Abstracts
An "in-house" RT-PCR method was developed that allows the simultaneous detection of the RNA of the Hepatitis C Virus (HCV) and an artificial RNA employed as an external control. Samples were analyzed in pools of 6-12 donations, each donation included in two pools, one horizontal and one vertical, permitting the immediate identification of a reactive donation, obviating the need for pool dismembering. The whole process took 6-8 hours per day and results were issued in parallel to serology. The method was shown to detect all six HCV genotypes and a sensitivity of 500 IU/mL was achieved (95% hit rate). Until July 2005, 139,678 donations were tested and 315 (0.23%) were found reactive for HCV-RNA. Except for five false-positives, all 310 presented the corresponding antibody as well, so the yield of NAT-only donations was zero, presenting a specificity of 99.83%. Detection of a window period donation, in the population studied, will probably demand testing of a larger number of donations. International experience is showing a rate of 1:200,000 - 1:500,000 of isolated HCV-RNA reactive donations.
Nucleic Acid Testing; Blood Donors; RT-PCR; Transfusional Risk; Hepatitis C Virus; HCV
Desenvolveu-se uma metodologia própria ("in-house") baseada em RT-PCR, que permite detectar simultaneamente o RNA do vírus HCV e de um RNA artificial empregado como controle externo. As amostras são analisadas em pools de 6-12 doações, cada doação sendo incluída em dois pools diferentes, um horizontal e um vertical, permitindo a identificação imediata de uma doação reativa, sem a necessidade de desmembrar-se um pool reativo. O processo todo consumiu de 6-8 horas diárias e os resultados foram emitidos em paralelo à sorologia. O método detectou os seis genótipos de HCV, com um limite de sensibilidade de 500 UI/mL (95% hit rate). Até julho de 2005 haviam sido testadas 139.678 doações com a detecção de 315 (0,23%) doações reativas para HCV-RNA. Exceto cinco falso-positivas, todas estas doações também apresentavam o respectivo anticorpo, portanto não se detectou nenhuma doação em janela imunológica. A especificidade foi de 99,83%. A detecção de amostra em janela imunológica, nesta população de doadores, provavelmente demandará a análise de um número maior de doações, espelhando-se na experiência internacional que tem mostrado a detecção de amostras HCV-RNA isoladas em 1:200.000 - 1:500.000 doações.
NUCLEIC ACID TESTING
Primary screening of blood donors by nat testing for HCV-RNA: development of an "in-house" method and results
Triagem primária de doadores de sangue por teste de ácidos nucléicos: desenvolvimento de um método não-comercial e resultados
Silvano WendelI, II; José Eduardo LeviI, II; Deise Tihe TakaokaII; Isabela Cristina SilvaII; Juliana Polachini de CastroII; Mário A. Torezan-FilhoI, II; Jorge GhanameIII; Romualdo GioachiniIV, V; Joselito BrandãoV; Edison Luis DurigonVI
IBanco de Sangue, Hospital Sírio Libanês, São Paulo, SP, Brasil
IICentro de Imunologia e Imunogenética, São Paulo, SP, Brasil
IIIBanco de Sangue, Hospital Nove de Julho, São Paulo, SP, Brasil
IVBanco de Sangue, Hospital Oswaldo Cruz, São Paulo, SP, Brasil
VBanco de Sangue, Hospital Evaldo Foz, São Paulo, SP, Brasil
VIDepartamento de Microbiologia, Instituto de Ciências Biomédicas, Universidade de São Paulo, São Paulo, SP, Brasil
Correspondence to Correspondence to: Silvano Wendel Rua Peixoto Gomide 515, 12º andar 01409-001 São Paulo, SP, Brasil Fax + 55 11 3284 5315 Phone + 55 11 3285 5999 E-mail snwendel@uninet.com.br
SUMMARY
An "in-house" RT-PCR method was developed that allows the simultaneous detection of the RNA of the Hepatitis C Virus (HCV) and an artificial RNA employed as an external control. Samples were analyzed in pools of 6-12 donations, each donation included in two pools, one horizontal and one vertical, permitting the immediate identification of a reactive donation, obviating the need for pool dismembering. The whole process took 6-8 hours per day and results were issued in parallel to serology. The method was shown to detect all six HCV genotypes and a sensitivity of 500 IU/mL was achieved (95% hit rate). Until July 2005, 139,678 donations were tested and 315 (0.23%) were found reactive for HCV-RNA. Except for five false-positives, all 310 presented the corresponding antibody as well, so the yield of NAT-only donations was zero, presenting a specificity of 99.83%. Detection of a window period donation, in the population studied, will probably demand testing of a larger number of donations. International experience is showing a rate of 1:200,000 - 1:500,000 of isolated HCV-RNA reactive donations.
Keywords: Nucleic Acid Testing; Blood Donors; RT-PCR; Transfusional Risk; Hepatitis C Virus; HCV.
RESUMO
Desenvolveu-se uma metodologia própria ("in-house") baseada em RT-PCR, que permite detectar simultaneamente o RNA do vírus HCV e de um RNA artificial empregado como controle externo. As amostras são analisadas em pools de 6-12 doações, cada doação sendo incluída em dois pools diferentes, um horizontal e um vertical, permitindo a identificação imediata de uma doação reativa, sem a necessidade de desmembrar-se um pool reativo. O processo todo consumiu de 6-8 horas diárias e os resultados foram emitidos em paralelo à sorologia. O método detectou os seis genótipos de HCV, com um limite de sensibilidade de 500 UI/mL (95% hit rate). Até julho de 2005 haviam sido testadas 139.678 doações com a detecção de 315 (0,23%) doações reativas para HCV-RNA. Exceto cinco falso-positivas, todas estas doações também apresentavam o respectivo anticorpo, portanto não se detectou nenhuma doação em janela imunológica. A especificidade foi de 99,83%. A detecção de amostra em janela imunológica, nesta população de doadores, provavelmente demandará a análise de um número maior de doações, espelhando-se na experiência internacional que tem mostrado a detecção de amostras HCV-RNA isoladas em 1:200.000 - 1:500.000 doações.
INTRODUCTION
Despite the excellent laboratorial tests in place today for blood screening, transmission of biological agents still occur as seen in the near past with the Hepatitis C Virus (HCV)1 and the Human Immunodeficiency Virus (HIV)22 and more recently in human encephalitis cases caused by West Nile Virus (WNV) transmitted by blood products and solid organ transplantation28.
In order to avoid transfusion of infectious units, blood banks rely primarily on clinical and epidemiological screening of blood donors, and secondly, on testing of their blood for markers of transmissible agents. The detection of antibodies and/or antigenic portions of these agents is the standard approach in blood screening. However, both suffer from inherent deficiencies: antibody tests may allow an infected unit to go undetected during the serological window period, the time span between infection and the development, by the potential donor, of detectable antibodies. Antigen detection may lead to false-negative results due to genetic variability of the microorganism, as illustrated by an p24 antigen escape mutant9, and, in practice, a diminished sensitivity of HIV p24 antigen and HCV core antigen in comparison to NAT has been observed7,34,36.
Molecular methods targeting fungi, bacteria and viruses nucleic acids have been introduced in the clinical pathology setting in the 1990s, driven in part by the successful clinical use of two quantitative viral load systems (HCV and HIV viral load testing, Amplicor Roche). The development of a tool able to reduce significantly the window period and providing a high-degree of specificity became the goal for all partners involved on the blood circuit12.
By 1997, the German Red Cross had already developed and implemented an 'in-house" PCR based test for this purpose29, soon followed by Japan14 and Scotland4. In parallel we were able to introduce nucleic acid testing (NAT) for the hepatitis C virus (HCV) RNA in March 199837.
NAT introduction in any country has to take in consideration the particularities of its blood system. The pool size, if done on donation pools, and the time taken for resolution of reactive primary pools is directly related to the availability of the blood supply. Other factors like the prevalence of reactive donors and financial resources are also important issues.
Our planning was based on two premises:
We decided to build-up a system of bi-dimensional pooling that would allow simultaneous processing of many samples and, when positive, it wouldn't result in the blockage of all individual sera contained in the pool. To our knowledge, this kind of pooling is not adopted on primary screening in any center elsewhere. The USA has been using single pools of 16-24 samples34 while Japan24 and Germany30 adopted pools of 48-96 donations.
The purpose of this publication is to describe the validation of a NAT method in place at our laboratory since 1998 and to report the results obtained up to July 2005.
MATERIAL AND METHODS
a) Donors and donations: Voluntary blood donors gave written informed consent for participation in this ongoing study. For this purpose a Serum Separation Tube (SST II, Becton-Dickinson, São Paulo, Brazil) was drawn right after collection of the blood bag. Tubes were centrifuged according to manufacturer instructions and transported to the molecular biology lab on the same day. Some donations were eventually kept refrigerated and transported at 4 - 8 ºC to the molecular biology lab on the following day. All donations were tested in parallel at the serology lab for the mandatory screening tests (anti-HCV, anti-HTLV-I/II, anti-HBc and HBsAg, anti-HIV 1 and 2, Chagas, Syphilis, and abnormal hemoglobin detection). Donations were collected between March 1st 1998 and July 31st 2005 at blood banks in São Paulo city (Hospital Sírio Libanês, Hospital Nove de Julho, Hospital Oswaldo Cruz, Hospital Evaldo Foz), Bragança Paulista (Núcleo de Hemoterapia de Bragança Paulista, São Paulo State) and Brasília (Hemolago, Federal District). More than 95% of the donors lived in São Paulo city at the time of donation. Frequency of reactivity to routine serological markers are shown on Table 1 below, as a general characterization of the enrolled donor population.
This study was approved by the ethical committee from the Instituto de Ciências Biomédicas, Microbiology Department from the University of São Paulo, São Paulo, Brazil.
b) Pooling: Pooling was performed on an automated device (Hamilton Microlab AT, Reno, NV, USA) by pipetting donor's serum to RNAse/DNAse free molecular biology grade microwell plates (NUNC, Roskilde, Denmark). Samples were grouped twice bidimensionally ("chessboard" format) on "minipools" of 6-12 donations (hereby generically denominated "pools"), in accordance to the daily number of samples to be tested, each donation contributing with 20.8 - 41.6 µL of serum, to a total of 250 µL. For example, if a pool of six samples was built, each donation contributed with 50 µL to a total of 300 µL. After homogenization, 250 µL was taken for the extraction process. If a pool of twelve was built, each donation contributed with 30 µL, and 250 µL removed from the 360 µL initially pooled. Every sample was represented on two "pools" and positioned in a way that allowed readily identification of a positive or inhibitory sample, obviating the need for dismembering reactive "pools". Pools were made both in the horizontal and vertical axis. Each homogenized pool was manually transferred to a conventional microtube. A scheme of pooling is depicted on Figure 1.
c) NAT METHOD:
c1) RNA extraction: RNA was extracted from 250 µL of serum pools with 750 µL of Trizol LS® (Invitrogen, São Paulo, Brazil), in the presence of 20 µg of glycogen as a carrier and 20,000 copies of an external control (EC) synthetic RNA (pAW 109 RNA, Applied Biosystems, Brazil).
c2) Reverse Transcription (RT): Total RNA was resuspended in 18 µL of Reverse Transcription mixture containing RNAse Inhibitor 1 U/µL (Invitrogen, São Paulo, Brazil), DTT 1 mM, 1x PCR buffer (10 mM Tris-HCl pH 8.3; 50 mM KCl; 1.5 mM MgCl2) 200 µM dNTPs, 3.5 mM MgCl2, and incubated for five minutes at 65 ºC , then centrifuged at 3,000Xg, 4 ºC for one minute. Moloney Murine Leukemia Virus Reverse Transcriptase (M-MLV RT, Invitrogen, São Paulo, Brazil) 2.5 U/µL and random hexamers 2.5 µM (Invitrogen, São Paulo, Brazil) as primers, were added to the mixture and subsequently incubated for 10 minutes at room temperature, then 30 minutes at 37 ºC and finally boiled for five minutes.
c3) HCV and external control primer choice: cDNA was synthesized from samples obtained from patients chronically infected by HCV and requiring HCV viral load testing for treatment monitoring. Samples were diluted with negative plasma in order to obtain viral loads of 50, 500, 5,000 and 50,000 copies/mL. These samples were processed as described above and submitted to amplification with four primer pair combinations, HC11/HC1831; SM3/HC18 (SM3 is a new sense primer here described); P32/P36 and P33/P48, these last two in a nested-PCR format where P32/P36 PCR product is used as a template for P33/P48 amplification, as described26. Primers sequences are given in Table 2 below.
c4) Amplification (1st generation method): This method was introduced in March 1st 1998 as a routine screening test for HCV-RNA and was replaced by a multiplex PCR mixture (item c5 below) in July 1st 2001, when HIV-RNA testing became a routine.
One fourth of the total cDNA (5 µL) corresponding to 5.2 - 10.4 µL of each donor serum, was directly applied on a 25 µL (final volume) Polymerase Chain Reaction (PCR) solution, containing 3.5 mM of MgCl2, 200 µM of dNTPs, 1.25 Units of Taq Polymerase (Pharmacia, Uppsala, Sweden), 500 nM of a primer pair (sense SM3 + antisense HC 18) spanning a fragment of 268 bp from the HCV 5' untranslated region (nucleotide - 267 to -1) and a primer pair (sense DM 151 + antisense DM 152) spanning 308 bp from the external control cDNA. Forty cycles of 94 ºC 30 seconds, 55 ºC 30 seconds and 72 ºC one minute were preceded by a 95 ºC five minutes incubation and followed by a final step of seven minutes at 72 ºC on a Perkin-Elmer 9600 Thermocycler.
c5) Amplification (2nd generation method): Multiplex PCR _ 10 µL of cDNA was added to a PCR mixture containing three primers pairs (HCV, HIV21 and External Control), glycerol 5%, cresol red 0.25 µg/µL, dNTPs 0.2 mM (A,C,G,U), 1.25 U of Platinum Taq (Invitrogen, São Paulo, Brazil), 0.03 U of Uracyl-N-Glycosilase (UNG, Amersham, São Paulo, Brazil) in a final volume of 25 µL. This mixture was submitted to the following thermocycling parameters: 95 ºC for five minutes then 40 cycles of 94 ºC /30 seconds; 55 ºC /30 seconds; 65 ºC / 1 minute, and a final incubation step at 65 ºC for seven minutes. The method was validated and performed on the following thermocyclers: Applied Biosystems models 2400, 9600 and 9700 and MJ Research model PTC-100.
c6) Electrophoresis: PCR products from the External Control (308 bp) and the HCV 5'untranslated region (268 bp) were run at 200mV/100mA for one hour on an ethidium bromide stained 3% agarose gel and directly observed under UV light on an UV Imager (Ultra-Violet Products, Cambridge, UK). All images were digitalized and stored allowing the traceability of the results. A legend was engraved on the gel image and a photo printed in a thermal printer, accompanying the daily working sheet.
c7) Controls: On each run four controls were included from the extraction; triplicates of a mix of HCV and HIV reconstituted international standards (available from WHO, National Institute for Biological Standards and Controls, Potters Bar, Herts, UK, HCV-RNA International Standard Cat. # 96/798 and HIV-RNA International Standard Cat. # 97/656 I) containing 500 IU/mL of both HCV and HIV-RNAs and one seronegative and NAT negative plasma. During the HCV-only screening phase, controls containing exclusively HCV were employed. An amplification control was included at the PCR step consisting of a "pool" of cDNAs from standards, previously shown to correctly amplify HCV and HIV. It was required two out of three replicates to amplify HCV and HIV RNA to validate the entire run. A negative result for HCV and HIV RNA on the negative control was mandatory; otherwise the run was invalid and repeated. The previously obtained cDNA "pool" from standards served to guide us in case of a run failure, i.e. if the run is completely negative but this amplification control was correct we started again from extraction; if this control failed we started from the PCR step onwards.
d) Validation of NAT: This method was validated for routine use by:
d1) Testing HCV quantitated plasma samples from donors and patients: Forty-two samples quantified by the referred HCV viral load test were tested neat and diluted to the depicted concentrations. At that time HCV viral load test adopted the copies/mL unit so sensitivity was estimated based on this. Results are shown on Table 3 below.
d2) Sensitivity: Lyophilized WHO standards, purchased from the NIBSC, were reconstituted in RNAse free-ddH2O and diluted with negative plasma to the desired concentrations. Twenty replicates were run three times at five dilution points namely 1,000 IU/mL; 500 IU/mL; 250 IU/mL; 50 IU/mL and 10 IU/mL of HCV-RNA. The detection limit of the method was 500 IU/mL of HCV-RNA (95% hit rate, Table 4).
d3) Specificity: On testing of 1,000 seronegative donations there was no sample assigned as "initially reactive". In quality control blind panels, no negative sample was assigned as reactive.
d4) Reproducibility: Assessed by running in parallel to serological routine for two months and by the performance of three distinct operators. The method was shown to be robust and reproducible, independent of operator, provided that the technician was properly trained.
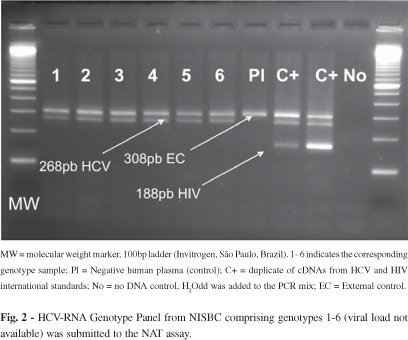
d5) Genotypic coverage: HCV RNA Genotype Reference Panel for NAT (NISBC code 02/202) was purchased from NISBC. HCV Panel contains genotypes 1-6. All samples were detected by the method both individually (Fig. 2) and in pools.
d6) External Assessment: Yearly participation on blind panels prepared for quality control on NAT labs (Viral Quality Control Program VQC, organized by CLB, Amsterdam, Netherlands). All samples containing more than 500 IU/mL of HCV-RNA were correctly assigned as reactive. No false-positives were observed.
e) Re-validation of NAT when switching from 1st to 2nd generation: All procedures above were repeated with identical results. Sensitivity was neither reduced nor improved, and specificity was superior to 99.9%, as before. Main modifications were: increase in the cDNA input from 5 µL to 10 µL; replacement of a conventional enzyme Taq Polymerase by an enzyme providing higher processivity and "hot-start" namely Platinum Taq (Invitrogen, São Paulo, Brazil); addition of glycerol and cresol red; adoption of an enzymatic control of contamination provided by amplification in the presence of dUTP instead of dTTP and addition of UNG21.
f) Confirmatory NAT assay: Repeatedly reactive samples were submitted to an alternative NAT test, either on the same sample or from a new one collected from the putative positive donor. HCV confirmatory test consisted of a nested-PCR targeting also the 5´ UTR, but with primers positioned in HCV genome in a way to avoid contamination from potential NAT-screening amplicon carryover. Five µL of the cDNA, produced as above, were included on a 25 µL final volume PCR, containing primers P32/P36. Five µL of this first-round PCR was transferred to a second tube containing primers P33/P48, as described26. Universal anti-contamination precautions were rigorously observed. Detection limit of this assay is 100 IU/mL (99% hit rate).
g) HCV genotyping: Genotyping of 33 unselected HCV positive donors was done by amplification of 310 base pairs from the 5'-Untranslated Region (5'UTR)31. PCR products were purified by filtration on a microspin column device (Microcon-30, Amicon Inc., USA) and quantified on gel electrophoresis by comparison to standards employing the GelWorks software (Ultra-Violet Products, UK). Cycle sequencing with the same PCR primers was performed with a dye-terminator DNA sequencing kit (Applied Biosystems, USA) and fragments were run on an automated DNA sequencing apparatus (ABI Prism 377 DNA Sequencer, Applied Biosystems, USA). Assignment of the genotype was based on type-specific motifs present on the 5'UTR33.
h) HCV-RNA viral load: HCV-RNA quantification was performed by use of the ROCHE HCV-Monitor v 1.0 kit, according to the manufacturer instructions. Upper limit of this method is 500,000 copies/mL.
i) HGV-RNA PCR: HGV-RNA was detected by amplifying a sequence from the 5'UTR in a single-step assay and also by a nested-PCR method targeting the NS3 region, as described20.
j) EIA: Anti-HCV testing (EIA) was performed either with ABBOTT, Axsym HCV 3.0 (Wiesbaden - Delkenheim, Germany) or ORTHO Anti-HCV EIA 3.0 (Raritan, New Jersey, USA) according to manufacturer's instructions. These solid phase enzyme immunoassays contain peptides and recombinant proteins attached, representing the core and non-structural proteins NS3, NS4 and NS5. An absorbance/cut-off (A/CO) ratio > 1 is considered reactive, according to the manufacturer criteria.
k) Immunoblot (IB): Samples repeatedly reactive to screening EIA, independent of NAT results, underwent a confirmatory step, using LiaTek HCV III (Innogenetics, Zwijndrecht, Belgium). Test strips are coated with synthetic recombinant HCV antigens from the E2/NS1, NS3, NS4, NS5 and core proteins. Binding of serum/plasma antibodies to one or more antigen lines results in color development and are interpreted as a reactive sample, according to manufacturer's instructions.
l) ALT levels: ALT level determination was systematically performed in an Alcyon 300/300i analyzer by a UV-kinetic method (Abbot Park, IL, USA), as an additional screening test until its discontinuation in 2004. Samples presenting values higher than 84 U/L were rejected, according to internal operational procedures.
RESULTS
a) Primer choice: Among the primer combinations tested, SM3/HC18 displayed the greatest sensitivity on single-step assays. Samples containing 500 copies/mL were readily detected by this system as well as by the second-round of the nested PCR (Fig. 3). At higher HCV-RNA concentrations, signals were similar for all combinations19.This analysis led to the choice of primer pair SM3/HC18 for our "in-house" NAT test.
b) Blood donors screening: The NAT method has been in use as a routine since March 1998 for HCV RNA and in July 2001 HIV RNA screening was added to the test, in a multiplex format (Fig. 4), replacing p24 Ag detection21. Until July 2005 139,678 donations were tested for HCV RNA, including 77,555 also tested for HIV RNA. 48,272 PCR reactions were performed, counting pools and controls. 550 samples were NAT initially reactive (0.39%) demanding their testing individually in duplicate on the next day routine. If they were found repeatedly reactive the donation was rejected and discarded, otherwise (non-reactive) it was released for transfusion. 1,114 samples were blocked for inadequacy (0.80%; external control non-reactive) demanding their inclusion in pools on the next day routine. Only one sample was repeatedly inadequate, both in pools and individually (Table 5). This donor was advised to wait for six months and then to donate again, which resulted in a negative result, therefore, acceptable for transfusion and he was subsequently reintegrated. By serology, 805/139,678 donations (0.58%) were rejected due to repeatedly reactive results while NAT rejected 315/139,678 (0.23%), where 310 (98.4%) were also reactive by the NAT confirmatory method using nested-PCR (Table 5).
In practice, the observed specificity of the method was 99.83% when considering initially reactive donations, derived from the 235 samples that were initially reactive but didn't confirm either when analyzed by the screening NAT test individually (230) or when submitted to the ancillary NAT confirmatory test (five samples), the nested-PCR described in item "f" of material and methods section. However, if evaluating rejection due to the initially reactive (pools)/ repeatedly reactive (individual sample) analysis, the specificity is > 99.9% since only five samples displayed this pattern and were further non reactive in the confirmatory assay. These five NAT false-positives amplicons were sequenced, revealing the presence of an HGV "Iowan" strain38 in four of them. Those four were also reactive for two distinct HGV-specific molecular assays, confirming their status of HGV carriers. The other NAT false-positive result was attributed to amplicon contamination of the index sample and/or the corresponding cDNA. Approximately 17% of the blot-reactive samples were NAT non-reactive (Table 6).
c) Genotypes and viral load: Genotypes detected among donors were 1a (13/33), 1b (11/33), 2b (1/33) and 3a (8/33). Observed viral load was generally very high (median = 500,000 copies/mL).
d) ALT levels and EIA A/CO rate in viremic (NAT RT) and non-viremic (NAT NRT) donors: Mean ALT values were calculated for donors serologically confirmed for HCV (blot RT) grouped according to NAT reactivity. Viremic donors displayed mean ALT values above the cut-off for this marker (84 U/mL), but the median for this group was only 54 U/mL, as seen in Table 7A. Accordingly, the mean A/CO rate is similar between NAT reactive and non-reactive donors (Table 7B).
DISCUSSION
This study describes our experience with HCV NAT testing by an "in-house" developed method for screening of blood donors. Development of our own methodology, which in 1997 was the unique option for those willing to NAT screen their blood supply, took us 18 months of research that culminated in the process described above. We were among the first blood banks to report NAT data in the literature18, however, at this time, due to the small number of samples analyzed, these results had low impact, in comparison to those obtained by large centralized blood banks processing hundreds of thousands donations per year. It became evident to us, that every blood bank must adapt their NAT process to the particularities of its own service, such as the prevalence of HCV and HIV, the demand for blood components for transfusion and the daily number of samples to be analyzed. Within these parameters, we developed this method in consonance to a routine of 50-200 donations/day and a maximum turnaround time of 12 hours. From March 1998 until July 2005 there was no single day that we were unable to release NAT results for any reason. Occasionally, NAT had to be repeated from the beginning, due to a complete failure of the process, and on one occasion, a strong contamination required extreme cleaning measures and delayed for additional eight hours the release of blood components, solved by use of alternative primers, which are part of our salve guard system. In general, the rate of repetitions for inadequacy (0.80%) or false-reactivity (0.16%) is similar to commercial NAT systems currently approved by regulatory agencies and in use worldwide. In Japan, using a Roche multiplex real-time PCR system, 0.17% of false-positive results were observed and 1.99% of the pools demanded repetition of the process due to internal control failure24. In Europe the combination of NucliSens extraction (Organon Teknika, Boxtel, Netherlands) and AmpliScreen (Roche Diagnostic Systems, Branchburg, NJ, USA) is adopted in some countries like the Netherlands. They reported a rate of 1.01% of invalid test results and 0.14% of initial false-positives15. The performance of commercial tests is variable. The rate of invalid runs for the Chiron TMA assay is 0.1 - 4% and for Roche Ampliscreen 0.1-1.6% while the specificity lies in between 99.6 - 99.9% for both assays, as reported in a NAT International Forum5.
The total number of screened samples here presented (139,678) may be found of low significance when compared to the expected yield of NAT isolated reactive donations (³ 1:200,000). Nevertheless, this is one of the largest series from Brazil presented so far, and the only one that depicts simultaneous results (screening serology/NAT). Since no withdrawal of serological screening was implemented, evaluating NAT and serology on a routine basis also provides a constant challenge and quality control of both laboratories, and has increased our confidence in the NAT "in-house" method.
It should be emphasized that the option for an "in-house" system is not based on economical grounds. Though it would be naive to deny this aspect as one of the parameters for developing our own method, several blood banks from wealthy nations (USA6, Japan14, Germany29 and Scotland4) used and some still use such "in-house" systems.
As Brazilian plasma has been sent abroad in exchange for hemoderivatives, NAT data on Brazilian plasma became available23; four cases of HCV NAT reactive donations were found among 1,159,241 donations tested (1:289,810). Such NAT reactive samples may be classified as actually window-period donations only after disclosing which brand and version of antibody assay was used to screen these donations (supposedly EIA non-reactive) and documented seroconversion of implied donors.
Due to the unknown percentage of repeat donors in this series, estimating the interdonation interval was not attempted, and consequently, the incidence/window-period model32 cannot be applied. In this situation, HIV incidence and the transfusional risk may be estimated by using a modification of the ELISA test called "detuned assay"17 but for HCV a similar assay has not been described, so, in practice, risk has to be derived by performing NAT mini-pool testing10.
The 5'-Untranslated Region is the most conserved genomic segment among HCV isolates. However, some variability can be found inside the 5'UTR affecting the primer binding for some isolates, and thus the amplification yield. We have tested several published primer combinations and a new sense primer, based on the comparison of the 5'UTR of several HCV isolates from all genotypes33. SMITH et al. (1995) has proposed two 20mers showing 100% nucleotide conservancy to all isolates studied, as theoretically ideal primers33. We have investigated one of these in combination to HC1831 as the reverse primer. Due to the contamination problem and time constraints we discarded the performance of "nested-PCR" and evaluated several primer pairs but always in one round reactions, taking 2-3 hours to complete. We tested also primer combinations that would allow the co-amplification of HCV and the control RNA without significant loss of sensitivity, in a multiplex format.
Sensitivity of the here described method is of 1,000 copies of HCV-RNA/mL (100% hit rate) and was estimated by testing CLB Pelipsy standard (Amsterdam, Netherlands) and samples quantified by the Roche HCV Monitor v1.0 (Roche, Brazil) individually and on pools. This means that the method is able, on minipools of 8-12, to detect a positive donor if serum contains > 6,000 copies/mL. After international standardization of HCV in International Units, and availability of working reagents to be used as calibrators, we challenged our method against NISBC HCV international standard, achieving a sensitivity of 500 IU/mL which is in agreement to the units/copies relationship16 (1U @ 1-3 copies). The sensitivity achieved, of 500 IU/mL for HCV is in consonance to the requirements of strict regulatory agencies such the FDA8 and the Paul-Ehrlich Institute27. In fact, these agencies stipulate a hit rate of 95% or higher for a donation containing at least 5,000 IU/mL, independently of the pool size. Enhancement of the sensitivity or processing of larger pool sizes can be achieved by concentration steps prior to RNA extraction, which we found unnecessary for the donation volume processed daily in our laboratory.
Seropositive (anti-HCV) donors also NAT-reactive have a trend to harbor high viral titers, median is > 500,000 copies/mL and only one sample depicted a titer (26,586 copies/mL) close to the cut-off but was readily detected by our PCR test19. Viremia in the window period starts at the day of the inoculum and rapidly increases due to the rapid doubling time of HCV, estimated as 10.8 hours11. Actually, viral load presented by NAT-only reactive donors crosses the detection threshold of NAT-minipool systems a few days after infection. This is illustrated by viral load determination on a series of 176 NAT HCV reactive donors detected in the US where only eight displayed a viral load inferior to 5,000 IU/mL (Dr Susan L Stramer, American Red Cross, Gaithersburg, Maryland, USA, personal communication). This situation of very low viremia occurs in the period called pre-ramp phase and is responsible for the still existing transfusional risk for NAT screened blood2. Unfortunately, it has been proved that even those donations with very low viremia transmit HCV to receptors at a very high rate25.
Genotypes observed on this donor population follow the pattern described for HCV patients from the same geographic region3; 73% genotype 1 (1a and 1b equally frequent), 24% genotype 3 (all 3a) and 3% genotype 2 (all 2b).
Among the PCR-HCV non-reactive donations there were HIV-RNA, HGV-RNA20 and HBV-DNA positive donors (data not shown), attesting the high specificity of the method (99.83%). Five donations presented an amplification product suggestive of HCV in both pools and subsequently, in individual reactions. However, the confirmatory nested-PCR was unable to corroborate that, since no amplification was observed. Sequencing of these amplicons (268 bp, NAT screening) depicted the presence of HGV sequences in four of them and authentic HCV in one. We interpreted this finding as PCR false-positives caused by the undesired capacity of the HCV primers to anneal to HGV genome. Unexpectedly, this spurious hybridization occurs at the NS3 gene, not at the HGV 5'UTR, where higher homology between both Flaviviruses do occur. Since these false-positives were only observed during the months of April and May of 2000, we speculate that a particular HGV strain was circulating at that time. Another possible explanation is that one of the reagents (primers, Taq polymerase etc.) may have presented some performance variability, causing a decrease in the reaction stringency, which we were unable neither to detect during quality assurance regular procedures nor to attribute to a particular component of the amplification mixture. The remaining false-positive may be ascribed to amplicon carry-over.
The frequency of truly inhibitory samples was very low, only one sample prevented the amplification of the external control on both "pools" and also individually. The donor was asked to return and a new sample didn't show inhibition so the donor was re-integrated while the inhibitory donation was discarded.
We observed 46 donors with a positive EIA and IB and a negative PCR result both on "pools" and individually (Table 6). We believe that these persons (46/268 = 17.16%) without detectable viremia (HCV-RNA) represent cases of previous exposure to HCV with further clearance of the virus or intermittent viremia. We noticed that ALT levels are significantly lower on this than on the PCR reactive group (Table 7a), indicating normal liver function, which is compatible to absence of hepatic viral replication. On the other hand, the anti-HCV absorbance/cut-off rate doesn't differ between the two groups (Table 7b). Moreover, their index sample and follow-up samples were negative on a more sensitive NAT assay (nested-PCR, sensitivity 100 IU/mL, 99% hit rate). Prospective testing of German blood donors for HCV by PCR29 has identified a rate of truly positive non-viremic donors of 13/77 (27%), similar to the one reported here. It is well established that truly antibody reactive-RNA negative persons correspond to 15-25% of all HCV infected individuals13.
In conclusion, it was shown that a reliable "in-house" method was developed and successfully implemented as a routine for several Brazilian blood banks. So far, the yield of isolated NAT reactive samples is zero, probably because of the still relatively small number of donors tested. In fact, in the largest series published35, 170 HCV NAT-reactive/anti-HCV non-reactive units were identified among 39,721,404 tested, giving a rate of 1:230,000.
The system in place is an alternative to more expensive commercial assays; is flexible and allows the fast introduction of new RNA targets, as shown for HGV20.
Received: 20 September 2005
Accepted: 24 November 2006
- 1. ARROJO, I.P.; PAREJA, M.O.; ORTA, M.D.R. et al. - Detection of a healthy carrier of HCV with no evidence of antibodies for over four years. Transfusion, 43: 953-957, 2003.
- 2. BUSCH, M.P.; TOBLER, L.H.; GERLICH, W.H. et al. - Very low level viremia in HCV infectious unit missed by NAT. Transfusion, 43: 1173-1174, 2003.
- 3. CAMPIOTTO, S.; PINHO, J.R.; CARRILHO, F.J. et al. - Geographic distribution of hepatitis C virus genotypes in Brazil. Braz. J. med. biol. Res, 38: 41-49, 2005.
- 4. CLELAND, A.; DAVIS, C.; ADAMS, N. et al - Development of multiplexed nucleic acid testing for human immunodeficiency virus type 1 and hepatitis C virus. Vox Sang. (Basel), 81: 93-101, 2001.
- 5. COSTE, J.; REESINK, H.W.; ENGELFRIET, C.P. et al. - Implementation of donor screening for infectious agents transmitted by blood by nucleic acid technology: update to 2003. Vox Sang. (Basel), 88: 289-303, 2005.
- 6. DELWART, E.L.; KALMIN, N.D.; JONES, T.S. et al. - First report of human immunodeficiency virus transmission via an RNA-screened blood donation. Vox Sang. (Basel), 86: 171-177, 2004.
- 7. DOW, B.C.; MUNRO, H.; BUCHANAN, I. et al - Acute hepatitis C virus seroconversion in a Scottish blood donor: HCV antigen is not comparable with HCV nucleic acid amplification technology screening. Vox Sang. (Basel), 86: 15-20, 2004.
-
8FDA/ Center for Biologics Evaluation and Research - Use of nucleic acid tests on pooled and individual samples from donors of whole blood and blood components for transfusion to adequately and appropriately reduce the risk of transmission of HIV-1 and HCV. Guidance for Industry, Draft guidance. March 2002. Available at http://www.fda.gov/cber/gdlns/hivhcvnatbld.pdf and http://www.fda.gov/cber/gdlns/hivnas.pdf
- 9. GAUDY, C.; MOREAU, A.; BRUNET, S. et al - Subtype B human immunodeficiency virus (HIV) type 1 mutant that escapes detection of fourth-generation immunoassay for HIV infection. J. clin. Microbiol., 42: 2847-2849, 2004.
- 10. GLYNN, S.A.; KLEINMAN, S.H.; WRIGHT, D.J.; BUSCH, M.P. & NHLBI RETROVIRUS EPIDEMIOLOGY DONOR STUDY - International application of the incidence rate/window period model. Transfusion, 42: 966-972, 2002.
- 11. GLYNN S.A.; WRIGHT, D.J.; KLEINMAN, S.H. et al - Dynamics of viremia in early hepatitis C virus infection. Transfusion, 45: 994-1002, 2005.
- 12. HEWLETT, I.K. & EPSTEIN, J.S. - Food and Drug Administration Conference on the feasibility of genetic technology to close the HIV window in donor screening (conference report). Transfusion, 37: 346-351, 1997.
- 13. HOOFNAGLE, J.H. - Course and outcome of hepatitis C. Hepatology, 36 (suppl.1): S21-S29, 2002.
- 14. JAPANESE RED CROSS NAT SCREENING RESEARCH GROUP - Nationwide nucleic acid amplification testing of hepatitis B virus, hepatitis C virus and human immunodeficiency virus type 1 for blood transfusion and follow-up study of nucleic acid amplification positive donors. Jap. J. infect. Dis., 53: 116-123, 2000.
- 15. JONGERIUS, J.M.; SJERPS, M.; CUIJPERS, T.M. et al. - Validation of the NucliSens Extractor combined with the AmpliScreen HIV version 1.5 and HCV version 2.0 test for application in NAT minipool screening. Transfusion, 42: 792-797, 2002.
- 16. KONNICK, E.Q.; ERALI, M.; ASHWOOD, E.R. & HILLYARD, D.R. - Performance characteristics of the COBAS Amplicor Hepatitis C Virus (HCV) Monitor, Version 2.0, International Unit assay and the National Genetics Institute HCV Superquant assay. J. clin. Microbiol, 40: 768-773, 2002.
- 17. KOTHE, D.; BYERS, R.H.; CAUDILL, S.P. et al. - Performance characteristics of a new less sensitive HIV-1 enzyme immunoassay for use in estimating HIV seroincidence. J. Acquir. Immune Defic. Syndr., 33: 625-634, 2003.
- 18. LEVI, J.E.; CONTRI, D.G.; TAKAOKA, D.T. & WENDEL, S. - PCR as a tool for primary screening of blood donors. Transfusion, 38(suppl. 10): 57, 1998.
- 19. LEVI, J.E. - Triagem de doadores de sangue por PCR: desenvolvimento e aplicação na detecção do vírus da hepatite C. São Paulo, 2000. (Dissertação de doutorado - Instituto de Ciências Biomédicas da Universidade de São Paulo).
- 20. LEVI, J.E.; CONTRI, D.G.; LIMA, L.P. et al - High prevalence of GB virus C/hepatitis G virus RNA among Brazilian blood donors. Rev. Inst. Med. trop. S. Paulo, 45: 75-78, 2003.
- 21. LEVI, J.E.; WENDEL, S.; TAKAOKA, D.T. et al - Replacement of HIV p24 Ag test by a multiplex RT-PCR method for primary screening of blood donors. Rev. Inst. Med. trop. S. Paulo, 49: 171-176, 2007.
- 22. LING, A.E.; ROBBINS, K.E.; BROWN, T.M. et al. - Failure of routine HIV-1 tests in a case involving transmission with preseroconversion blood components during the infectious window period. J. Amer. Med. Ass., 284: 210-214, 2000.
- 23. MAC DOWELL, B.; AMORIM, L.; SABACK, F.L.; MELO, H. & MENDES, A. - Resultados dos testes de biologia molecular (NAT) para os vírus HIV, hepatite B (HBV) e hepatite C (HCV) em doadores de sangue. In: CONGRESSO BRASILEIRO DE EPIDEMIOLOGIA, VI, Recife, 2004. Anais.
- 24. MINE, H.; EMURA, H.; MIYAMOTO, M. et al. - High throughput screening of 16 million serologically negative blood donors for hepatitis B virus, hepatitis C virus and human immunodeficiency type-1 by nucleic acid amplification testing with specific and sensitive multiplex reagent in Japan. J. virol. Meth, 112: 145-151, 2003.
- 25. OPERSKALSKI, E.A.; MOSLEY, J.W.; TOBLER, L.H. et al. - HCV viral load in anti-HCV-reactive donors and infectivity for their recipients. Transfusion, 43: 1433-1441, 2003.
- 26. OKAMOTO, H.; OKADA, S.; SUGIYAMA, Y. et al. - Detection of hepatitis C virus RNA by a two-stage polymerase chain reaction with two pairs of primers deduced from the 5'-noncoding region. Jap. J. exp. Med, 60: 215-222, 1990.
-
27PAUL-EHRLICH INSTITUTE - Requirements for the validation and routine operation of the HCV-NAT in the blood collection service. Available at http://www.pei.de/english/valid_engl.pdf
- 28. PEALER, L.N.; MARFIN, A.A.; PETERSEN, L.R. et al - Transmission of West Nile virus through blood transfusion in the United States in 2002. New. Engl. J. Med., 349: 1236-1245, 2004.
- 29. ROTH, W.K.; WEBER, M. & SEIFRIED, E. - Feasibility and efficacy of routine PCR screening of blood donations for hepatitis C virus, hepatitis B virus, and HIV-1 in a blood-bank setting. Lancet, 353: 359-363, 1999.
- 30. ROTH. W.K. & SEIFRIED, E. - The German experience with NAT. Transfus. Med, 12: 255-258, 2002.
- 31. SALDANHA, J. & MINOR, P. - A sensitive PCR method for detecting HCV RNA in plasma pools, blood products and single donations. J. med. Virol., 43: 72-76, 1994.
- 32. SCHREIBER, G.B.; BUSCH, M.P.; KLEINMAN, S.H. & KORELITZ, J.J. - The risk of transfusion-transmitted viral infections. The Retrovirus Epidemiology Donor Study. New Engl. J. Med, 334: 1685-1690, 1996.
- 33. SMITH, D.B.; MELLOR, J.; JARVIS, L.M. et al. - Variation of the hepatitis C virus 5' non-coding region: implications for secondary structure, virus detection and typing. The International HCV collaborative study group. J. gen. Virol., 76: 1749-1761, 1995.
- 34. STRAMER, S.L.; CAGLIOTI, S. & STRONG, D.M. - NAT of the United States and Canadian blood supply. Transfusion, 40: 1165-1168, 2000.
- 35. STRAMER, S.L.; GLYNN, S.A.; KLEINMAN, S.H. et al - Detection of HIV-1 and HCV infections among antibody-negative blood donors by nucleic acid-amplification testing. New Engl. J. Med, 351: 760-768, 2004.
- 36. TOBLER, L.H.; STRAMER, S.L.; LEE, S.R. et al. - Performance of ORTHO HCV core antigen and trak-C assays for detection of viraemia in pre-seroconversion plasma and whole blood donors. Vox Sang. (Basel), 89: 201-207, 2005.
- 37. WENDEL, S.; CONTRI, D.G.; TAKAOKA, D.T. et al. - Screening of blood donors for HCV-RNA: development of a PCR test and results after 20 months. Vox Sang. (Basel), 78(suppl. 1): 429, 2000.
- 38. XIANG, J.; WUNSCHMANN, S.; SCHMIDT, W.; SHAO, J. & STAPLETON, J.T. - Full-length GB virus C (Hepatitits G virus) RNA transcripts are infectious in primary CD4-positive T cells. J. Virol 74: 9125-9133, 2000.
Correspondence to:
Publication Dates
-
Publication in this collection
29 June 2007 -
Date of issue
June 2007
History
-
Received
20 Sept 2005 -
Accepted
24 Nov 2006