Abstracts
OBJECTIVE: To estimate the additional number of affected individuals based on the prevalence of sickle-cell syndromes among relatives of index cases. METHODS: Cross-sectional study of relatives of a random sample of index cases identified through a neonatal screening program in Northeastern Brazil, between 2001 and 2005. The extended family trial model included 463 relatives of 21 index cases. Relatives were classified as nuclear family (NF: father, mother, and siblings); first degree extended family (N1: grandparents, uncles and aunts, and first cousins); second degree extended family (N2: children of first cousins); extended family (NA: NF+N1+N2); and extended nuclear family (NA1: NF+N1). The presence of HBB*S and other abnormal hemoglobins was confirmed by high-performance liquid chromatography. The association between the presence of HBB*S and other variables was calculated using prevalence ratios and their respective 95% confidence intervals, and differences between means were calculated using Student's t test with a 5% significance level. RESULTS: Of relatives, 81% had no knowledge of sickle-cell anemia and HBB*S was present in 114 family members. A total of 53.3% of the studied population was considered as of reproductive age, and 80% of HBB*S carriers had already had children. Frequency was higher among NF (69%), but was also high in N1 (22.8%). NA1 screening resulted in the detection of 69 carriers additional (a 172% increase). CONCLUSIONS: These results indicate that family screening for the identification of sickle-cell carriers should be extended to first degree relatives.
Anemia, Sickle Cell; Sickle Cell Trait; Heterozygote Detection; Neonatal Screening; Genetic Screening; Cross-Sectional Studies
OBJETIVO: Estimar o incremento no número adicional de afetados com base na prevalência de síndromes falciformes em familiares de casos-índice. MÉTODOS: Estudo transversal em familiares de amostra aleatória dos casos-índice identificados por programa de triagem neonatal em Pernambuco, no período de 2001 a 2005. O modelo de triagem familiar ampliado incluiu 463 membros familiares de 21 casos-índice. Os familiares foram categorizados como: núcleo reduzido (NR -pai, mãe e irmãos); de primeiro grau (N1 - avós, tios e primos de primeiro grau); de segundo grau (N2 - filhos dos primos de primeiro grau); ampliado (NA - NR+N1+N2) e ampliado de primeiro grau (NA1 -NR+N1). A confirmação da presença de HBB*S e detecção de hemoglobinas anormais foram realizadas por meio da High Performance Liquid Chromathgraphy. A associação entre a presença de HBB*S e variáveis foi testada pelo cálculo da razão de prevalência e respectivos IC 95% e a diferença entre médias verificadas pelo teste t de Student, ao nível de significância de 5%. RESULTADOS: A anemia falciforme era desconhecida por 81% dos familiares; o gene HBB*S esteve presente em 114 familiares. Observou-se que 53,3% da população estudada estava na faixa considerada reprodutiva e 80% das pessoas portadoras do gene HBB*S já tinham gerado filhos. A freqüência foi maior no núcleo NR (69%), mas também elevada no N1 (22,8%). O NA1 resultou na detecção de 69 portadores adicionais (aumento de 172%). CONCLUSÕES: Os resultados indicam que a triagem familiar para identificação de portadores de síndrome falciforme deve ser estendida para os familiares até o primeiro grau.
Anemia Falciforme; Traço Falciforme; Detecção de Heterozigoto; Triagem Neonatal; Triagem Genética; Estudos Transversais
ORIGINAL ARTICLES
Family screening for HBB*S gene and detection of new cases of sickle cell trait in Northeastern Brazil
Flavia Miranda Gomes C BandeiraI; Magnun Nueldo Nunes SantosI; Marcos André C BezerraI; Yara M GomesII; Aderson Silva AraujoI; Maria Cynthia BragaII; Wayner Vieira SouzaII; Frederico G C AbathII, In memoriam
IFundação de Hematologia e Hemoterapia de Pernambuco. Recife, PE, Brasil
IICentro de Pesquisas Aggeu Magalhães. Fundação Oswaldo Cruz. Recife,PE, Brasil
Correspondence Correspondence: Yara M Gomes Departamento de Imunologia Centro de Pesquisas Aggeu Magalhães/ FIOCRUZ Av. Morais Rego, s/n Cidade Universitária 50670-420 Recife, PE, Brasil E-mail: yara@cpqam.fiocruz.br
ABSTRACT
OBJECTIVE: To estimate the additional number of affected individuals based on the prevalence of sickle-cell syndromes among relatives of index cases.
METHODS: Cross-sectional study of relatives of a random sample of index cases identified through a neonatal screening program in Northeastern Brazil, between 2001 and 2005. The extended family trial model included 463 relatives of 21 index cases. Relatives were classified as nuclear family (NF: father, mother, and siblings); first degree extended family (N1: grandparents, uncles and aunts, and first cousins); second degree extended family (N2: children of first cousins); extended family (NA: NF+N1+N2); and extended nuclear family (NA1: NF+N1). The presence of HBB*S and other abnormal hemoglobins was confirmed by high-performance liquid chromatography. The association between the presence of HBB*S and other variables was calculated using prevalence ratios and their respective 95% confidence intervals, and differences between means were calculated using Student's t test with a 5% significance level.
RESULTS: Of relatives, 81% had no knowledge of sickle-cell anemia and HBB*S was present in 114 family members. A total of 53.3% of the studied population was considered as of reproductive age, and 80% of HBB*S carriers had already had children. Frequency was higher among NF (69%), but was also high in N1 (22.8%). NA1 screening resulted in the detection of 69 carriers additional (a 172% increase).
CONCLUSIONS: These results indicate that family screening for the identification of sickle-cell carriers should be extended to first degree relatives.
Descriptors: Anemia, Sickle Cell, epidemiology. Sickle Cell Trait, epidemiology. Heterozygote Detection. Neonatal Screening. Genetic Screening. Cross-Sectional Studies.
INTRODUCTION
Sickle-cell anemia and disease are characterized as hereditary hemolytic conditions that evolve chronically, leading to physical and emotional damage in afflicted individuals. To date, the only curative treatment available for sickle-cell disease is bone-marrow transplantation, which is still under evaluation in clinical trials. Worldwide, about 270 million individuals carry alleles that encode abnormal hemoglobins, and 300-400 thousand live born children have either sickle-cell anemia or some form of severe thalassemia.22
In Brazil, sickle-cell anemia is a matter of public health and of epidemiological importance due to its high prevalence. Prevalence varies from 0.1% to 0.3%, depending on the group and region under study20 and on morbidity/mortality.13 Prevalence of the sickle-cell trait varies from 2.7% to 6%.7,17,20 The Brazilian Southeast and Northeast Regions show the greatest prevalence of affected individuals and carriers. Sickle-cell anemia is predominant among populations of African origin,20 with a trend to affect an ever increasing share of the population due to the level of miscegenation in Brazil.
Neonatal screening for sickle-cell syndromes, especially sickle-cell anemia, is necessary due to the high mortality associated with septicemia caused by encapsulated bacteria during the first five years of life.15
Major clinical characteristics of sickle-cell anemia include vaso-occlusive episodes, splenic sequestration, thoracic syndrome, and neurological complications, such as ischemic and hemorrhagic vascular accidents. These outcomes predominate in different forms and intensities according to age, hemoglobin F (Hb F) levels, leukocyte counts, basal hemoglobin levels, and haplotype, directly affecting the quality of life of patients and relatives.15
The high morbidity and mortality and the economic difficulties generated by the disease raise the need for community programs for early diagnosis, medical, social, and psychological orientation, and genetic counseling for couples carrying the sickle-cell trait.13
Detection of hemoglobin S (Hb S) during the neonatal period is a marker for one genetic risk group. Neonatal hemoglobinopathy screening is essential for early diagnosis and for the implementation of preventive and health-promoting measures, especially for sickle-cell anemia.2
The city of Campinas (Southeastern Brazil) was among the first places to implement routine hemoglobinopathy screening, which is mandatory by law in this municipality since October 1997. The Brazilian Ministry of Health created the Programa Nacional de Triagem Neonatal (PNTN National Neonatal Screening Program), by Statute GM/MS no. 822, from 6 July 2001. The specific aims of this program were to increase coverage to 100% of live births, implement active search for screened patients, and provide adequate diagnostic confirmation, follow-up, and treatment to identified patients.* * Ministério da Saúde. Programa Nacional de Triagem Neonatal. [acesso em 3 julho 2007]. Disponível em: http://portal.saude.gov.br/portal/saude/visualizar_texto.cfm?idtxt=24915&janela=1
In the state of Pernambuco (Northeastern Brazil), neonatal hemoglobinopathy screening was instituted by the Ministry of health by Statute GM/MS 452 of October 2001. State capital Recife, in response to the pleas of hemoglobinopathy patients and to the power exerted by social control, instituted the Sickle-cell Anemia Program by Statute 16635/2001.
PNTN proposes the screening of parents following the identification of heterozygote children,* * Ministério da Saúde. Programa Nacional de Triagem Neonatal. [acesso em 3 julho 2007]. Disponível em: http://portal.saude.gov.br/portal/saude/visualizar_texto.cfm?idtxt=24915&janela=1 and does not recommend that screening be extended to other family members.
The aim of the present study was to estimate the increase in the additional number of affected cases based on the prevalence of sickle-cell syndromes among the relatives of index cases.
METHODS
We carried out a cross-sectional study in a state reference hospital for the treatment of hematological diseases in the state of Pernambuco, which located in Recife. This hospital is part of the state's Neonatal Screening Reference Service (Serviços de Referência em Triagem Neonatal SRTN/PE), and provides care to individuals with hemoglobinopathies since June 2001.
We studied relatives of index cases of sickle-cell anemia identified between 2001 and 2005.
Index cases were defined according to the presence of hemoglobin S (HBB*S) in homo or heterozygosis. Cases were identified by collection facilities for heel-prick tests, cities in the Recife metropolitan area, and the remaining cities in the state, notified by SRTN/PE, and enrolled in a neonatal screening outpatient service. Index cases, registered numerically in order of enrollment in the Service, were randomly selected and categorized as SS, SX, or AS, where X represents Hb C (hemoglobin C) or b Thal (beta-thalassemia).
After introducing and explaining the project to the relatives of 21 randomly selected index cases, 508 individuals showed interest in participating in the study. Of these, 463 (91%) were effectively recruited. Compliance among relatives of index cases of SS, SX, and AS was 80%, 100%, and 92.3%, respectively.
Based on the index cases, the following family nuclei were defined: nuclear family (NF), composed of father, mother, and siblings; first degree extended family (N1), including grandparents, uncles and aunts, and first cousins; second degree extended family (N2) composed of the children of first cousins; extended family (NE) defined as NF+N1+N2; and extended nuclear family (NE1) defined as NF+N1.
Selected families were informed about the aims of the survey. At this time, educational material on the disease and on the condition of carrier of the HBB*S gene was provided. Family members up to the second generation were invited to participate in the study. After reading and signing a term of informed consent, participants were evaluated by a physician at the neonatal screening facility, under the support of psychologists, medical geneticists, and social workers.
Participating families were interviewed using a semi-structured questionnaire. During this interview, blood samples were collected by venous puncture using disposable material into EDTA-coated 5 ml vacuum tubes. Blood from children under age 2 years was collected using 2 ml tubes. These samples were then transported to the hemoglobinopathy laboratory for Hb S detection. Samples were also used for erythrograms, Hb electrophoresis at alkaline pH, solubility tests for confirmation of presence of HBB*S in children older than 6 months, and detection of abnormal hemoglobins by high performance liquid chromatography (HPLC) using Variant (Bio-Rad, CA, USA) equipment. Test results were delivered in person to each family member; those diagnosed as carriers of the HBB*S allele were given personalized genetic orientation, according to the international norms for this procedure.11 Subjects showing other hematological alterations were referred for further investigation and outpatient follow-up at the facility.
The following variables were investigated: presence of Hb S, detected by Hb electrophoresis and solubility test, and confirmed by HPLC; degree of relatedness to index cases: parent, sibling, uncle/aunt, cousin, grandparent; skin color/race (classified as black, white, mixed, Asian, and Amerindian), defined by the interviewer according to criteria of the Brazilian Institute for Geography and Statistics** ** IBGE. Censo Demográfico 2000: Características da População e dos Domicílios: Resultados do universo. [acesso em 23 mar 2006]. Disponível em: http://www.ibge.gov.br/home/estatistica/populacao/censo2000/default.shtm (Instituto Brasileiro de Geografia e Estatística IBGE); consanguinity among parents, classified as positive or negative; willingness of relatives to undergo testing; presence of hemoglobinopathies among tested relatives; number of individuals analyzed based on each index case; knowledge of sickle-cell syndromes; and intention to use the genetic information provided. For demographic characterization, we also obtained information on age, sex, and schooling of relatives.
Data entry and analysis were carried out using EpiInfo 3.32 software. For the evaluation of receptiveness and efficiency of the proposal of extended family screening, we analyzed the following indicators, adapted from Teixeira & Ramalho21 (1994):
-
proportion of receptivity to the test;
-
rate of positivity among tested relatives,
-
percentage of index case family memgers willing to be tested,
-
mean number of individuals tested based on each index case.
Prevalence and 95% confidence intervals were estimated, and differences in proportions were analyzed by chi-square test.
The association between presence of HBB*S and the variables considered (sex, age, reproductive age, skin color/race) was tested by calculating prevalence ratios and respective 95%CI. Differences in means were analyzed by Student's t-test. Differences were considered as significant when p<0.05.
The study was approved by the Research Ethics Committee of CPqAM-Fiocruz and Fundação Hemope.
RESULTS
Index cases were divided into the following groups: S homozygotes (HbSS; N=7), double heterozygotes (5 HbSC and 3 HbSb), and HbAS carriers (N=6) (Table 1). The mean number of subjects evaluated per index case was 22.
The age of relatives carrying the HBB*S allele in homo or heterozygosis ranged from six months to 81 years (median=22 years). The sample included 171 (36.9%) males and 292 (63.1%) females. Distribution in terms of schooling was as follows: 203 (44%) with complete elementary school, 85 (18.3%) with complete secondary school, 2 (0.4%) with higher education, and 25 (5.4%) illiterate. Of the 463 family members studied, 30 did not provide information on schooling and 118 had not yet reached school age. Black or mixed subjects were predominant (405/463, 87.5%).
Prevalence of HBB*S carriers was significantly higher in the SS group than in the AS and SX groups (p=0.006 and p=0.02, respectively) (Table 1). These findings can be extrapolated only within their strata, and are not inferences regarding the general population of the state of Pernambuco, given that the sample was not allocated proportionally to the size of the SS, SX, and AS groups in the general population.
When subjects were asked about whether they had previous knowledge sickle-cell anemia, 265 family members (81%) responded negatively, 60 (18.3%) reported previous knowledge of the problem, and two (0.61%) were unable to reply.
When asked whether they would consider their carrier condition when planning a family, 235 family members (82%) responded positively, 37 (13%) negatively, 4 (1.4%) were unable to reply, and 10 (3.5%) did not answer the question.
The HBB*S allele was present in 114/463 (24.4%) of subjects. Of these, 66 (57.9%) were females and 48 (42.1%) were males. There was no statistically significant difference in distribution of HBB*S according to sex (p=0.23). Analysis of the distribution of abnormal hemoglobin detection profiles showed a frequency of AS heterozygotes of 23.1%. Frequencies of other hemoglobinopathies were as follows: AS heterozygotes 14/463 (3.0%), b-thalassemia trait 11/463 (2.4%), sickle-cell anemia (HbSS) 3/463 (0.6%), Sb-thalassemia interaction 3/463 (0.6%). Only seven HBB*S carriers had consanguineous partners.
Table 2 shows, for each index case, the total number of relatives tested along with their respective phenotypes and Hb S frequencies. Mean number of carriers in the NF, N1, N2, NE1, and NE2 groups was 3.29, 0.24, 5.19, and 5.43, respectively. These differences were significant when NF was compared to NE1 or NE2 (p<0.0001). However, mean number of carriers was not different between NE1 and NE2 (p=0.7924).
Table 3 presents the distribution of HBB*S in the family nuclei evaluated. Prevalence of sickle-cell trait was higher in NF (69%), but was also high in N1 (22.8%). Prevalence of N2 was similar to that of the general population (4.9%), which does not justify the inclusion of this group in screening programs. When analyzed collectively, these results indicate that an important number of sickle-cell trait carriers would be detected by inclusion of N1. NE1 screening resulted in the detection of 69 additional carriers (approximately 172% additional cases) (Tables 2 and 3).
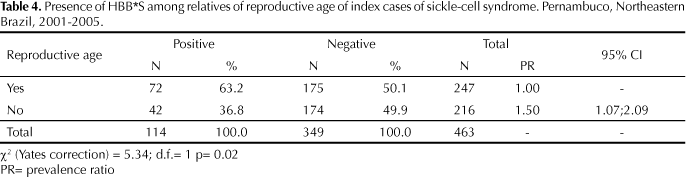
Table 4 addresses the implications of the presence of the allele in the studied group when analyzed in terms of possibility of reproduction, defined as a function of age. We considered as of reproductive age subjects aged 14-45 years, which corresponded to 53.3% of the studied population (p=0.04).
From the perspective of genetic orientation, 80% of subjects carrying HBB*S had already had children, and had therefore already been exposed to the risk of mating with a partner with high risk for sickle-cell disease (p=0.01). Table 5 shows the proportion of subjects with children, and its association with presence of HBB*S.
DISCUSSION
Despite the large number of studies that investigate hemoglobinopathies, especially sickle-cell syndromes, from the standpoint of frequency and clinical characteristics,1 only recently have aspects related to public health in this population been addressed.7,9,18
The response rate in the present study was high (91%). In a survey published by Ramalho et al18 (1999), response was high among pregnant women and blood donors (100%), but lower among students (54%). The high rate in the present study was similar to that found by Ramalho et al18 among pregnant women and blood donors. This indicates that the success of community programs aimed at diagnosing hemoglobinopathies is directly linked to the community's receptiveness to such programs.
In a similar study, Al-Ahmed et al2 (2002) examined a greater number of subjects from families with high levels of consanguinity in Pakistan. Factors such as larger families and the frequent consanguineous marriages in that region may explain the differences in sample size.
Consanguineous marriages are uncommon in the Pernambuco region, which may explain the lower frequency of the allele among the studied family members. The population of the present study was mostly young and of reproductive age, which should be taken into consideration when designing screening programs for hemoglobinopathy.
Several studies discuss what is the best age for this type of genetic screening. While some studies5 argue that screening should be carried out during adolescence, others10 argue for screening during antenatal care. Some studies do not recommend screening for carriers during childhood, given that the efficacy of the program at this stage has been questioned.3 The American Academy of Pediatrics4 (2000) emphasizes the fact that underage children may not be able to provide informed consent for screening. Certain issues must be considered when carrying out such screens, in order to justify their use in underage subjects:4 1. the need for an intervention that provides immediate benefit to the subject, such as the prevention or delay of a complication identifiable by testing; 2. when the screening may eventually benefit another family member without, in principle, leading to any disadvantage to the screened child. The American Academy of Pediatrics also recommends that screening for reproductive purposes or by parental request should be delayed until the child has the ability to provide consent.3
According to the 2000 Census, 45.3% of the Brazilian population was considered as black or mixed; this proportion is 70.1% in the Northeast Region, was 87.5% in the present study. Several factors can explain this difference, including the socioeconomic level of the studied population and the predominance of the HBB*S allele among black/mixed individuals. Ethnicity is important in the historical context of the dissemination of HBB*S, given the frequency of this mutation among Africans brought to Brazil to work as slaves.8 The predominance of African descendents in the population of the present study may have had a positive influence of the occurrence of this allele. Such distribution should have an influence on the discussion of who should or should not be screened. Certain studies postulate targeting screening to high-risk populations, whereas others argue that such populations should be screened universally, and that governments should develop programs for control and management.6 According to Peckham & Dezateux14 (1998), screening programs are justified for certain conditions considered as public health problems and for which therapeutic or control measures are available.19
The discussion of what is the ideal model for family screening and its implementation must involve not only health care authorities, but also patient and community associations. Carrier screening and genetic orientation or counseling are important because these allow for the discussion of reproductive life in a conscientious and informed manner. Nevertheless, it is essential that the difference between carrier and patient be clarified, since there is a risk of errors of interpretation and stigmatization of the person under screening.12
Despite the low number of consanguineous marriages in the present study when compared to communities where such marriages are frequent,2 the issue remains relevant because of the lack of knowledge of their carrier status among the population. The finding that 81% of the studied population were unaware of the existence of sickle-cell anemia shows that any intervention involving screening and genetic orientation in a population should be followed by information and debate within the community itself. Strategies such as lectures and the direct involvement of health workers in orienting the population may be jointly employed for clarifying this condition, thus allowing for greater autonomy on making decisions regarding procreation.
The level of schooling among the population will also be important both for the degree of knowledge of this population and for the introduction of control measures for a given genetic condition.
The finding of an increase in the number of HBB*S carriers detected subsidizes a proposal to expand screening among the risk population. When the distribution of all hemoglobinopathies in the sample is considered, it can be noted that other mutations, such as the presence of HbC or of the b-thalassemia trait follow the same mode of inheritance. The lack of knowledge of carrier status among these individuals, and the consequent absence of genetic counseling and of a conscious decision as to family planning are reason for concern.
The prevalence detected in the family nuclei should be taken into account when planning hemoglobinopathy screening programs, especially those for sickle-cell syndromes.
Finally, the data obtained in the present study suggest that:
-
neonatal screening for hemoglobinopathies should continue to be universal;
-
extended family screening, when aimed at detecting sickle-cell syndrome carriers, should be extended to all first-degree relatives;
-
it is recommended that educational initiatives regarding sickle-cell syndromes be adopted in a systematic manner;
-
greater involvement of the primary health care system will be required in order to multiply knowledge of these syndromes, given that primary health care workers are ever more frequently the first to contact these families.
REFERENCES
Received: 1/17/2007
Reviewed: 9/17/2007
YM Gomes and WV Souza are supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq Proc. no. 306.427/2006-0 and 307.484/2007-6, respectively; productivity grant); MAC Bezerra was supported by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES master's degree scholarship); MNN Santos was supported by Fundação de Amparo à Ciência e Tecnologia do Estado de Pernambuco (FACEPE Proc. no. 2345-4.01/04; trainee grant).
Article based on the doctoral thesis by FMGC Bandeira, presented at the Programa de Pós-Graduação em Saúde Pública do Centro de Pesquisas Aggeu Magalhães/Fiocruz, in 2006.
- 1. Adekile AD. Mild phenotype sickle cell disease: molecular basis clinical presentation and managenent recommendations. Curr Paediatr. 2005;15(1):57-61.
- 2. Al-Ahmed S, Saleem M, Modell B, Petrou M. Screening extended families for genetic hemoglobin disorders in Pakistan. N England J Med. 2002;347(15):1162-8.
- 3. American Academy of Pediatrics. Comittee on Genetics. Health supervision for children with sickle cell diseases and their families. Pediatrics. 1996;98(3 Pt 1):467-72.
- 4. American Academy of Pediatrics. Committee on Genetics. Molecular genetic testing in pediatric practice: a subject review. Pediatrics. 2000;106(6):1494-7.
- 5. ASHG (American Society of Human Genetics) Board of Directors and the ACMG (American College of Medical Genetics) Board of Directors. Points to consider: ethical, legal, and psychological implications of genetic testing in children and adolescents. Am J Hum Genet 1995;57:1233-41.
- 6. Aspinall P. The use of ethnicity to identify the population at risk in pre-operative sickle cell screening. Anaesthesia. 2003;58(11):1121-3.
- 7. Bandeira FMGC, Leal MC, Souza RR, Furtado VC, Gomes YM, Marques NM. Características de recém-nascidos portadores de hemoglobina "S" detectados através de triagem em sangue de cordão umbilical. J Pediatr (Rio J). 1999;75(3):167-71.
- 8. Bezerra MAC, Santos MNN, Araújo AS, Gomes YM, Abath FGC, Bandeira FMGC. Molecular variations linked to the grouping of b- and a-globin genes in neonatal patients with sickle cell disease in the state of Pernambuco, Brazil. Hemoglobin. 2007;31(1):83-8.
- 9. Brandalise S, Pinheiro V, Gabetta CS,/ Hambleton I,/ Serjeant B,/ Serjeant G. Newborn screening for sickle cell disease in Brasil: the Campinas experience. Clin Lab Haematol. 2004;26(1):15-9.
- 10. Cunniff C. Prenatal screening and diagnosis for pediatricians. Pediatrics 2004;114:889-94.
- 11. Harper PS. Insurance and genetic testing. Lancet. 1993;341(8839):224-7.
- 12. Nelson RM, Botkjin JR, Kodish ED, Levetown M, Truman JT, Wilfond BS, et al. Ethical issues with genetic testing in pediatrics. Pediatrics. 2001;107(6):1451-4.
- 13. Paiva e Silva RB, Ramalho AS, Cassorla RMS. A anemia falciforme como problema de saúde pública no Brasil. Rev Saude Publica. 1993;27(1):54-8.
- 14. Peckham CS, Dezateux C. Issues underlying the evaluation of screening programmes. British Med Bull. 1998;54(4):767-78.
- 15. Powars D, Overturf G, Weiss J, Lee S, Chan L. Pneumococcal septicemia in children with sickle cell anemia. JAMA. 1981;245(18):1839-42.
- 16. Ramalho AS, Nassim-Jorge R, Oliveira JA, Pereira DA. Hemoglobina S em recém-nascidos brasileiros. J Pediatr (Rio J). 1976;41(1):9-10.
- 17. Ramalho AS. Talassemia minor, traço falciforme e deficiência de G6PD: dados de prevalência e de morbidade na região de Campinas, SP. Bol Soc Bras Hematol Hemot. 1985;7(134):133-6.
- 18. Ramalho AS, Silva RBP, Teixeira RC, Compri MB. Hemoglobin screening: response of a Brasilian community to opitional programs. Cad Saude Publica. 1999;15(3):591-5.
- 19. Ramalho AS, Magna LA, Paiva-e-Silva RB. A Portaria nş 822/01 do Ministério da Saúde e as peculiaridades das hemoglobinopatias em saúde pública no Brasil. Cad Saude Publica. 2003;19(4):1195-9.
- 20. Ruiz MA, Guerra CC, Naoum PC. Detecção de hemoglobinas anormais em sangue de cordão de recém-nascidos na cidade de Santos, SP, através de eletroforese em gel de ágar amido. Bol Soc Bras Hematol Hemot. 1986;8(137):8-13.
- 21. Teixeira RC, Ramalho AS. Genetics and public health: response of Brazilian population to an optimal hemoglobinopathy program. Rev Bras Gen. 1994;17(4):435-8.
-
22World Health Organization. Hereditary diseases programme. Guidelines for the control of haemoglobin disorders. Geneva; 1994.
Publication Dates
-
Publication in this collection
29 Feb 2008 -
Date of issue
Apr 2008
History
-
Reviewed
17 Sept 2007 -
Received
17 Jan 2007 -
Accepted
15 Sept 2007