ABSTRACT
Leptospirosis is an acute bacterial septicemic febrile disease caused by pathogenic leptospires, which affect humans and animals in all parts of the world. Transmission can occur by direct contact with infected animals or, more commonly, through indirect contact with water or soil contaminated with urine from infected animals. Leptospires enter the body by penetrating mucous membranes or skin abrasions and disseminate through the hematogenic route. In humans, leptospirosis may cause a wide spectrum of symptoms. Most cases have a biphasic clinical presentation, which begins with the septicemic phase followed by immune manifestations. The severe forms of the disease may be life threatening with multisystem damage including renal failure, hepatic dysfunction, vascular damage, pulmonary hemorrhage and muscle lesions. In this review, we present and discuss the pathogenesis of the human disease and the mechanisms of cell membrane injuries, which occur mainly due to the presence of leptospires and/or their antigen/s in the host tissues.
Leptospira; Leptospirosis; Cadherins; Weil syndrome; Acute liver and kidney injury
INTRODUCTION
Leptospirosis is an acute bacterial septicemic febrile disease caused by pathogenic species of Leptospira, which affect humans and animals in all parts of the world11. Costa F, Hagan JE, Calcagno J, Kane M, Torgerson P, Martinez-Silveira MS, et al. Global morbidity and mortality of leptospirosis: a systematic review. PLoS Negl Trop Dis. 2015;9:e0003898.. It is a zoonosis associated with chronically infected carrier animals22. Bharti AR, Nally JE, Ricaldi JN, Matthias MA, Diaz MM, Lovett MA, et al. Leptospirosis: a zoonotic disease of global importance. Lancet Infect Dis. 2003;3:757-71.. In humans, leptospirosis has a biphasic clinical presentation, which begins with the septicemic phase followed by immune manifestations33. Haake DA, Levett PN. Leptospirosis in humans. In: Adler B, editor. Leptospira and leptospirosis. Berlin: Springer-Verlag; 2015. p.65-97.. The most severe form of the disease, with multisystem damage, including vascular, hepatic, renal, pulmonary and skeletal muscles injury, is known as the Weil syndrome44. Plank R, Dean D. Overview of the epidemiology, microbiology, and pathogenesis of Leptospira spp. in humans. Microbes Infect. 2000;2:1265-76.
5. De Brito T, Menezes LF, Lima DM, Lourenço S, Silva AM, Alves VA. Immunohistochemical and in situ hybridization studies of the liver and kidney in human leptospirosis. Virchows Arch. 2006;448:576-83.
6. Del Carlo Bernardi F, Ctenas B, da Silva LF, Nicodemo AC, Saldiva PH, Dolhnikoff M, et al. Immune receptors and adhesion molecules in human pulmonary leptospirosis. Hum Pathol. 2012;43:1601-10.-77. Goris MG, Kikken V, Straetemans M, Alba S, Goeijenbier M, van Gorp EC, et al. Towards the burden of human leptospirosis: duration of acute illness and occurrence of post-leptospirosis symptoms of patients in the Netherlands. PLoS One. 2013;8:e76549.. Many cases of leptospirosis exhibit a non-specific picture of a febrile disease, however the vascular manifestation of the disease may be present as a hemorrhagic conjunctivitis33. Haake DA, Levett PN. Leptospirosis in humans. In: Adler B, editor. Leptospira and leptospirosis. Berlin: Springer-Verlag; 2015. p.65-97.,88. Wagenaar JF, Goris MG, Partiningrum DL, Isbandrio B, Hartskeerl RA, Brandjes DP, et al. Coagulation disorders in patients with severe leptospirosis are associated with severe bleeding and mortality. Trop Med Int Health. 2010;15:152-9.. It has been estimated that the annual mortality rate of leptospirosis is 0.84 deaths per 100,000 people, globally11. Costa F, Hagan JE, Calcagno J, Kane M, Torgerson P, Martinez-Silveira MS, et al. Global morbidity and mortality of leptospirosis: a systematic review. PLoS Negl Trop Dis. 2015;9:e0003898..
Pathogenic leptospires are widespread in nature and their life cycles keep them in the environment thanks to their hematogenous and intercellular dissemination to the proximal kidney tubules of the various reservoir hosts. Potential gateways to leptospire are usually skin abrasions and mucous membranes of the oral cavity and conjunctiva. Transmission may occur by direct contact with an infected host, but more commonly occurs via soil, from contaminated water where bacteria are known to persist, and from the urine of infected animals. Therefore, occupational activities involving infected animals and contaminated soil and water are risk factors for developing leptospirosis33. Haake DA, Levett PN. Leptospirosis in humans. In: Adler B, editor. Leptospira and leptospirosis. Berlin: Springer-Verlag; 2015. p.65-97.,99. Levett PN. Leptospirosis. Clin Microbiol Rev. 2001;14:296-326.. Humans are accidental hosts, and commonly exhibit urinary shedding of leptospires for short durations33. Haake DA, Levett PN. Leptospirosis in humans. In: Adler B, editor. Leptospira and leptospirosis. Berlin: Springer-Verlag; 2015. p.65-97.,99. Levett PN. Leptospirosis. Clin Microbiol Rev. 2001;14:296-326.. In the proximal tubules, the leptospires colonize the reservoir’s brush border over long periods, which does not produce apparent adverse effects in the reservoir host but acts to disseminate the microorganism1010. Adler B, de la Peña-Moctezuma A. Leptospira and leptospirosis. Vet Microbiol. 2010;140:287-96.. However, we should remember that during human and animal infections, proximal tubules are the main site of important lesions, which determine the brush border of morphological and immunoenzymatic changes, among other alterations1111. De Brito T, Freymüller E, Hoshino S, Penna DO. Pathology of the kidney and liver in the experimental leptospirosis of the guinea-pig. Alight and electron microscopy study. Virchows Arch Pathol Anat Physiol Klin Med. 1966;341:64-78.. It should be expected that the presence of the microorganism in the reservoir host might be accompanied, at least in certain cases, by discrete pathophysiological manifestations. In this context, a study of asymptomatic renal colonization of humans by leptospires has shown that excretion of leptospires can occur for periods exceeding a year. Moreover, it was speculated that in regions with high disease transmission like the one studied, “humans can develop some clinical and serological characteristics of asymptomatic urinary carriers” as observed in reservoir hosts1212. Ganoza CA, Matthias MA, Saito M, Cespedes M, Gotuzzo E, Vinetz JM. Asymptomatic renal colonization of humans in the Peruvian Amazon by Leptospira. PLoS Negl Trop Dis. 2010;4:e612..
An important study was carried out by RamachandraRao et al.1313. RamachandraRao SP, Matthias MA, Kokoy-Mondragon C, Aghania E, Park C, Kong C, et al. Proteomic analysis of urine exosomes reveals renal tubule response to leptospiral colonization in experimentally infected rats. PLoS Negl Trop Dis. 2015;9:e0003640. in rats, which are animal carrier hosts for leptospires. Both female and male rats were experimentally infected with leptospires and urine exosome analysis was carried out in the kidneys. Exosomes are nanovesicles that are released from cells and mediate intercellular communication1414. Edgar JR. Q&A: what are exosomes, exactly? BMC Biol. 2016;14:46.. Exosome analysis can identify important information related to kidney function such as the protein content of urine. It was found that the urine from infected rats showed a marked increase in protein content and many of the proteins were unique to the urine of infected male or female rats. Moreover, the top discriminator between control and infected rats was an alanyl aminopeptidase of membrane origin, also known as neutral aminopeptidase or CD13, expressed on the kidney brush border of the proximal tubules. Additionally, the Tamm-Horsfall protein, which is secreted by the transitional epithelium of the thick ascending Henle loop and distal tubules, was significantly decreased in the infected rat urine, suggesting a lesion in this part of the nephron.
Once inside the host, the hematogenous dissemination of leptospires through capillaries occurs and persists there during the septicemic phase of the disease, which lasts for the first eight days of fever1515. Agampodi SB, Matthias MA, Moreno AC, Vinetz JM. Utility of quantitative polymerase chain reaction in leptospirosis diagnosis: association of level of leptospiremia and clinical manifestations in Sri Lanka. Clin Infect Dis. 2012;54:1249-55.. Experimental studies of leptospirosis suggest that afterwards, leptospires find their way to the interstitium. This usually occurs either when the leptospires pass between endothelial cells, as suggested by a study in guinea pigs using colloidal carbon as capillary marker1616. De Brito T, Böhm GM, Yasuda PH. Vascular damage in acute experimental leptospirosis of the guinea-pig. J Pathol. 1979;128:177-82. or after other kind of insult to the cell such as direct damage to the endothelial membrane. Additionally, leptospires can pass between epithelial cells, including the ones lining the renal tubules, and this could be an important way to reach and colonize the lumen of the proximal tubules. It is interesting to speculate about the interplay between leptospires and cell brush borders, where the microorganisms apparently find a relatively safe environment to persist for a long time. However, morphological lesions, such as the disappearance of the PAS (periodic acid–Schiff reaction) and alkaline phosphatase histochemical reactions, are present in this segment of the nephron during the infection, suggesting that the presence of the microorganism can damage the host during active disease1111. De Brito T, Freymüller E, Hoshino S, Penna DO. Pathology of the kidney and liver in the experimental leptospirosis of the guinea-pig. Alight and electron microscopy study. Virchows Arch Pathol Anat Physiol Klin Med. 1966;341:64-78..
Liver cell plate disarray is a major histopathologic finding in human autopsies of leptospirosis, receiving attention since the early pathologic studies of the disease during the first World War (1914 - 1918)1717. Beitzke H. Ueber die pathologische Anatomie des ansteckenden Gelbsucht (Weilsche Krankheit). Berliner Klin Wochenschr. 1916;53:188-91,1818. Pick L. Zur pathologischen Anatomie des infektiösen Icterus. Klin Wochenschr. 1917;54:451-5.. They are also present in experimental leptospirosis and are accompanied by antigen deposits over the cell membrane, as demonstrated by immunohistochemistry1919. Alves VA, Gayotto LC, De Brito T, Santos RT, Wakamatsu A, Vianna MR, et al. Leptospiral antigens in the liver of experimentally infected guinea pig and their relation to the morphogenesis of liver damage. Exp Toxicol Pathol. 1992;44:425-34. and immunoelectron microscopy2020. De Brito T, Prado MJ, Negreiros VA, Nicastri AL, Sakata EE, Yasuda PH, et al. Detection of leptospiral antigen (L. interrogans serovar Copenhageni serogroup Icterohaemorrhagiae) by immunoelectron microscopy in the liver and kidney of experimentally infected guinea-pigs. Int J Exp Pathol. 1992;73:633-42.. Liver cell disarray is absent in human biopsies of leptospirosis cases when studied by light microscopy2121. De Brito T, Machado MM, Montans SD, Hoshino S, Freymüller E. Liver biopsy in human leptospirosis: a light and electron microscopy study. Virchows Arch Pathol Anat Physiol Klin Med. 1967;342:61-9.,2222. Alves VA, Vianna MR, Yasuda PH, De Brito T. Detection of leptospiral antigen in the human liver and kidney using an immunoperoxidase staining procedure. J Pathol. 1987;151:125-31.. However, it must be remembered that since the biopsy procedure is only possible when the hemorrhagic phase of the disease subsides and patients are recovering, it is expected that at least some of the acute lesions are no longer present. In accordance with this, electron microscopy studies of human liver biopsies in leptospirosis show cellular junctions practically restored but with a few enlarged intercellular spaces2121. De Brito T, Machado MM, Montans SD, Hoshino S, Freymüller E. Liver biopsy in human leptospirosis: a light and electron microscopy study. Virchows Arch Pathol Anat Physiol Klin Med. 1967;342:61-9.,2323. Brito de T, Penna DO, Hoshino S, Pereira VG, Caldas AC, Rothstein W. Cholestasis in human leptospirosis: a clinical, histochemical, biochemical and electron microscopy study based on liver biopsies. Beitr Pathol Anat. 1970;140:345-61.. Moreover, although most of the electron microscopy findings are non-specific and might be related to the septicemic manifestations of the disease, the altered microvilli, both at the sinusoidal and the biliary pole, suggest a marked cell membrane disturbance. It was speculated at the time that the damage to cell membranes could be the result of hypothetical toxins produced by pathogenic leptospires, since it has been shown that leptospires have genes encoding for proteases and other products that can cause host cell membrane degradation2424. Kassegne K, Hu W, Ojcius DM, Sun D, Ge Y, Zhao J, et al. Identification of collagenase as a critical virulence factor for invasiveness and transmission of pathogenic Leptospira species. J Infect Dis. 2014;209:1105-15.,2525. Amamura TA, Fraga TR, Vasconcellos SA, Barbosa AS, Isaac L. Pathogenic leptospira secreted proteases target the membrane attack complex: a potential role for thermolysin in complement inhibition. Front Microbiol. 2017;8:958.
CADHERINS
Our previous autopsy work on human leptospirosis2626. De Brito T, Menezes LF, Lima DM, Lourenço S, Silva AM, Alves VA. Immunohistochemical and in situ hybridization studies of the liver and kidney in human leptospirosis. Virchows Arch. 2006;448:576-83. has shown the importance of loss of E-cadherin membrane expression in the pathogenesis of liver plate disarray. E-cadherin is a 120 kDa glycoprotein that is highly expressed during development and is the predominant cadherin of the liver epithelium, where it is concentrated in the adherens junction and interacts with the actin filaments of the cell. This duality, including an extracellular domain that mediates cell-cell adhesion interactions and a cytoplasmic tail associated with catenins, and therefore with the cytoskeleton, produces its fundamental roles in the organization and maintenance of the cell junction. In leptospirosis, the expression of E-cadherin in liver cells is diminished and/or absent in areas of the lobule, thus contributing to the lack of a stable intercellular adhesion (Figures 1A and 1B). Human biopsy studies of the liver in leptospirosis demonstrate a robust recovery of E-cadherin expression in the liver, since, as noted earlier, biopsy is possible only during the recovery phase of the disease (Figures 1C and 1D). Moreover, the definite signs of hepatocyte regeneration demonstrate an important previous insult to the liver during the acute stage of the disease. The data corroborate, as previously noted (De Brito et al.)55. De Brito T, Menezes LF, Lima DM, Lourenço S, Silva AM, Alves VA. Immunohistochemical and in situ hybridization studies of the liver and kidney in human leptospirosis. Virchows Arch. 2006;448:576-83., leptospirosis as a disease of the cell membrane.
- Immunohistochemistry and histology analyses. A) Expression of E-cadherin in lobule of normal human liver; B) Areas of diminished and/or absent E-cadherin in liver in human leptospirosis; C) Liver biopsy in human leptospirosis. Hepatic cells show slight variation in size and occasional more marked hyaline cytoplasmatic change. Portal space is unremarkable; D) Human liver biopsy in leptospirosis showing areas of dense reticulin framework. Variations of size and shape of liver cells is more evident with reticulin staining; E) VE-cadherin in normal human lung; F) VE-cadherin in lung in leptospirosis. Marked intra-alveolar hemorrhage. Dilated and discontinuous capillary walls with irregular staining
Leptospirosis can also be regarded as a hemorrhagic septicemia, therefore, the main findings involving vessels, chiefly those of the microcirculatory circulation, are essential for its pathogenesis.
Evangelista et al.2727. Evangelista K, Franco R, Schwab A, Coburn J. Leptospira interrogans binds to cadherins. PLoS Negl Trop Dis. 2014;8:e2672,2828. Evangelista KV, Hahn B, Wunder EA Jr, Ko AI, Haake DA, Coburn J. Identification of cell-binding adhesins of Leptospira interrogans. PLoS Negl Trop Dis. 2014;8:e3215. focused their work on vascular VE-cadherin, a major component of endothelial adherence junctions, which maintains vascular integrity, and found that pathogenic leptospires bind to endothelial cells lining blood vessels and to VE-cadherin of human mammalian cell culture. The binding of bacteria was reduced in the presence of antibodies against VE-cadherin, supporting the importance of this surface protein in the integrity of the endothelium. Non-pathogenic leptospires such as L. biflexa were less efficient, compared with L. interrogans, in attaching to endothelia and to fibronectin. The binding of pathogenic leptospires to host cells through receptors, such as cadherins, is not followed by a definite intracellular invasion. However, experimental and autopsy data suggest an altered cell membrane permeability1616. De Brito T, Böhm GM, Yasuda PH. Vascular damage in acute experimental leptospirosis of the guinea-pig. J Pathol. 1979;128:177-82.,2626. De Brito T, Menezes LF, Lima DM, Lourenço S, Silva AM, Alves VA. Immunohistochemical and in situ hybridization studies of the liver and kidney in human leptospirosis. Virchows Arch. 2006;448:576-83. with the presence of leptospira remnants and/or antigens in the cytoplasm and even in the nuclei of endothelial and human hepatic cells.
Therefore, VE-cadherin is a quite important previously identified receptor for pathogenic leptospires. The binding of bacteria to VE-cadherin is mediated by adhesins, proteins and lipoproteins, which have been identified in sera from leptospirosis patients2727. Evangelista K, Franco R, Schwab A, Coburn J. Leptospira interrogans binds to cadherins. PLoS Negl Trop Dis. 2014;8:e2672,2828. Evangelista KV, Hahn B, Wunder EA Jr, Ko AI, Haake DA, Coburn J. Identification of cell-binding adhesins of Leptospira interrogans. PLoS Negl Trop Dis. 2014;8:e3215..
Martinez-Lopez et al.2929. Martinez-Lopez DG, Fahey M, Coburn J. Responses of human endothelial cells to pathogenic and non-pathogenic Leptospira species. PLoS Negl Trop Dis. 2010;4:e918. demonstrated that pathogenic strains of Leptospira did not cause apoptosis or necrosis of the cells even after prolonged incubation periods, but significantly disrupted the endothelial cell layers, which may be interpreted as a main contributor to the hemorrhagic manifestations of the disease.
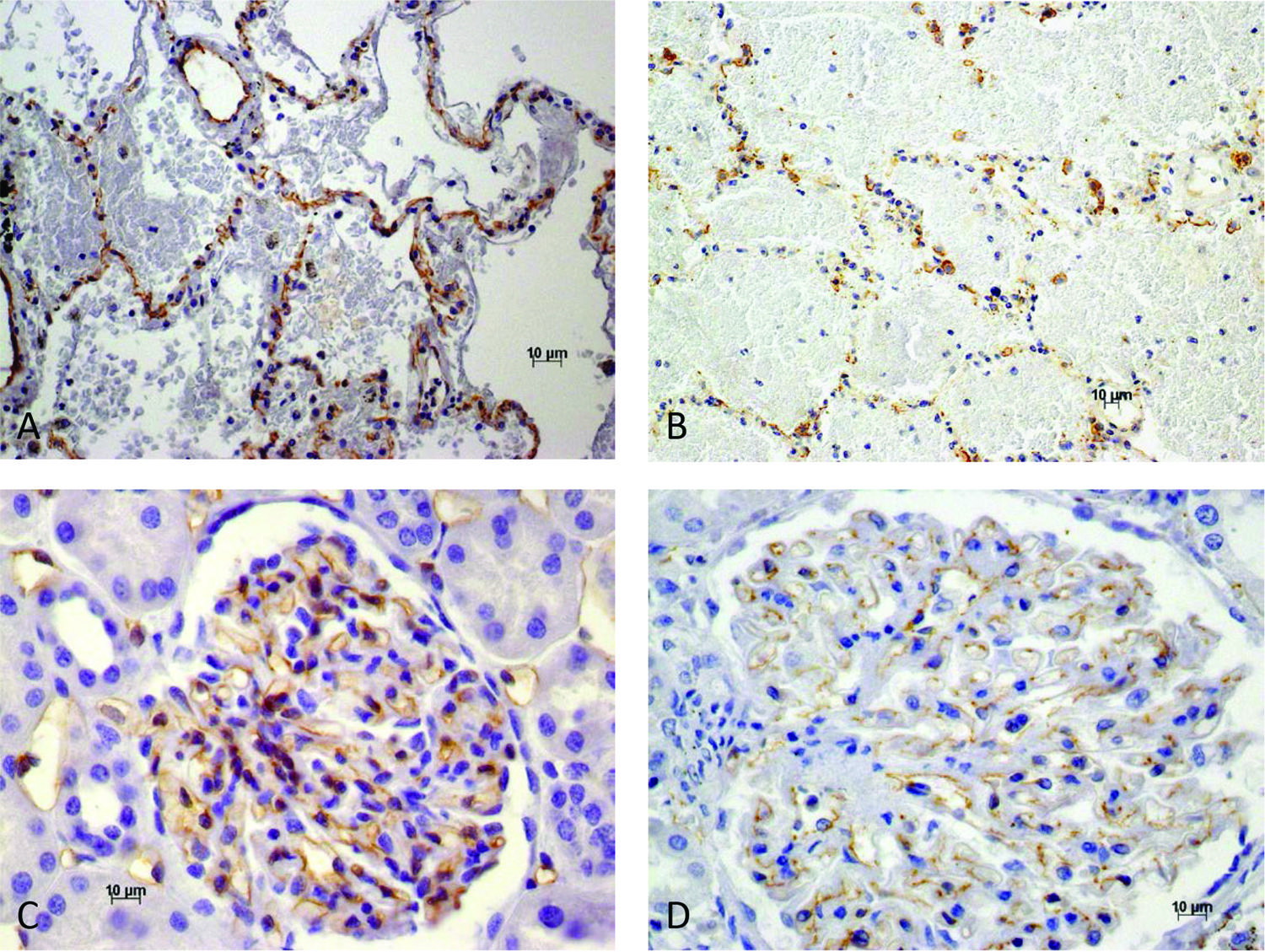
Our previous observations, using CD343030. De Brito T, Aiello VD, da Silva LF, Gonçalves da Silva AM, Ferreira da Silva WL, Castelli JB, et al. Human hemorrhagic pulmonary leptospirosis: pathological findings and pathophysiological correlations. PLoS One. 2013;8:e71743. and now VE-cadherin3131. Herwig MC, Müller KM, Müller AM. Endothelial VE-cadherin expression in human lungs. Pathol Res Pract. 2008;204:725-30. immunohistochemical detection in the pulmonary circulation of autopsied patients who manifested marked pulmonary hemorrhage, showed morphological alterations which, particularly in the case of VE-cadherin, can explain the increased vascular permeability seen in conditions affecting the adherens junction organization3131. Herwig MC, Müller KM, Müller AM. Endothelial VE-cadherin expression in human lungs. Pathol Res Pract. 2008;204:725-30.,3232. Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci. 2008;121:2115-22. As previously noted3030. De Brito T, Aiello VD, da Silva LF, Gonçalves da Silva AM, Ferreira da Silva WL, Castelli JB, et al. Human hemorrhagic pulmonary leptospirosis: pathological findings and pathophysiological correlations. PLoS One. 2013;8:e71743., the hemorrhage is due to a non-inflammatory vasculopathy. VE-cadherin immunohistochemical detection shows disruption of endothelial cell-cell junctions, cell retraction and the consequent opening of intercellular gaps, thereby explaining the marked increase in paracellular permeability that is responsible for both pulmonary edema and hemorrhage (Figures 1E and 1F and Figures 2A and 2B). Areas of capillary dilation are present and there is an irregular expression of CD34 in capillary walls by immunohistochemistry. As expected, the findings are in accordance with the marked clinical manifestations of acute pulmonary failure.
– Immunohistochemistry and histology analyses. A) VE-cadherin in lung in human leptospirosis. Small openings in the alveolar lining. Areas of intra-alveolar hemorrhage; B) Low view of VE-cadherin in human lung in leptospirosis. Intra-alveolar hemorrhage and diminished and/or absent staining of capillary walls; C) Normal expression of VE cadherin in normal human glomerular capillaries; D) VE-cadherin expression is diminished and/or absent in focal areas of glomerular capillaries in human leptospirosis
A recent study by Sato and Coburn3333. Sato H, Coburn J. Leptospira interrogans causes quantitative and morphological disturbances in adherens junctions and other biological groups of proteins in human endothelial cells. PLoS Negl Trop Dis. 2017;11: e0005830. showed that the most important finding seen in L. interrogans was the disruption of adherens junctions due to protein alterations. VE-cadherins, p120 catenin, alpha and beta catenins are specifically involved in the process. In contrast to the disturbance of the adherens junction, there were no changes in the tight junctional transmembrane, except for mislocalization of ZO-1. Moreover, infection by leptospires altered other host proteins of different classes.
However, VE-cadherin detected by immunohistochemistry is not the only component of the endothelial cells of the microcirculation involved in the altered vascular permeability seen in leptospirosis. The immunohistochemical detection of endothelial transmembrane glycoproteins by CD343434. Ordóñez NG. Immunohistochemical endothelial markers: a review. Adv Anat Pathol. 2012;19:281-95., which also plays a role in cell adhesion, showed a reduction of focal expression in the capillaries of the lung microcirculation. Gaps of different sizes were present, which were interpreted either as sections of twisted dilated capillaries or disrupted cell junctions3030. De Brito T, Aiello VD, da Silva LF, Gonçalves da Silva AM, Ferreira da Silva WL, Castelli JB, et al. Human hemorrhagic pulmonary leptospirosis: pathological findings and pathophysiological correlations. PLoS One. 2013;8:e71743.. Moreover, an increased reactive expression of vascular intercellular adhesion molecules and Toll-like receptor was found in the lungs of leptospirosis cases3535. Del Carlo Bernardi F, Ctenas B, da Silva LF, Nicodemo AC, Saldiva PH, Dolhnikoff M, et al. Immune receptors and adhesion molecules in human pulmonary leptospirosis. Hum Pathol. 2012;43:1601-10..
Recently, glycocalyx, another important component of the endothelium, was found to be damaged in a study of acute kidney injury in human leptospirosis3636. Libório AB, Braz MB, Seguro AC, Meneses GC, Neves FM, Pedrosa DC, et al. Endothelial glycocalyx damage is associated with leptospirosis acute kidney injury. Am J Trop Med Hyg. 2015;92:611-6.. Glycocalyx has antiadhesive and anticoagulant properties that are essential for the endothelium to maintain the barrier function. In this study, leptospirosis was found to be associated with higher levels of intercellular adhesion molecules, particularly ICAM-1 and syndecan-13636. Libório AB, Braz MB, Seguro AC, Meneses GC, Neves FM, Pedrosa DC, et al. Endothelial glycocalyx damage is associated with leptospirosis acute kidney injury. Am J Trop Med Hyg. 2015;92:611-6..
Taking the above data together, we suggest that the endothelial cell is injured in leptospirosis, and this includes not only the main important lesion at the adherens junction but also alterations of the cell membrane, leading to altered permeability.
The lung in leptospirosis showed a surprising amount of preservation of the immunohistochemical phenotype for cells of the alveolar lining, including those in areas of edema and hemorrhage3030. De Brito T, Aiello VD, da Silva LF, Gonçalves da Silva AM, Ferreira da Silva WL, Castelli JB, et al. Human hemorrhagic pulmonary leptospirosis: pathological findings and pathophysiological correlations. PLoS One. 2013;8:e71743.. Usually, rare cell necrosis and/or no definite cell necrosis was observed. Connexin 43 expression seen in the epithelial cells inside areas of edema and hemorrhage might be interpreted as evidence of cytoplasmic communication between less damaged cells. This finding suggests previous cell disarray, as observed in other organs such as the liver, and therefore strengthens the possibility of a predominant lesion of the cell adherens junction, thus following a common pattern of cell membrane lesion. In addition, the immunohistochemical aspects of VE-cadherin detection, which mediates the paracellular pathway of endothelial permeability in the pulmonary capillaries, is in accordance with a similar pathogenesis.
MUSCULAR LESIONS
Even though this broad approach to the pathogenesis of aspects of the disease does not apply to muscular lesions in leptospirosis, the most severely involved skeletal muscles are those of the calf, pectoral muscles may also be, although less frequently, damaged3737. Jeghers HJ, Houghton JB, Foley JA. Weil’s disease: report of a case with postmortem observations and review of recent literature. Arch Pathol. 1935;20:447-76.. As seen microscopically, the process selects isolated or small groups of muscle fibers, which may exhibit loss of striation, and round lumps of hyaline material, interpreted as isolated muscle fiber necrosis. A peripheral inflammatory infiltrate made up mostly of mononuclear cells was seen close to the damaged muscle fiber. Leptospira antigens were detected by immunohistochemistry, particularly in the macrophages but also occasionally inside the muscle as granular deposits3838. Uip DE, Amato Neto V, Duarte MS. Diagnóstico precoce da leptospirose por demonstração de antígenos através de exame imuno-histoquímico em músculo da panturrilha. Rev Inst Med Trop Sao Paulo. 1992;34:375-81..
Recently, it was also shown, in normal guinea pigs that Lp25, a surface protein of pathogenic leptospires, was partially responsible for hyperkalemic pre-renal acute kidney manifestations induced by rhabdomyolysis3939. Abreu PA, Seguro AC, Canale D, Silva AM, Matos LD, Gotti TB, et al. Lp25 membrane protein from pathogenic Leptospira spp. is associated with rhabdomyolysis and oliguric acute kidney injury in a guinea pig model of leptospirosis. PLoS Negl Trop Dis. 2017;11:e0005615.. Pathology demonstrated in this experiment, that there is no wide range of muscle fiber sizes nor small or large groups of atrophic or hypertrophic fibers. Internal sarcolemma nuclei were not detected in isolated muscle fibers. However, focal muscle lesions were present, ranging from hyaline cytoplasmic changes, which may progress to excessive hypercontraction of isolated fibers and necrosis prior to phagocytosis, or to intermediate damage characterized by staining changes of myofibrils, producing pale necrotic cells (“ghost” cells), which can be demonstrated by usual staining, but chiefly by the Gomori trichrome dye. Small vacuoles might be detected in muscle fibers near irregular areas of muscle necrosis, which are the result of these changes. Mild inflammatory infiltrates, made up mostly of macrophages, may be found around this group of cells. Immunohistochemistry with rabbit anti-Lp25 marked isolated or small groups of muscle fibers with antigen deposits delineating partially isolated myocyte membranes and occasionally spreading to the cytoplasm below. The inflammatory infiltrate, which is important in human muscular disease4040. Tidball JG. Inflammatory processes in muscle injury and repair. Am J Physiol Regul Integr Comp Physiol. 2005;288: R345-53., was usually discrete and was made up mostly of small groups of monocytes. These monocytes were present as focal isolated interstitial groups or as a part of small groups of isolated muscle fibers, frequently with antigenic linear deposits partially circumscribing muscle fibers or even inside their cytoplasms. Rarely, when the inflammatory infiltrate was more conspicuous, cytoplasmic antigenic granules were detected also in the mononuclear phagocytic cells.
The subsurface lipoprotein LipL32, which is abundant in pathogenic leptospires, was not initially considered important for the pathogenesis of the skeletal muscle lesions. However, LipL32 causes similar, but apparently less frequent, focal muscle lesions, characterized chiefly by small linear areas of necrosis. The focal isolated muscle lesions, as described after Lp25 administration, were less frequent when LipL32 was used. Immunohistochemistry with rabbit anti LipL32 was positive in isolated muscle fibers. Scarce mononuclear inflammatory interstitial reactions close to the damaged muscle were also present. In summary, both LipL32 and Lp25 can induce skeletal muscle damage, but lesions after Lp25 administration appear to be more diffuse and more accentuated than those seen with LipL32, in accordance with the pathophysiological findings described in the experiment3939. Abreu PA, Seguro AC, Canale D, Silva AM, Matos LD, Gotti TB, et al. Lp25 membrane protein from pathogenic Leptospira spp. is associated with rhabdomyolysis and oliguric acute kidney injury in a guinea pig model of leptospirosis. PLoS Negl Trop Dis. 2017;11:e0005615..
The human skeletal muscle disease showed more accentuated findings, but also as seen in the experimental design, Lp25 was demonstrated over the muscle cell membrane and in the underlying cytoplasm. The focal inflammatory infiltrate was more marked when compared to the experimental results, but again was made up mostly of mononuclear cells. Antigens were demonstrated not only in phagocytic cells but also in the muscle sarcoplasm3939. Abreu PA, Seguro AC, Canale D, Silva AM, Matos LD, Gotti TB, et al. Lp25 membrane protein from pathogenic Leptospira spp. is associated with rhabdomyolysis and oliguric acute kidney injury in a guinea pig model of leptospirosis. PLoS Negl Trop Dis. 2017;11:e0005615.. However, it is necessary to note that in this instance, we are dealing with patients with leptospirosis, and the immune serum can also recognize the microorganism antigens which, as previously demonstrated3838. Uip DE, Amato Neto V, Duarte MS. Diagnóstico precoce da leptospirose por demonstração de antígenos através de exame imuno-histoquímico em músculo da panturrilha. Rev Inst Med Trop Sao Paulo. 1992;34:375-81., may be present in the muscle.
LIVER IN LEPTOSPIROSIS
It is well known that the most severe form of leptospirosis, the Weil’s disease, is mainly characterized by hemorrhage, hepatic and renal manifestations33. Haake DA, Levett PN. Leptospirosis in humans. In: Adler B, editor. Leptospira and leptospirosis. Berlin: Springer-Verlag; 2015. p.65-97.. Jaundice is an important manifestation of hepatic dysfunction, but its mechanism in leptospirosis remains not completely elucidated. Serum biochemical studies have demonstrated that the concentrations of transaminases and to a lesser degree of alkaline phosphatase, are moderately increased in human leptospirosis. Jaundice is mainly due to increased conjugated bilirubin and disturbances of bile excretion with intrahepatic cholestasis, which appeared at the time to be its main cause.
Previous observations in patients recovering from leptospirosis, therefore out of the hemorrhagic phase of the disease, showed a non-specific hepatocellular damage affecting the sinusoidal pole, endoplasmic reticulum, mitochondria and the bile secretory apparatus2121. De Brito T, Machado MM, Montans SD, Hoshino S, Freymüller E. Liver biopsy in human leptospirosis: a light and electron microscopy study. Virchows Arch Pathol Anat Physiol Klin Med. 1967;342:61-9.,2323. Brito de T, Penna DO, Hoshino S, Pereira VG, Caldas AC, Rothstein W. Cholestasis in human leptospirosis: a clinical, histochemical, biochemical and electron microscopy study based on liver biopsies. Beitr Pathol Anat. 1970;140:345-61.. The sinusoidal pole showed altered microvilli, and the intercellular spaces were frequently widened with secondary microvilli and bile canaliculi dilated with microvilli partially or completely absent. Tight junctions were usually, but not always preserved. Golgi complexes might disclose dilated vesicles with an electron dense material inside. Hepatocytes showed glycogen depletion, microbodies and, in more severe cases, a predominance of the smooth endoplasmic reticulum. Mitochondrial alterations were present, correlating with a histochemical low demonstration of respiratory enzymes, probably related to the previous hemorrhagic phase of the disease. Remnants of leptospires, and probably bile pigment, were inside the widened intercellular spaces, between the hepatocytes. It is worth mentioning that, in guinea pigs, seemingly intact leptospires were seen by electron microscopy in the intercellular space between the hepatocytes in the experimental disease, sometimes close to the cell junctions1111. De Brito T, Freymüller E, Hoshino S, Penna DO. Pathology of the kidney and liver in the experimental leptospirosis of the guinea-pig. Alight and electron microscopy study. Virchows Arch Pathol Anat Physiol Klin Med. 1966;341:64-78..
Therefore, cholestasis in leptospirosis was interpreted, at the time, as the result of disturbances to the bile secretory apparatus, which comprises the smooth endoplasmic reticulum, Golgi apparatus and biliary capillaries. Unknown factors, among them a currently hypothetical circulating toxin (or toxins), along with cell membrane damage, were thought to explain the pathological findings. These pathological findings include the presence of possible biliary products in the widened intercellular spaces, which exhibited microvilli on their surfaces, suggesting an abnormal paracellular conjugated bile excretory route through the altered cell membrane2323. Brito de T, Penna DO, Hoshino S, Pereira VG, Caldas AC, Rothstein W. Cholestasis in human leptospirosis: a clinical, histochemical, biochemical and electron microscopy study based on liver biopsies. Beitr Pathol Anat. 1970;140:345-61..
Recent report from Miyhara et al.4141. Miyahara S, Saito M, Kanemaru T, Villanueva SY, Gloriani NG, Yoshida S. Destruction of the hepatocyte junction by intercellular invasion of Leptospira causes jaundice in a hamster model of Weil’s disease. Int J Exp Pathol. 2014;95:271-81. in a hamster model of experimental leptospirosis proposes the destruction of the hepatocytic intercellular junction directly by leptospire intercellular migration, as cause of jaundice in the disease. Using scanning electron microscopy together with freeze-cracking and cross-cutting methods for liver samples, three-dimensional images were obtained. Leptospires were found to migrate from the sinusoids to the biliary capillaries after disruption of the cellular junctions. Bile leaks from the biliary capillaries to the blood circulation during the induction of jaundice. This is a novel mechanism of jaundice caused by leptospiral infection, based on a previous electron microscopy study of the liver in a hamster experimental model of leptospirosis4242. Miller NG, Wilson RB. Electron microscopy of the liver of the hamster during acute and chronic leptospirosis. Am J Vet Res. 1966;27:1071-81..
The mechanism of jaundice proposed by Miyhara et al.4141. Miyahara S, Saito M, Kanemaru T, Villanueva SY, Gloriani NG, Yoshida S. Destruction of the hepatocyte junction by intercellular invasion of Leptospira causes jaundice in a hamster model of Weil’s disease. Int J Exp Pathol. 2014;95:271-81. is supported by the electron microscopy finding for leptospires and/or their remnants in the intercellular spaces of hepatocytes in humans2323. Brito de T, Penna DO, Hoshino S, Pereira VG, Caldas AC, Rothstein W. Cholestasis in human leptospirosis: a clinical, histochemical, biochemical and electron microscopy study based on liver biopsies. Beitr Pathol Anat. 1970;140:345-61., and particularly in guinea pigs experimental disease1111. De Brito T, Freymüller E, Hoshino S, Penna DO. Pathology of the kidney and liver in the experimental leptospirosis of the guinea-pig. Alight and electron microscopy study. Virchows Arch Pathol Anat Physiol Klin Med. 1966;341:64-78..
However, despite the electron microscopy findings showing that human and experimental leptospirosis is non-specific and differs only in degree from findings associated with viral hepatitis2323. Brito de T, Penna DO, Hoshino S, Pereira VG, Caldas AC, Rothstein W. Cholestasis in human leptospirosis: a clinical, histochemical, biochemical and electron microscopy study based on liver biopsies. Beitr Pathol Anat. 1970;140:345-61., they show the importance of altered hepatocyte transport and chiefly the excretion of bile in leptospirosis. Most likely, both mechanisms are at work in the pathogenesis of jaundice in leptospirosis, but the mechanism suggested by Miyhara et al.4141. Miyahara S, Saito M, Kanemaru T, Villanueva SY, Gloriani NG, Yoshida S. Destruction of the hepatocyte junction by intercellular invasion of Leptospira causes jaundice in a hamster model of Weil’s disease. Int J Exp Pathol. 2014;95:271-81. has strong experimental support and is in accordance with leptospirosis being a disease with important cell membrane damage.
A biopsy study of the liver in leptospirosis during recovery2121. De Brito T, Machado MM, Montans SD, Hoshino S, Freymüller E. Liver biopsy in human leptospirosis: a light and electron microscopy study. Virchows Arch Pathol Anat Physiol Klin Med. 1967;342:61-9. showed that the liver architecture is preserved but infrequent mitotic figures are present in hepatocytes, along with multinucleation and hepatic cells with acidophilic cytoplasm suggesting apoptosis, and even the presence of occasional Councilman-like corpuscles. Observations on the repair of the E-cadherin showed a robust proliferation of fibers and the presence of frequent binucleated hepatocytes with variations in size and shape. This shows the importance of a previous general hepatocyte injury in leptospirosis, which cannot be neglected when the pathogenesis of jaundice is discussed.
KIDNEY IN LEPTOSPIROSIS
The kidney is a common target organ in leptospirosis, and tubule-interstitial nephritis is the most common clinical and pathological manifestation. Clinically, non-oliguric acute kidney injury with hypokalemia, sodium and magnesium wasting frequently occurs in human leptospirosis. Immunohistochemical exams in autopsy material4343. Araujo ER, Seguro AC, Spichler A, Magaldi AJ, Volpini RA, De Brito T. Acute kidney injury in human leptospirosis: an immunohistochemical study with pathophysiological correlation. Virchows Arch. 2010;456:367-75. confirmed a primary lesion of the proximal convoluted tubules when a decrease of the endogenous sodium/hydrogen exchanger isoform 3 (NH3), aquaporin 1 and 2 and α-Na+ K+ ATPase are found. Sodium and water transport were particularly affected with increased distal potassium excretion and consequent hypokalemia and polyuria. Previous electron microscopy results also showed definite lesions, chiefly of the brush border of the convoluted proximal tubules, both in the human disease4444. De Brito T, Penna DO, Pereira VC, Hoshino S. Kidney biopsies in human leptospirosis: a biochemical and electron microscopy study. Virchows Arch Pathol Anat Physiol Klin Med. 1967;343:124-35. and in experimental infection of guinea pigs1111. De Brito T, Freymüller E, Hoshino S, Penna DO. Pathology of the kidney and liver in the experimental leptospirosis of the guinea-pig. Alight and electron microscopy study. Virchows Arch Pathol Anat Physiol Klin Med. 1966;341:64-78.. Glomeruli, both in humans and experimental models, showed few alterations, usually manifested by areas of foot processes fusion, focal swelling of the basal membrane and endothelial cells. VE-cadherin immunohistochemical expression in glomerular capillaries was diminished and even focally absent (Figures 2C and 2D). VE-cadherin is part of the slit diaphragm protein complex of the glomerular podocytes, which are composed of proteins common to most cell-cell junctions. The possible role of VE-cadherin for the functional renal filtration barrier in the podocyte is so far unknown4545. Patrakka J, Tryggvason K. Molecular make-up of the glomerular filtration barrier. Biochem Biophys Res Commun. 2010;396:164-9.. These glomerular alterations might be related to the proteinuria observed in a few leptospirosis patients4646. Mori M, Van Esbroeck M, Depoorter S, Decaluwe W, Vandecasteele SJ, Fretin D, et al. Outbreak of leptospirosis during a scout camp in the Luxembourg Belgian province, Belgium, summer 2012. Epidemiol Infect. 2015;143:1761-6.. Mesangial cell hypertrophy, also focally detected, is a non-specific reactive manifestation commonly seen in septicemias.
OTHER MANIFESTATIONS OF LEPTOSPIROSIS
Leptospirosis is a septicemic disease and manifestations such as leptomeningitis and myocarditis are expected during the disease. Myocarditis in leptospirosis was reported initially by Arean in 1957 in 61.5% of the hearts studied4747. Arean VM. Leptospiral myocarditis. Lab Invest. 1957;6:462-71.. Our data4848. De Brito T, Morais CF, Yasuda PH, Lancellotti CP, Hoshino-Shimizu S, Yamashiro E, et al. Cardiovascular involvement in human and experimental leptospirosis: pathologic findings and immunohistochemical detection of leptospiral antigen. Ann Trop Med Parasitol. 1987;81:207-14. found myocarditis in 50% of the hearts, a figure close to the one previously reported. The pathological findings were essentially the same as the ones reported by Arean4747. Arean VM. Leptospiral myocarditis. Lab Invest. 1957;6:462-71.,4949. Arean VM. The pathologic anatomy and pathogenesis of fatal human leptospirosis (Weil’s disease). Am J Pathol. 1962;40:393-423., except that we detected the involvement of the conductive system of the heart in our cases, which might be clinically relevant.
Vascular injury is present in the heart in leptospirosis and usually characterized by segmental lesions in on the walls of the coronary artery and their branches. Deposits of leptospiral antigens are usually revealed across groups of endothelial cells. Small intramyocardial branches might present subintimal edema and a perivascular non-specific inflammatory infiltrate, usually consisting of mononuclear cells. Occasionally, a peculiar mononuclear focal response of Anitchkow-like cells, resembling Aschoff bodies of rheumatic fever, was described by Arean4747. Arean VM. Leptospiral myocarditis. Lab Invest. 1957;6:462-71. close to the medium-sized coronary branches. They were also seen by us both in humans and in experimental animals4848. De Brito T, Morais CF, Yasuda PH, Lancellotti CP, Hoshino-Shimizu S, Yamashiro E, et al. Cardiovascular involvement in human and experimental leptospirosis: pathologic findings and immunohistochemical detection of leptospiral antigen. Ann Trop Med Parasitol. 1987;81:207-14..
Another interesting vascular lesion seen in leptospirosis is aortitis, found in 57.8% of cases4848. De Brito T, Morais CF, Yasuda PH, Lancellotti CP, Hoshino-Shimizu S, Yamashiro E, et al. Cardiovascular involvement in human and experimental leptospirosis: pathologic findings and immunohistochemical detection of leptospiral antigen. Ann Trop Med Parasitol. 1987;81:207-14.. First described by Arean4949. Arean VM. The pathologic anatomy and pathogenesis of fatal human leptospirosis (Weil’s disease). Am J Pathol. 1962;40:393-423., it resembles syphilitic aortitis but, unlike syphilis, the vasa vasorum fail to show obstructive vasculitis and there are no changes in the aortic media other than the extension of adventitial inflammation.
Therefore, we suggest that the most common and important lesion in leptospirosis is the vascular damage, which affects the endothelial cell as a whole. Moreover, the altered adhesion between endothelial cells is quite important. It is mediated chiefly by VE-cadherin but also by CD34 and other endothelial cell components, which permits endothelial permeability with massive loss of blood. Immunohistochemical studies, when demonstrating alterations to the glycoproteins of the endothelial cells membrane, add transcellular permeability to the hemorrhagic manifestations of the disease.
Leptospires and their antigens are frequently in close contact with the cell membrane and it has been demonstrated that the leptospire can penetrate the cell. Their antigens can be found in the cytoplasm, and rarely, inside the nucleus55. De Brito T, Menezes LF, Lima DM, Lourenço S, Silva AM, Alves VA. Immunohistochemical and in situ hybridization studies of the liver and kidney in human leptospirosis. Virchows Arch. 2006;448:576-83..
Recently, a leptospiral lipase was characterized (Abreu et al., unpublished results) and its immunohistochemistry on hepatocytes from human autopsies showed a definite presence of the enzyme in the cell membrane in leptospirosis. Lipase is probably secreted by leptospires during cell adhesion and is probably important in the pathogenesis of cellular disruption, as seen chiefly in the liver and possibly in other organs during leptospiral infection.
Deposits of leptospiral antigen (or antigens) on the host cell membrane and the loss and/or alteration of cadherin expression might corroborate suggestions that cell membrane damage is the primary lesion in leptospirosis, which is mediated by unknown factors, among them glycol and/or lipoproteins of the leptospiral surface. Vinh et al.5050. Vinh T, Adler B, Faine S. Glycolipoprotein cytotoxin from Leptospira interrogans serovar copenhageni. J Gen Microbiol. 1986;132:111-23. suggested that the adherence of leptospires and/or their products might lead the intercalation of the microorganism’s fatty acids into the cell membrane. We may, however, provide an alternative to Vinh’s speculation and consider that the major carbon and energy sources of leptospires are long-chain fatty acids derived chiefly from beta-oxidation. Therefore, the adherence to the cell membrane is a mechanism to fulfill the microorganism’s energy requirements, which would be provided by the cell membrane fatty acids55. De Brito T, Menezes LF, Lima DM, Lourenço S, Silva AM, Alves VA. Immunohistochemical and in situ hybridization studies of the liver and kidney in human leptospirosis. Virchows Arch. 2006;448:576-83..
Therefore, the general pattern of altered cell adhesion with partial or total disappearance of cadherins is also present in microcirculatory vessels.
In the context of a definite lesion of the cell membrane, the pathogenesis of the muscular damage, as seen chiefly in the experimental work, suggests a toxin-like action of a protein from the subsurface of the leptospiral membrane as an important factor in the mechanism of the disease.
CONCLUSIONS
In summary, the primary lesion in leptospirosis seems to be the cell membrane damage mediated by unknown factors, possibly leptospiral proteins and/or toxic cellular components. Injuries to the cell membrane may result in severe consequences to the host, such as loss of vascular integrity, ischemia and necrosis, leading to significant organ dysfunctions. Understanding the molecular mechanisms involved in the pathogenesis and pathology of leptospirosis is of utmost importance for prognostic and therapeutic applications.
REFERENCES
-
1Costa F, Hagan JE, Calcagno J, Kane M, Torgerson P, Martinez-Silveira MS, et al. Global morbidity and mortality of leptospirosis: a systematic review. PLoS Negl Trop Dis. 2015;9:e0003898.
-
2Bharti AR, Nally JE, Ricaldi JN, Matthias MA, Diaz MM, Lovett MA, et al. Leptospirosis: a zoonotic disease of global importance. Lancet Infect Dis. 2003;3:757-71.
-
3Haake DA, Levett PN. Leptospirosis in humans. In: Adler B, editor. Leptospira and leptospirosis. Berlin: Springer-Verlag; 2015. p.65-97.
-
4Plank R, Dean D. Overview of the epidemiology, microbiology, and pathogenesis of Leptospira spp. in humans. Microbes Infect. 2000;2:1265-76.
-
5De Brito T, Menezes LF, Lima DM, Lourenço S, Silva AM, Alves VA. Immunohistochemical and in situ hybridization studies of the liver and kidney in human leptospirosis. Virchows Arch. 2006;448:576-83.
-
6Del Carlo Bernardi F, Ctenas B, da Silva LF, Nicodemo AC, Saldiva PH, Dolhnikoff M, et al. Immune receptors and adhesion molecules in human pulmonary leptospirosis. Hum Pathol. 2012;43:1601-10.
-
7Goris MG, Kikken V, Straetemans M, Alba S, Goeijenbier M, van Gorp EC, et al. Towards the burden of human leptospirosis: duration of acute illness and occurrence of post-leptospirosis symptoms of patients in the Netherlands. PLoS One. 2013;8:e76549.
-
8Wagenaar JF, Goris MG, Partiningrum DL, Isbandrio B, Hartskeerl RA, Brandjes DP, et al. Coagulation disorders in patients with severe leptospirosis are associated with severe bleeding and mortality. Trop Med Int Health. 2010;15:152-9.
-
9Levett PN. Leptospirosis. Clin Microbiol Rev. 2001;14:296-326.
-
10Adler B, de la Peña-Moctezuma A. Leptospira and leptospirosis. Vet Microbiol. 2010;140:287-96.
-
11De Brito T, Freymüller E, Hoshino S, Penna DO. Pathology of the kidney and liver in the experimental leptospirosis of the guinea-pig. Alight and electron microscopy study. Virchows Arch Pathol Anat Physiol Klin Med. 1966;341:64-78.
-
12Ganoza CA, Matthias MA, Saito M, Cespedes M, Gotuzzo E, Vinetz JM. Asymptomatic renal colonization of humans in the Peruvian Amazon by Leptospira. PLoS Negl Trop Dis. 2010;4:e612.
-
13RamachandraRao SP, Matthias MA, Kokoy-Mondragon C, Aghania E, Park C, Kong C, et al. Proteomic analysis of urine exosomes reveals renal tubule response to leptospiral colonization in experimentally infected rats. PLoS Negl Trop Dis. 2015;9:e0003640.
-
14Edgar JR. Q&A: what are exosomes, exactly? BMC Biol. 2016;14:46.
-
15Agampodi SB, Matthias MA, Moreno AC, Vinetz JM. Utility of quantitative polymerase chain reaction in leptospirosis diagnosis: association of level of leptospiremia and clinical manifestations in Sri Lanka. Clin Infect Dis. 2012;54:1249-55.
-
16De Brito T, Böhm GM, Yasuda PH. Vascular damage in acute experimental leptospirosis of the guinea-pig. J Pathol. 1979;128:177-82.
-
17Beitzke H. Ueber die pathologische Anatomie des ansteckenden Gelbsucht (Weilsche Krankheit). Berliner Klin Wochenschr. 1916;53:188-91
-
18Pick L. Zur pathologischen Anatomie des infektiösen Icterus. Klin Wochenschr. 1917;54:451-5.
-
19Alves VA, Gayotto LC, De Brito T, Santos RT, Wakamatsu A, Vianna MR, et al. Leptospiral antigens in the liver of experimentally infected guinea pig and their relation to the morphogenesis of liver damage. Exp Toxicol Pathol. 1992;44:425-34.
-
20De Brito T, Prado MJ, Negreiros VA, Nicastri AL, Sakata EE, Yasuda PH, et al. Detection of leptospiral antigen (L. interrogans serovar Copenhageni serogroup Icterohaemorrhagiae) by immunoelectron microscopy in the liver and kidney of experimentally infected guinea-pigs. Int J Exp Pathol. 1992;73:633-42.
-
21De Brito T, Machado MM, Montans SD, Hoshino S, Freymüller E. Liver biopsy in human leptospirosis: a light and electron microscopy study. Virchows Arch Pathol Anat Physiol Klin Med. 1967;342:61-9.
-
22Alves VA, Vianna MR, Yasuda PH, De Brito T. Detection of leptospiral antigen in the human liver and kidney using an immunoperoxidase staining procedure. J Pathol. 1987;151:125-31.
-
23Brito de T, Penna DO, Hoshino S, Pereira VG, Caldas AC, Rothstein W. Cholestasis in human leptospirosis: a clinical, histochemical, biochemical and electron microscopy study based on liver biopsies. Beitr Pathol Anat. 1970;140:345-61.
-
24Kassegne K, Hu W, Ojcius DM, Sun D, Ge Y, Zhao J, et al. Identification of collagenase as a critical virulence factor for invasiveness and transmission of pathogenic Leptospira species. J Infect Dis. 2014;209:1105-15.
-
25Amamura TA, Fraga TR, Vasconcellos SA, Barbosa AS, Isaac L. Pathogenic leptospira secreted proteases target the membrane attack complex: a potential role for thermolysin in complement inhibition. Front Microbiol. 2017;8:958
-
26De Brito T, Menezes LF, Lima DM, Lourenço S, Silva AM, Alves VA. Immunohistochemical and in situ hybridization studies of the liver and kidney in human leptospirosis. Virchows Arch. 2006;448:576-83.
-
27Evangelista K, Franco R, Schwab A, Coburn J. Leptospira interrogans binds to cadherins. PLoS Negl Trop Dis. 2014;8:e2672
-
28Evangelista KV, Hahn B, Wunder EA Jr, Ko AI, Haake DA, Coburn J. Identification of cell-binding adhesins of Leptospira interrogans. PLoS Negl Trop Dis. 2014;8:e3215.
-
29Martinez-Lopez DG, Fahey M, Coburn J. Responses of human endothelial cells to pathogenic and non-pathogenic Leptospira species. PLoS Negl Trop Dis. 2010;4:e918.
-
30De Brito T, Aiello VD, da Silva LF, Gonçalves da Silva AM, Ferreira da Silva WL, Castelli JB, et al. Human hemorrhagic pulmonary leptospirosis: pathological findings and pathophysiological correlations. PLoS One. 2013;8:e71743.
-
31Herwig MC, Müller KM, Müller AM. Endothelial VE-cadherin expression in human lungs. Pathol Res Pract. 2008;204:725-30.
-
32Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci. 2008;121:2115-22
-
33Sato H, Coburn J. Leptospira interrogans causes quantitative and morphological disturbances in adherens junctions and other biological groups of proteins in human endothelial cells. PLoS Negl Trop Dis. 2017;11: e0005830.
-
34Ordóñez NG. Immunohistochemical endothelial markers: a review. Adv Anat Pathol. 2012;19:281-95.
-
35Del Carlo Bernardi F, Ctenas B, da Silva LF, Nicodemo AC, Saldiva PH, Dolhnikoff M, et al. Immune receptors and adhesion molecules in human pulmonary leptospirosis. Hum Pathol. 2012;43:1601-10.
-
36Libório AB, Braz MB, Seguro AC, Meneses GC, Neves FM, Pedrosa DC, et al. Endothelial glycocalyx damage is associated with leptospirosis acute kidney injury. Am J Trop Med Hyg. 2015;92:611-6.
-
37Jeghers HJ, Houghton JB, Foley JA. Weil’s disease: report of a case with postmortem observations and review of recent literature. Arch Pathol. 1935;20:447-76.
-
38Uip DE, Amato Neto V, Duarte MS. Diagnóstico precoce da leptospirose por demonstração de antígenos através de exame imuno-histoquímico em músculo da panturrilha. Rev Inst Med Trop Sao Paulo. 1992;34:375-81.
-
39Abreu PA, Seguro AC, Canale D, Silva AM, Matos LD, Gotti TB, et al. Lp25 membrane protein from pathogenic Leptospira spp. is associated with rhabdomyolysis and oliguric acute kidney injury in a guinea pig model of leptospirosis. PLoS Negl Trop Dis. 2017;11:e0005615.
-
40Tidball JG. Inflammatory processes in muscle injury and repair. Am J Physiol Regul Integr Comp Physiol. 2005;288: R345-53.
-
41Miyahara S, Saito M, Kanemaru T, Villanueva SY, Gloriani NG, Yoshida S. Destruction of the hepatocyte junction by intercellular invasion of Leptospira causes jaundice in a hamster model of Weil’s disease. Int J Exp Pathol. 2014;95:271-81.
-
42Miller NG, Wilson RB. Electron microscopy of the liver of the hamster during acute and chronic leptospirosis. Am J Vet Res. 1966;27:1071-81.
-
43Araujo ER, Seguro AC, Spichler A, Magaldi AJ, Volpini RA, De Brito T. Acute kidney injury in human leptospirosis: an immunohistochemical study with pathophysiological correlation. Virchows Arch. 2010;456:367-75.
-
44De Brito T, Penna DO, Pereira VC, Hoshino S. Kidney biopsies in human leptospirosis: a biochemical and electron microscopy study. Virchows Arch Pathol Anat Physiol Klin Med. 1967;343:124-35.
-
45Patrakka J, Tryggvason K. Molecular make-up of the glomerular filtration barrier. Biochem Biophys Res Commun. 2010;396:164-9.
-
46Mori M, Van Esbroeck M, Depoorter S, Decaluwe W, Vandecasteele SJ, Fretin D, et al. Outbreak of leptospirosis during a scout camp in the Luxembourg Belgian province, Belgium, summer 2012. Epidemiol Infect. 2015;143:1761-6.
-
47Arean VM. Leptospiral myocarditis. Lab Invest. 1957;6:462-71.
-
48De Brito T, Morais CF, Yasuda PH, Lancellotti CP, Hoshino-Shimizu S, Yamashiro E, et al. Cardiovascular involvement in human and experimental leptospirosis: pathologic findings and immunohistochemical detection of leptospiral antigen. Ann Trop Med Parasitol. 1987;81:207-14.
-
49Arean VM. The pathologic anatomy and pathogenesis of fatal human leptospirosis (Weil’s disease). Am J Pathol. 1962;40:393-423.
-
50Vinh T, Adler B, Faine S. Glycolipoprotein cytotoxin from Leptospira interrogans serovar copenhageni. J Gen Microbiol. 1986;132:111-23.
-
ERRATUM
Page 5, 1st column, line 49, where it reads:Recently, it was also shown, in normal guinea pigs that Lp25, a surface protein of pathogenic leptospires, was partially responsible for hyperkalemic pre-renal acute kidney manifestations induced by rhabdomyolysis.Should be read:Recently, it was also shown, in normal guinea pigs that Lp25, a surface protein of pathogenic leptospires, was partially responsible for hyperkalemic and oliguric acute kidney manifestations induced by rhabdomyolysis.On page 7, 1st column, line 39, where it reads:Immunohistochemical exams in autopsy material confirmed a primary lesion of the proximal convoluted tubules when a decrease of the endogenous sodium/hydrogen exchanger isoform 3 (NH3), aquaporin 1 and 2 and α-Na+ K+ ATPase are found.Should be read:Immunohistochemical exams in autopsy material confirmed a primary lesion of the proximal convoluted tubules when a decrease of the endogenous sodium/hydrogen exchanger isoform 3 (NH3), aquaporin 1 and α-Na+ K+ ATPase are found.
Publication Dates
-
Publication in this collection
28 May 2018 -
Date of issue
2018
History
-
Received
4 Jan 2018 -
Accepted
23 Apr 2018