Abstracts
Report of a study of a family with a remarkable combination of endothelial corneal dystrophy, and anterior chamber dysgenesis classified as Axenfeld-Rieger anomaly. A 56 year-old female had blurred vision complaints. A slit lamp evaluation showed a bilateral posterior embryotoxon and guttata with corneal edema, Descemet membrane's folds, characterizing Fuchs endothelial dystrophy. A 27 year-old daughter was asymptomatic. The biomicroscopic exam disclosed a bilateral nasal and temporal posterior embryotoxon and a central guttata. Gonioscopy showed iridocorneal strands with a prominent Schwalbe´s line. The intraocular pressure was normal. Her sister, a 25 year-old female presented very similar abnormalities along with her brother (22 years old). A 19 year-old female sister presented the iridocorneal angle alterations, without Fuchs endothelial dystrophy, until this moment. This paper presents a family with a central cornea guttata or Fuchs dystrophy associated with Axenfeld-Rieger anomaly. Two different inherited diseases appearing together in an entire family may suggest a single molecular and genetic etiology, with high penetrance.
Eye abnormalities; Fuchs endothelial dystrophy; Anterior eye segment; Cornea; Syndrome; Case reports
Relato dos resultados do estudo de uma família com distrofia corneana endotelial associada a uma disgenesia do segmento anterior, a anomalia de Axenfeld-Rieger. Paciente de 56 anos, feminina, queixava-se de visão borrada. O exame na lâmpada de fenda evidenciou embriotoxo posterior bilateral e guttata no centro da córnea, edema estromal e dobras na membrana de Descemet, caracterizando distrofia endothelial de Fuchs. A filha de 27 anos estava assintomática. A biomicroscopia revelou embriotoxo posterior nasal e temporal bilateral e guttata central. A gonioscopia evidenciou processos iridocorneanos até a linha de Schwalbe, que se encontrava anteriorizada e proeminente. A pressão intra-ocular estava normal. Não foram observadas alterações sistêmicas. Sua irmã, 25 anos, sexo feminino, apresentava alterações muito similares, assim como seu irmão (22 anos). Outra irmã de 19 anos apresentava alterações do ângulo iridocorneano, porém sem distrofia de Fuchs, até o presente. Este trabalho relata os resultados de estudo de uma família com córnea guttata central ou distrofia de Fuchs associada à anomalia de Axenfeld-Rieger. Duas doenças hereditárias com expressão simultânea em toda a família sugerem uma etiologia molecular e genética única, com alta penetrância.
Anormalidades do olho; Distrofia endotelial de Fuchs; Segmento anterior do olho; Córnea; Síndrome; Relatos de casos
RELATO DE CASO
Axenfeld-Rieger anomaly and corneal endothelial dystrophy: a case series
Anomalia de Axenfeld-Rieger e distrofia corneana endotelial: uma série de casos
Mariana Borges OliveiraI; Roberto dos Santos MitraudII; Riuitiro YamaneIII
IResidente do Terceiro Ano do Hospital dos Servidores do Estado do Rio de Janeiro - HSE - Rio de Janeiro (RJ), Brasil
IIMédico Oftalmologista do Instituto de Hematologia Arthur Siqueira de Cavalcanti - Hemorio - Rio de Janeiro (RJ), Brasil
IIIProfessor Titular do Departamento de Oftamolologia da Faculdade de Ciências Médicas da Universidade do Estado do Rio de Janeiro - UERJ - Rio de Janeiro (RJ), Brasil
Corresponding authors Corresponding authors: E-mail: oliveiramb@gmail.com
ABSTRACT
Report of a study of a family with a remarkable combination of endothelial corneal dystrophy, and anterior chamber dysgenesis classified as Axenfeld-Rieger anomaly. A 56 year-old female had blurred vision complaints. A slit lamp evaluation showed a bilateral posterior embryotoxon and guttata with corneal edema, Descemet membrane's folds, characterizing Fuchs endothelial dystrophy. A 27 year-old daughter was asymptomatic. The biomicroscopic exam disclosed a bilateral nasal and temporal posterior embryotoxon and a central guttata. Gonioscopy showed iridocorneal strands with a prominent Schwalbe´s line. The intraocular pressure was normal. Her sister, a 25 year-old female presented very similar abnormalities along with her brother (22 years old). A 19 year-old female sister presented the iridocorneal angle alterations, without Fuchs endothelial dystrophy, until this moment. This paper presents a family with a central cornea guttata or Fuchs dystrophy associated with Axenfeld-Rieger anomaly. Two different inherited diseases appearing together in an entire family may suggest a single molecular and genetic etiology, with high penetrance.
Keywords: Eye abnormalities/genetics; Fuchs endothelial dystrophy; Anterior eye segment/abnormalities; Cornea/pathology; Syndrome; Case reports
RESUMO
Relato dos resultados do estudo de uma família com distrofia corneana endotelial associada a uma disgenesia do segmento anterior, a anomalia de Axenfeld-Rieger. Paciente de 56 anos, feminina, queixava-se de visão borrada. O exame na lâmpada de fenda evidenciou embriotoxo posterior bilateral e guttata no centro da córnea, edema estromal e dobras na membrana de Descemet, caracterizando distrofia endothelial de Fuchs. A filha de 27 anos estava assintomática. A biomicroscopia revelou embriotoxo posterior nasal e temporal bilateral e guttata central. A gonioscopia evidenciou processos iridocorneanos até a linha de Schwalbe, que se encontrava anteriorizada e proeminente. A pressão intra-ocular estava normal. Não foram observadas alterações sistêmicas. Sua irmã, 25 anos, sexo feminino, apresentava alterações muito similares, assim como seu irmão (22 anos). Outra irmã de 19 anos apresentava alterações do ângulo iridocorneano, porém sem distrofia de Fuchs, até o presente. Este trabalho relata os resultados de estudo de uma família com córnea guttata central ou distrofia de Fuchs associada à anomalia de Axenfeld-Rieger. Duas doenças hereditárias com expressão simultânea em toda a família sugerem uma etiologia molecular e genética única, com alta penetrância.
Descritores: Anormalidades do olho/anormalidades; Distrofia endotelial de Fuchs; Segmento anterior do olho/anormalidades; Córnea/patologia; Síndrome; Relatos de casos
INTRODUCTION
Axenfeld-Rieger (AR) syndrome (MIM 180500) is an inherited autosomal dominant disorder with high penetrance and variable expressivity. It is included in a heterogeneous spectrum of overlapping phenotypes referred to as an anterior segment dysgenesis (ASD)1-3 and is characterized by posterior embryotoxon, iridocorneal strands, iris changes associated with systemic signs, like oligodontia, microdontia, cranial and facial defects. The AR anomaly presents only ocular changes3. Glaucoma is associated with half of the cases in each subtype, with exception of the isolated posterior embryotoxon3. There are other genetically related phenotypes that share many features with AR syndrome, like iridogoniodysgenesis (IGD) anomaly and syndrome as well as familial glaucoma iridogoniodysplasia. For this reason, some authors suggest that they might be considered part of the same group3. Three genetic loci are linked to AR syndrome and related phenotypes, on chromosomes 4q25 (PITX2 gene), 6p25 (FOX-C1 gene), 13q14 (unrecognized gene) and recently, it was also linked to chromosome 11p13 (PAX6 gene).1 These genes are involved in the regulation of eye embryogenesis, and codify transcription factors expressed during this phase1. Shields has postulated that both the ocular and systemic phenotypes result from the disruption of migration and/or the differentiation process of the neural crest-derived tissues, which include the corneal endothelium, the anterior chamber angle (trabecular meshwork, Schlemm´s canal) and the iris.4-5
Fuchs endothelial dystrophy of the cornea (FECD, MIM 136800) is a relatively common and slowly progressive corneal dystrophy; adult-onset corneal degeneration begins with an axially (central or paracentral) arranged guttae. It becomes clinically evident when epithelial and stromal edemas produce painful bullae, which can lead to corneal swelling and severely reduced vision6. The pathogenesis of Fuchs dystrophy is not entirely understood yet. There are clues suggesting a disturbance of regulation of apoptosis7, while other studies suggest that the disturbance occurs on terminal differentiation of the neural crest-derived corneal endothelium8-9. In this report, we describe a family (mother and four children) having a combination of endothelial corneal dystrophy with anterior chamber dysgenesis classified as Axenfeld-Rieger anomaly.
Case Reports
The family was examined in order to find clinical signs of anterior chamber dysgenesis, cornea guttata or Fuchs endothelial dystrophy. The findings are listed on Table1 and their heredogram is shown on Figure1.
Case 1
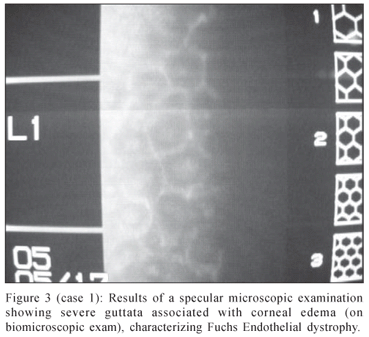
A 56 year-old female (mother) had blurred vision complaints. Her best visual acuity was 20/25 in the right eye and 20/50 in the left eye. The biomicroscopic exam showed bilateral posterior embryotoxon (Figure 2) and guttata with corneal edema and Descemet membrane's folds, characterizing Fuchs endothelial dystrophy. Gonioscopic view was not possible because of the corneal edema. The specular microscopic examination revealed that the endothelial cells were almost absent, and it was not possible to execute the cell count (Figure 3). The intraocular pressure (aplannation tonometry) was 12 mmHg in the right eye and 14 mmHg in the left eye. The fundoscopy was normal and revealed no signs of glaucomatous optic nerve damage. The rest of the ocular examination was normal. This case was indicated to corneal transplant.
Case 2
A 27 year-old Caucasian female patient (daughter) had the best visual acuity equal to 20/20 in both eyes. Slit lamp evaluation disclosed a bilateral nasal and temporal posterior embryotoxon (Figure 4). No systemic signs were observed. The intraocular pressure (aplannation tonometry) was 15 mmHg in the right eye and 16 mmHg in the left eye. Gonioscopy showed iridocorneal strands with a prominent Schwalbe´s line (Figure 5). Specular microscopic examination demonstrated bilateral dark bodies (guttae) and the endothelial cells showed pleomorphism and polymegathism (Figure 6). Central corneal thickness was 571 ìm in the right eye and 587ìm in the left eye. The fundoscopy was normal and revealed no signs of glaucomatous optic nerve damage.
Case 3
A 25 year-old Caucasian daughter was examined. The best visual acuity was equal to 20/20 in both eyes. Slit lamp evaluation presented bilateral nasal and temporal posterior embryotoxon. This may be an incomplete form of Axenfeld-Rieger anomaly. No sectorial iris hypoplasia or atrophy, corectopy, polycoria or systemic signs and symptoms were detected.
The intraocular pressure (aplannation tonometry) was 14 mmHg in the right eye and 15 mmHg in the left eye. Specular microscopic examination was similar to case 2. The fundoscopy was normal and revealed no signs of glaucomatous optic nerve damage.
Case 4
A 21 year-old Caucasian son was examined. The best visual acuity was equal to 20/20 in both eyes. Slit lamp evaluation presented bilateral nasal and temporal posterior embryotoxon. This may be an incomplete form of Axenfeld-Rieger anomaly. No sectorial iris hypoplasia or atrophy, corectopy, polycoria or systemic signs and symptoms were detected.
for The intraocular pressure (applannation tonometry) was 16 mmHg in the right eye and 15 mmHg in the left eye. Specular microscopic examination was similar to case 2. The fundoscopy was normal and revealed no signs of glaucomatous optic nerve damage.
Case 5
A 19 year-old Caucasian daughter was examined and considered asymptomatic. The best visual acuity was equal to 20/20 in both eyes. Slit lamp evaluation disclosed bilateral nasal and temporal posterior embryotoxon and sectorial iris atrophy with mild corectopy. No polycoria or systemic signs was observed. The intraocular pressure (applanation tonometry) was 13 mmHg in the right eye and 12 mmHg in the left eye. Specular microscopic examination was normal as well as the rest of the ocular examination. The fundoscopy was normal and revealed no signs of glaucomatous optic nerve damage.
DISCUSSION
This is a remarkable case series in which a family, including two generations presented Fuchs endothelial dystrophy or central cornea guttata associated with Axenfeld-Rieger anomaly. The trait appeared to be autosomal dominant with high penetrance (100%), considering that all siblings inherited the abnormalities of the mother. Interestingly, Hwang et al.10 described a case in which mother and son had a combination of corectopy, congenital corneal opacities and cornea guttata (MIM 608484), with an overlap with Peters anomaly (characterized by central corneal opacities and absence of Descemet membrane), but it was not associated with posterior embryotoxon, polycoria or iris stromal hypoplasia.
There are two genes encoding transcription factors and three loci that are associated with the Axenfeld-Rieger malformation. The gene PITX2 ("pituitary homeobox transcription factor 2", RIEG1) was cloned on chromosome 4p25 and regulates the expression of other genes during embryonic development11. Several pathological mutations (deletions, translocations or spot mutations) in PITX2 have been described causing different phenotypes, including AR malformation11, iridogoniodysgenesis 12, iris hypoplasia13 and rarely Peters' anomaly14. A second gene associated with AR syndrome is FOXC1 ("Forkhead Box C1"), on chromosome 6p2515. During the embryogenesis, FOXC1 is expressed in craniofacial structures, in periocular and pre-endothelial mesenchyma, sclera and cornea and further, in the pos-fetal phase, in trabecular meshwork, iris and conjuntival epithelium. The mutations include duplications, insertions, deletions and missense types affecting the DNA ligation stability, activation of specific genes and transcriptional regulation. These mutations produce a very large spectrum of phenotypes of anterior segment dysgenesis, mild to severe form, including or not systemic manifestations.15-17 Other locus for Rieger syndrome was mapped on 13q14 (RIEG2)13, but no gene was identified18.
Besides the genes described previously, there are evidences of the involvement of PAX6 gene ("Paired Box Gene 6") in 11p13 chromosome and MAF gene ("V-MAF Avian Musculoaponeurotic Fibrosarcoma Oncogene Homolog") in 16q24 chromosome on AR malformations4.
In the Fuchs dystrophy, the mechanism of progressive dysfunction of corneal endothelium and the collagen deposition remains unknown, and it is not clear whether it is a primary or secondary effect. Several theories have been proposed to explain the etiology of FECD. Biswas et al.8 described missense mutations in COL8A2, a gene of the a2 chain of type VII collagen in association with FECD, suggesting that this molecule may be related with a disturbance in the terminal differentiation of the neural crest derived endothelial cells8. There are studies suggesting that apoptosis plays an important role in endothelial cell degeneration7. It is possible that a modification of the basement membrane composition (Descemet membrane) or the loss of contact between endothelial cell and Descemet membrane may cause apoptosis of these cells in Fuchs' Dystrophy. Further studies are needed to investigate this hypothesis.
The mechanism of the ocular bad development has been widely studied and discussed3-5. Often the factors that result in disturbance of neural crest may act at more than one phase of this process, producing anomalies that involve more than one tissue derived from neural crest.
This case series was the first description of this combination of two ocular abnormalities together (Fuchs dystrophy and Axenfeld-Rieger anomaly) and was reported on world ophthalmology congress in 200619. Later on, Kniestedt et al. showed very similar ocular findings presented in a five generation family with a PITX2 mutation and polymorphism20.
CONCLUSION
Two different inherited diseases appearing together in an entire family may suggest a single molecular and genetic etiology, but additional studies are necessary to explain the relationship among these overlapping phenotypes and their inheritance. Perhaps, in future, it would be possible to develop alternative therapeutic tools for the treatment of Fuchs dystrophy and glaucoma, based on the knowledge of their etiology.
Recebido para publicação em: 18/9/2008
Aceito para publicação em 22/12/2008
- 1. Espinoza H, Cox CJ, Semina EV, Amendt BA. A molecular basis for differential developmental anomalies in Axenfeld-Rieger syndrome. Hum Mol Genet. 2002; 11(7):743-53.
- 2. Gould DB and John, SWM. Anterior segment dysgenesis and the developmental glaucomas are complex traits. Hum Mol Genet. 2002;11(10):1185-93.
- 3. Alward WL. Axenfeld-Rieger syndrome in the age of molecular genetics. Am J Ophthalmol. 2000;130(1):107-15.
- 4. Lines MA, Kozlowski K, Walter MA. Molecular genetics of Axenfeld-Rieger malformations. Hum Mol Genet. 2002; 1 (10): 1177-84.
- 5. Shields MB. Axenfeld-Rieger syndrome: a theory of mechanism and distinctions from the iridocorneal endothelial syndrome. Trans Am Ophthalmol Soc. 1983; 81: 736-84.
- 6. Bergmanson JP, Sheldon TM, Goosey JD. Fuchs´ endothelial dystrophy: a fresh look at an aging disease. Ophthalmic Physiol Opt.1999; 19(3): 210-22.
- 7. Borderie VM, Baudrimont M, Vallée A, Ereau TL, Gray F, Laroche L. Corneal endothelial cell apoptosis in patients with Fuchs' dystrophy. Invest Ophthalmol Vis Sci. 2000;41(9):2501-5.
- 8. Biswas S, Munier FL, Yardley J, Hart-Holden N, Perveen R, Cousin P, et al. Missense mutations in COL8A2, the gene encoding the alpha2 chain of type VIII collagen, cause two forms of corneal endothelial dystrophy. Hum Mol Genet. 2001;10(21):2415-23.
- 9. Bahn CF, Falls HF, Varley GA, Meyer RF, Edelhauser HF, Bourne WM. Classification of corneal endothelial disorders based on neural crest origin. Ophthalmology. 1984; 9 (6):558-63.
- 10. Hwang JM, Chung DC, Traboulsi EI. A new syndrome of hereditary congenital corneal opacities, cornea guttata, and corectopia. Arch Ophthalmol. 2003; 121(7): 1053- 4.
- 11. Semina EV, Reiter R, Leysens NJ, Alward WL, Small KW, Datson NA, Siegel-Bartelt J, et al. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat Genet. 1996; 14(4):392-9.
- 12. Kulak SC, Kozlowski K, Semina EV, Pearce WG, Walter MA. Mutation in the RIEG1 gene in patients with iridogoniodysgenesis syndrome. Hum. Mol. Genet. 1998;7(7):1113-7.
- 13. Alward WL, Semina EV, Kalenak JW, Heon E, Sheth BP, Stone EM, Murray JC. Autosomal dominant iris hypoplasia is caused by a mutation in the Rieger syndrome (RIEG/PITX2) gene. Am J Ophthalmol.1998;125(1):98-100.
- 14. Doward W, Perveen R, Lloyd LC, Ridgway AE, Wilson L Black GC. Mutation in the RIEG1 gene associated with Peters' anomaly. J Med Genet. 1999;36(2):152- 5.
- 15. Mears AJ, Jordan T, Mirzayans F, Dubois S, Kume T, Parlee M, et al. Mutations of the forkhead/winged-helix gene, FKHL7, in patients with Axenfeld-Rieger anomaly. Am J Hum Genet. 1998;63(5):1316-28.
- 16. Honkanen RA, Nishimura DY, Swiderski RE, Bennett SR, Hong S, Kwon YH. A Family with Axenfeld-Rieger syndrome and Peters anomaly caused by a point mutation (Phe112Ser) in the FOXC1 gene. Am J Ophthalmol. 2003;135(3):368-75.
- 17. Lehmann OJ, Ebenezer ND, Ekong R, Ocaka L, Mungall AJ, Fraser S, McGill JI, et al. Ocular developmental abnormalities and glaucoma associated with intersticial 6p25 duplications and deletions. Invest Ophthalmol Vis Sci. 2002; 43(6):1843-9.
- 18. Phillips JC, del Bono EA, Haines JL, Pralea AM, Cohen JS, Greff LJ, Wiggs JL. A second locus for Rieger syndrome maps to chromosome 13q14. Am J Hum Genet. 1996;59(3):613-9.
- 19. Oliveira MB, Mitraud RS, Cunha AL, Yamane R. Axenfeld-Rieger Anomaly and corneal endothelial dystrophy: a case series. In: World Ophthalmology Congress, February, 2006. São Paulo (Brazil). [Poster CO227]
- 20. Kniestedt C, Taralczak M, Thiel MA, Stuermer J, Baumer A, Gloor BP. A novel PITX2 mutation and a polymorphism in a 5-generation family with Axenfeld-Rieger anomaly and coexisting Fuchs' endothelial dystrophy. Ophthalmology. 2006;113(10):1791-98.
Publication Dates
-
Publication in this collection
30 Jan 2009 -
Date of issue
Dec 2008
History
-
Accepted
22 Dec 2008 -
Received
18 Sept 2008