Developmental changes in arginase expression and activity in the lung. Am J Physiol Lung Cell Mol Physiol. 2008;294:L498-504.
- 6. Schmidt HH, Schmidt PM, Stasch JP. NO- and haem-independent soluble guanylate cyclase activators. Handb Exp Pharmacol. 2009;309-39.
- 7. Stasch JP, Hobbs AJ. NO-independent, haem-dependent soluble guanylate cyclase stimulators. Handb Exp Pharmacol. 2009;277-308.
- 8. Behrends S, Kempfert J, Mietens A, Koglin M, Scholz H, Middendorff R. Developmental changes of nitric oxidesensitive guanylyl cyclase expression in pulmonary arteries. Biochem Biophys Res Commun. 2001;283:883-7.
- 9. Behrends S, Mietens A, Kempfert J, Koglin M, Scholz H, Middendorff R. The expression pattern of nitric oxide-sensitive guanylyl cyclase in the rat heart changes during postnatal development. J Histochem Cytochem. 2002;50:1325-32.
- 10. Belik J, Hehne N, Pan J, Behrends S. Soluble guanylate cyclase-dependent relaxation is reduced in the adult rat bronchial smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2007;292:L699-703.
- 11. Bloch KD, Filippov G, Sanchez LS, Nakane M, de la Monte SM. Pulmonary soluble guanylate cyclase, a nitric oxide receptor, is increased during the perinatal period. Am J Physiol 1997;272:L400-6.
- 12. Zhu B, Strada SJ. The novel functions of cGMP-specific phosphodiesterase 5 and its inhibitors in carcinoma cells and pulmonary/cardiovascular vessels. Curr Top Med Chem. 2007;7:437-54.
- 13. Patel MD, Katz SD. Phosphodiesterase 5 inhibition in chronic heart failure and pulmonary hypertension. Am J Cardiol. 2005;96:47M-51.
- 14. Hanson KA, Ziegler JW, Rybalkin SD, Miller JW, Abman SH, Clarke WR. Chronic pulmonary hypertension increases fetal lung cGMP phosphodiesterase activity. Am J Physiol. 1998;275:L931-41.
- 15. Reffelmann T, Kloner RA. Cardiovascular effects of phosphodiesterase 5 inhibitors. Curr Pharm Des. 2006;12:3485-94.
- 16. Juliana AE, Abbad FC. Severe persistent pulmonary hypertension of the newborn in a setting where limited resources exclude the use of inhaled nitric oxide: successful treatment with sildenafil. Eur J Pediatr. 2005;164:626-9.
- 17. Sood BG, Delaney-Black V, Aranda JV, Shankaran S. Aerosolized PGE1: a selective pulmonary vasodilator in neonatal hypoxemic respiratory failure results of a Phase I/II open label clinical trial. Pediatr Res. 2004;56:579-85.
- 18. Murray F, MacLean MR, Pyne NJ. Increased expression of the cGMP-inhibited cAMP-specific (PDE3) and cGMP binding cGMP-specific (PDE5) phosphodiesterases in models of pulmonary hypertension. Br J Pharmacol. 2002;137:1187-94.
- 19. Jankov RP, Luo X, Cabacungan J, Belcastro R, Frndova H, Lye SJ, et al. Endothelin-1 and O2-mediated pulmonary hypertension in neonatal rats: a role for products of lipid peroxidation. Pediatr Res. 2000;48:289-98.
- 20. Munzel T, Sinning C, Post F, Warnholtz A, Schulz E. Pathophysiology, diagnosis and prognostic implications of endothelial dysfunction. Ann Med. 2008;40:180-96.
- 21. Belik J, Jankov RP, Pan J, Tanswell AK. Peroxynitrite inhibits relaxation and induces pulmonary artery muscle contraction in the newborn rat. Free Radic Biol Med. 2004;37:1384-92.
- 22. Grover TR, Zenge JP, Parker TA, Abman SH. Vascular endothelial growth factor causes pulmonary vasodilation through activation of the phosphatidylinositol-3-kinase-nitric oxide pathway in the late-gestation ovine fetus. Pediatr Res.2002;52:907-12.
- 23. Le Cras TD, Markham NE, Tuder RM, Voelkel NF, Abman SH. Treatment of newborn rats with a VEGF receptor inhibitor causes pulmonary hypertension and abnormal lung structure. Am J Physiol Lung Cell Mol Physiol. 2002;283:L555-62.
- 24. Papaioannou AI, Kostikas K, Kollia P, Gourgoulianis KI. Clinical implications for vascular endothelial growth factor in the lung: friend or foe? Respir Res. 2006;7:128.
- 25. Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc MG, Waltenberger J, et al. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001;15:427-38.
- 26. Schermuly RT, Stasch JP, Pullamsetti SS, Middendorff R, Muller D, Schluter KD, et al. Expression and function of soluble guanylate cyclase in pulmonary arterial hypertension. Eur Respir J. 2008;32:881-91.
- 27. Braam B, Verhaar MC. Understanding eNOS for pharmacological modulation of endothelial function: a translational view. Curr Pharm Des. 2007;13:1727-40.
- 28. Belik J, Kerc E, Pato MD. Rat pulmonary arterial smooth muscle myosin light chain kinase and phosphatase activities decrease with age. Am J Physiol Lung Cell Mol Physiol. 2006;290:L509-16.
- 29. Fagan KA, Oka M, Bauer NR, Gebb SA, Ivy DD, Morris KG, et al. Attenuation of acute hypoxic pulmonary vasoconstriction and hypoxic pulmonary hypertension in mice by inhibition of Rho-kinase. Am J Physiol Lung Cell Mol Physiol. 2004;287:L656-64.
- 30. Fukumoto Y, Tawara S, Shimokawa H. Recent progress in the treatment of pulmonary arterial hypertension: expectation for rho-kinase inhibitors. Tohoku J Exp Med. 2007;211:309-20.
- 31. Hyvelin JM, Howell K, Nichol A, Costello CM, Preston RJ, McLoughlin P. Inhibition of Rho-kinase attenuates hypoxia-induced angiogenesis in the pulmonary circulation. Cir Res. 2005;97:185-91.
- 32. McNamara PJ, Murthy P, Kantores C, Teixeira L, Engelberts D, van VlieT, et al. Acute vasodilator effects of Rho-kinase inhibitors in neonatal rats with pulmonary hypertension unresponsive to nitric oxide. Am J Physiol Lung Cell Mol Physiol. 2008;294:L205-13.
- 33. Cassin S, Tyler T, Wallis R. The effects of prostaglandin E on fetal pulmonary vascular resistance (38588). Proc Soc Exp Biol Med. 1975;148:584-7.
- 34. Leffler CW, Hessler JR, Green RS. The onset of breathing at birth stimulates pulmonary vascular prostacyclin synthesis. Pediatr Res. 1984;18:938-42.
- 35. Danhaive O, Margossian R, Geva T, Kourembanas S. Pulmonary hypertension and right ventricular dysfunction in growth-restricted, extremely low birth weight neonates. J Perinatol. 2005;25:495-9.
- 36. Walsh-Sukys MC, Tyson JE, Wright LL, Bauer CR, Korones SB, Stevenson DK, et al. Persistent pulmonary hypertension of the newborn in the era before nitric oxide: practice variation and outcomes. Pediatrics. 2000;105:14-20.
- 37. Boden G, Bennett C. The management of persistent pulmonary hypertension of the newborn. Current Paediatrics. 2004;14:290-7.
- 38. Hernández-Diaz S, Van Marter LJ, Werler MM, Louik C, Mitchell AA. Risk Factors for Persistent Pulmonary Hypertension of the Newborn. Pediatrics. 2007;120:e272-82.
- 39. Konduri GG, Solimano A, Sokol GM, Singer J, Ehrenkranz RA, Singhal N, et al. A randomized trial of early versus standard inhaled nitric oxide therapy in term and near-term newborn infants with hypoxic respiratory failure. Pediatrics. 2004;113:559-64.
- 40. Ramachandrappa A, Jain L. Elective cesarean section: its impact on neonatal respiratory outcome. Clin Perinatol. 2008;35:373-93.
- 41. Jain L, Eaton DC. Physiology of fetal lung fluid clearance and the effect of labor. Semin Perinatol. 2006;30:34-43.
- 42. Belik J, Halayko AJ, Rao K, Stephens NL. Fetal ductus arteriosus ligation. Pulmonary vascular smooth muscle biochemical and mechanical changes. Cir Res. 1993;72:588-96.
- 43. Abman SH, Accurso FJ. Acute effects of partial compression of ductus arteriosus on fetal pulmonary circulation. Am J Physiol. 1989;257:H626-34.
- 44. Levy R, Matitiau A, Ben AA, Milman D, Or Y, Hagay Z. Indomethacin and corticosteroids: an additive constrictive effect on the fetal ductus arteriosus. Am J Perinatol. 1999;16:379-83.
- 45. Weiss H, Cooper B, Brook M, Schlueter M, Clyman R. Factors determining reopening of the ductus arteriosus after successful clinical closure with indomethacin. J Pediatr. 1995;127:466-71.
- 46. Respondek M, Weil SR, Huhta JC. Fetal echocardiography during indomethacin treatment. Ultrasound Obstet Gynecol. 1995;5:86-9.
- 47. Wooltorton E. Persistent pulmonary hypertension of the newborn and maternal use of SSRIs. CMAJ. 2006;174:1555-6.
- 48. Kallen B, Olausson PO. Maternal use of selective serotonin re-uptake inhibitors and persistent pulmonary hypertension of the newborn. Pharmacoepidemiol Drug Saf. 2008;17:801-6.
- 49. Chambers CD, Hernandez-Diaz S, Van Marter LJ, Werler MM, Louik C, Jones KL, et al. Selective serotonin-reuptake inhibitors and risk of persistent pulmonary hypertension of the newborn. N Engl J Med. 2006;354:579-87.
- 50. Fornaro E, Li D, Pan J, Belik J. Prenatal exposure to fluoxetine induces fetal pulmonary hypertension in the rat. Am J Respir Crit Care Med. 2007;176:1035-40.
- 51. Ziegler JW, Ivy DD, Kinsella JP, Abman SH. The role of nitric oxide, endothelin, and prostaglandins in the transition of the pulmonary circulation. Clin Perinatol. 1995;22:387-403.
- 52. Stenmark KR, Gerasimovskaya E, Nemenoff RA, Das M. Hypoxic activation of adventitial fibroblasts: role in vascular remodeling. Chest. 2002;122:326S-34.
- 53. Keller RL, Moore P, Teitel D, Hawgood S, McQuitty J, Fineman JR. Abnormal vascular tone in infants and children with lung hypoplasia: Findings from cardiac catheterization and the response to chronic therapy. Pediatr Crit Care Med. 2006;7:589-94.
- 54. Belik J, Davidge ST, Zhang W, Pan J, Greer JJ. Airway smooth muscle changes in the nitrofen-induced congenital diaphragmatic hernia rat model. Pediatr Res. 2003;53:737-43.
- 55. Kunig AM, Parker TA, Nogee LM, Abman SH, Kinsella JP. ABCA3 deficiency presenting as persistent pulmonary hypertension of the newborn. J Pediatr. 2007;151:322-4.
- 56. Singh SA, Ibrahim T, Clark DJ, Taylor RS, George DH. Persistent pulmonary hypertension of newborn due to congenital capillary alveolar dysplasia. Pediatr Pulmonol. 2005;40:349-53.
- 57. Plat G, Rouquette I, Marcoux MO, Bloom MC, Acar P, Dulac Y. Alveolar capillary dysplasia and persistent pulmonary hypertension of the newborn. Arch Mal Coeur Vaiss. 2007;100:458-61.
- 58. Tessler R, Wu S, Fiori R, Macgowan CK, Belik J. Sildenafil acutely reverses the hypoxic pulmonary vasoconstriction response of the newborn pig. Pediatr Res. 2008;64:251-5.
- 59. Belik J, Light RB. Effect of increased afterload on right ventricular function in newborn pigs. J Appl Physiol. 1989;66:863-9.
- 60. Peterson AL, Deatsman S, Frommelt MA, Mussatto K, Frommelt PC. Correlation of echocardiographic markers and therapy in persistent pulmonary hypertension of the newborn. Pediatr Cardiol. 2009;30:160-5.
- 61. Fabris VE, Pato MD, Belik J. Progressive lung and cardiac changes associated with pulmonary hypertension in the fetal rat. Pediatr Pulmonol. 2001;31:344-53.
- 62. Ricachinevsky CP, Amantea SL. Treatment of pulmonary arterial hypertension. J Pediatr (Rio J). 2006;82:S153-65.
- 63. Roze JC, Tohier C, Maingueneau C, Lefevre M, Mouzard A. Response to dobutamine and dopamine in the hypotensive very preterm infant. Arch Dis Child. 1993;69:59-63.
- 64. Dempsey EM, Barrington KJ. Evaluation and treatment of hypotension in the preterm infant. Clin Perinatol. 2009;36:75-85.
- 65. Evans N. Which inotrope for which baby? Arch Dis Child Fetal Neonatal Ed. 2006;91:F213-20.
- 66. Tourneux P, Rakza T, Bouissou A, Krim G, Storme L. Pulmonary circulatory effects of norepinephrine in newborn infants with persistent pulmonary hypertension. J Pediatr. 2008;153:345-9.
- 67. Jaillard S, Houfflin-Debarge V, Riou Y, Rakza T, Klosowski S, Lequien P, et al. Effects of catecholamines on the pulmonary circulation in the ovine fetus. Am J Physiol Regul Integr Comp Physiol. 2001;281:R607-14.
- 68. Magnenant E, Jaillard S, Deruelle P, Houfflin-Debarge V, Riou Y, Klosowski S, et al. Role of the alpha2-adrenoceptors on the pulmonary circulation in the ovine fetus. Pediatr Res. 2003;54:44-51.
- 69. Drummond WH, Gregory GA, Heymann MA, Phibbs RA. The independent effects of hyperventilation, tolazoline, and dopamine on infants with persistent pulmonary hypertension. J Pediatr. 1981;98:603-11.
- 70. Schreiber MD, Heymann MA, Soifer SJ. Increased arterial pH, not decreased PaCO2, attenuates hypoxia-induced pulmonary vasoconstriction in newborn lambs. Pediatr Res. 1986;20:113-7.
- 71. The Neonatal Inhaled Nitric Oxide Study Group. Inhaled Nitric Oxide in Full-Term and Nearly Full-Term Infants with Hypoxic Respiratory Failure. N Engl J Med. 1997;336:597-604.
- 72. Wessel DL, Adatia I, Van Marter LJ, Thompson JE, Kane JW, Stark AR, et al. Improved oxygenation in a randomized trial of inhaled nitric oxide for persistent pulmonary hypertension of the newborn. Pediatrics. 1997;100:E7.
- 73. Committee on Fetus and Newborn. Use of Inhaled Nitric Oxide. Pediatrics. 2000;106:344-5.
- 74. Cornfield DN, Maynard RC, deRegnier RA, Guiang SF, III, Barbato JE, Milla CE. Randomized, controlled trial of low-dose inhaled nitric oxide in the treatment of term and near-term infants with respiratory failure and pulmonary hypertension. Pediatrics. 1999;104:1089-94.
- 75. Davidson D, Barefield ES, Kattwinkel J, Dudell G, Damask M, Straube R, et al. Inhaled nitric oxide for the early treatment of persistent pulmonary hypertension of the term newborn: a randomized, double-masked, placebo-controlled, dose-response, multicenter study. The I-NO/PPHN Study Group. Pediatrics. 1998;101:325-34.
- 76. Clark RH, Kueser TJ, Walker MW, Southgate WM, Huckaby JL, Perez JA, et al. Low-dose nitric oxide therapy for persistent pulmonary hypertension of the newborn. Clinical Inhaled Nitric Oxide Research Group. N Engl J Med. 2000;342:469-74.
- 77. Albert J, Harbut P, Zielinski S, Ryniak S, Gillis-Haegerstrand C, Lindwall R, et al. Prolonged exposure to inhaled nitric oxide does not affect haemostasis in piglets. Intensive Care Med. 2007;33:1594-601.
- 78. Beghetti M, Sparling C, Cox PN, Stephens D, Adatia I. Inhaled NO inhibits platelet aggregation and elevates plasma but not intraplatelet cGMP in healthy human volunteers. Am J Physiol Heart Circ Physiol. 2003;285:H637-42.
- 79. Bos AP, Tibboel D, Koot VC, Hazebroek FW, Molenaar JC. Persistent pulmonary hypertension in high-risk congenital diaphragmatic hernia patients: incidence and vasodilator therapy. J Pediatr Surg. 1993;28:1463-5.
- 80. Nakayama T, Shimada H, Takatsuki S, Hoshida H, Ishikita T, Matsuura H, et al. Efficacy and limitations of continuous intravenous epoprostenol therapy for idiopathic pulmonary arterial hypertension in Japanese children. Circ J. 2007;71:1785-90.
- 81. Ewert R, Schaper C, Halank M, Glaser S, Opitz CF. Inhalative iloprost - pharmacology and clinical application. Expert Opin Pharmacother. 2009;10:2195-207.
- 82. Leuchte HH, Behr J. Iloprost for idiopathic pulmonary arterial hypertension. Expert Rev Cardiovasc Ther. 2005;3:215-23.
- 83. Ehlen M, Wiebe B. Iloprost in persistent pulmonary hypertension of the newborn. Cardiol Young. 2003;13:361-3.
- 84. Chotigeat U, Jaratwashirakul S. Inhaled iloprost for severe persistent pulmonary hypertension of the newborn. J Med Assoc Thai. 2007;90:167-70.
- 85. Krishnan U. Management of pulmonary arterial hypertension in the neonatal unit. Cardiol Rev. 2010;18:73-5.
- 86. Vargas-Origel A, Gomez-Rodriguez G, Aldana-Valenzuela C, Vela-Huerta MM, Alarcon-Santos SB, Amador-Licona N. The use of sildenafil in persistent pulmonary hypertension of the newborn. Am J Perinatol. 2010;27:225-30.
- 87. Steinhorn RH, Kinsella JP, Pierce C, Butrous G, Dilleen M, Oakes M, et al. Intravenous sildenafil in the treatment of neonates with persistent pulmonary hypertension. J Pediatr. 2009;155:841-7.
- 88. Bentlin MR, Saito A, De Luca AK, Bossolan G, Bonatto RC, Martins AS, et al. [Sildenafil for pulmonary hypertension treatment after cardiac surgery]. J Pediatr (Rio J). 2005; 81:175-8.
- 89. Oliveira EC, Amaral CF. Sildenafil in the management of idiopathic pulmonary arterial hypertension in children and adolescents. J Pediatr (Rio J). 2005;81:390-4.
- 90. Leibovitch L, Matok I, Paret G. Therapeutic applications of sildenafil citrate in the management of paediatric pulmonary hypertension. Drugs. 2007;67:57-73.
- 91. Stocker C, Penny DJ, Brizard CP, Cochrane AD, Soto R, Shekerdemian LS. Intravenous sildenafil and inhaled nitric oxide: a randomised trial in infants after cardiac surgery. Intensive Care Med. 2003;29:1996-2003.
- 92. Shah P, Ohlsson A. Sildenafil for pulmonary hypertension in neonates. Cochrane Database Syst Rev. 2007;CD005494.
- 93. Tessler RB, Zadinello M, Fiori H, Colvero M, Belik J, Fiori RM. Tadalafil improves oxygenation in a model of newborn pulmonary hypertension. Pediatr Crit Care Med. 2008;9: 330-2.
- 94. Lakshminrusimha S, Porta NF, Farrow KN, Chen B, Gugino SF, Kumar VH, et al. Milrinone enhances relaxation to prostacyclin and iloprost in pulmonary arteries isolated from lambs with persistent pulmonary hypertension of the newborn. Pediatr Crit Care Med. 2009;9:106-12.
- 95. McNamara PJ, Laique F, Muang-In S, Whyte HE. Milrinone improves oxygenation in neonates with severe persistent pulmonary hypertension of the newborn. J Crit Care. 2006;21:217-22.
- 96. Bassler D, Choong K, McNamara P, Kirpalani H. Neonatal persistent pulmonary hypertension treated with milrinone: four case reports. Biol Neonate. 2006;1:1-5.
- 97. Rosenberg AA, Kennaugh J, Koppenhafer SL, Loomis M, Chatfield BA, Abman SH. Elevated immunoreactive endothelin-1 levels in newborn infants with persistent pulmonary hypertension. J Pediatr. 1993;123:109-14.
- 98. Goissen C, Ghyselen L, Tourneux P, Krim G, Storme L, Bou P, et al. Persistent pulmonary hypertension of the newborn with transposition of the great arteries: successful treatment with bosentan. Eur J Pediatr. 2008;167:437-40.
- 99. Nakwan N, Choksuchat D, Saksawad R, Thammachote P. Successful treatment of persistent pulmonary hypertension of the newborn with bosentan. Acta Paediatr. 2009;98:1683-5.
- 100. Lakshminrusimha S, Russell JA, Wedgwood S, Gugino SF, Kazzaz JA, Davis JM, et al. Superoxide dismutase improves oxygenation and reduces oxidation in neonatal pulmonary hypertension. Am J Respir Crit Care Med. 2006;174:1370-7.
- 101. Konduri GG, Garcia DC, Kazzi NJ, Shankaran S. Adenosine infusion improves oxygenation in term infants with respiratory failure. Pediatrics. 1996;97:295-300.
- 102. Ng C, Franklin O, Vaidya M, Pierce C, Petros A. Adenosine infusion for the management of persistent pulmonary hypertension of the newborn. Pediatr Crit Care Med. 2004;5:10-3.
- 103. Motti A, Tissot C, Rimensberger PC, Prina-Rousso A, Aggoun Y, Berner M, et al. Intravenous adenosine for refractory pulmonary hypertension in a low-weight premature newborn: a potential new drug for rescue therapy. Pediatr Crit Care Med. 2006;7:380-2.
- 104. Raimondi F, Migliaro F, Capasso L, Ausanio G, Bisceglia M, Giliberti P, et al. Intravenous magnesium sulphate vs. inhaled nitric oxide for moderate, persistent pulmonary hypertension of the newborn. A Multicentre, retrospective study. J Trop Pediatr. 2008;54:196-9.
- 105. Ho JJ, Rasa G. Magnesium sulfate for persistent pulmonary hypertension of the newborn. Cochrane Database Syst Rev. 2007;CD005588.
- 106. Kinsella JP, Abman SH. High-frequency oscillatory ventilation augments the response to inhaled nitric oxide in persistent pulmonary hypertension of the newborn: Nitric Oxide Study Group. Chest. 1998;114:105.
- 107. Findlay RD, Taeusch HW, Walther FJ. Surfactant replacement therapy for meconium aspiration syndrome. Pediatrics. 1996;97:48-52.
- 108. Lotze A, Mitchell BR, Bulas DI, Zola EM, Shalwitz RA, Gunkel JH. Multicenter study of surfactant (beractant) use in the treatment of term infants with severe respiratory failure. Survanta in Term Infants Study Group. J Pediatr. 1998;132:40-7.
Persistet pulmonary hypertension of the newborn: recent advances in pathophysiology and treatment
Publication Dates
-
Publication in this collection
03 July 2013 -
Date of issue
June 2013
History
-
Received
25 Oct 2012 -
Accepted
08 Nov 2012