Abstract
INTRODUCTION:
Pulmonary arterial hypertension (PAH) is a serious pulmonary circulation disease caused by several etiologies, including schistosomiasis. The present study retrospectively evaluated the clinical and hemodynamic characteristics of patients with schistosomal PAH (PAH-Sch) compared to those of non-Sch PAH patients (non-Sch PAH).
METHODS:
Patients treated at the Pronto-Socorro Cardiológico de Pernambuco and diagnosed by right cardiac catheterization were divided into PAH-Sch and non-Sch PAH groups. Their socio-demographic and clinical characteristics, N-terminal-pro B-type natriuretic peptide (NT-proBNP), and echocardiography and hemodynamic parameters were retrospectively reviewed.
RESULTS:
Among the included 98 patients (mean age, 45 ± 14 years; 68 women [69.4%]), we found 56 PAH-Sch and 42 non-Sch PAH. The age distribution was heterogeneous in the PAH-Sch group, with patients predominantly ranging from 50-59 (p <0.004). Dyspnea was the most common symptom, reported by 92 patients (93.8%), and commonly present for over two years prior to diagnosis. Clinical symptoms were similar in both groups, with no differences in functional class, pulmonary artery systolic pressure (p = 0.102), 6-minute walk test score (p = 0.234), NT-proBNP serum levels (p = 0.081), or hemodynamic parameters.
CONCLUSIONS:
Patients with PAH-Sch present clinical, laboratory, and hemodynamic profiles similar to those with PAH resulting from other etiologies of poor prognosis. PAH is an important manifestation of schistosomiasis in endemic regions that is often diagnosed late.
Keywords:
Pulmonary hypertension; Schistosomiasis; Cardiopulmonary disease; Heart failure; Right ventricle
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a serious disease of the pulmonary circulation characterized by changes in the vascular wall, remodeling, vasoconstriction, and in situ thrombosis, with lesions primarily located in the pre-capillary segments of the pulmonary vasculature11. Rubin LJ. Primary pulmonary hypertension. N Engl J Med. 1997;336(2):111-7. that can lead to a progressive increase in pulmonary vascular resistance (PVR), right ventricular failure, and early death. Hemodynamically, this syndrome is defined by a mean pulmonary artery pressure (MPAP) ≥25 mmHg and a PVR >3 Wood units with pulmonary capillary pressure ≤15 mmHg22. Badesch DB, Champion HC, Gomez SMA, Hoeper MM, Loyd JE, Manes A, et al. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(1):S55-S66..
Infection caused by Schistosoma mansoni currently affects approximately 207 million people in 75 tropical and subtropical countries, severely in 20 million, and approximately 779 million people are at risk of contracting this disease33. Steinmann P, Keiser J, Bos R, Tanner M, Utzinger J. Schistosomiasis and water resources development: systematic review, meta-analysis, and estimates of people at risk. Lancet Infect Dis. 2006;6(7):411-25.. The prevalence of PAH associated with schistosomiasis (PAH-Sch) in patients with hepatosplenic schistosomiasis with hemodynamic confirmation ranges from 7.7-10.7%44. Lapa M, Dias B, Jardim C, Fernandes CJC, Dourado PMM, Figueiredo M, et al. Cardiopulmonary manifestations of hepatosplenic schistosomiasis. Circulation 2009;119(11):1518-23.,55. Ferreira RCS, Domingues ALC, Bandeira AP, Markman Filho B, Albuquerque Filho ES, Correia ACC, et al. Prevalence of pulmonary hypertension in patients with schistosomal liver fibrosis. Ann Trop Med Parasitol. 2009;103(2):129-43.. According to current PAH classification66. Simonneau G, Gatzoulis MA, Adatia I, Celemajer D, Denton C, Ghofrani A, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol . 2013;62(25 Suppl):D34-41., the schistosomiasis etiology is considered a part of Group 1.
The diagnosis of PAH has changed dramatically in recent years, as has its treatment, but early clinical recognition remains a challenge. The symptoms are often non-specific (i.e., cough, dyspnea, syncope, fatigue, and precordial pain) and are associated with significant morbidity and mortality, especially when identified in more advanced disease stages77. Engels D, Chitsulo L, Montresor A, Savioli L. The global epidemiological situation of schistosomiasis and new approaches to control and research. Acta Trop. 2002;82(2):139-46.,88. Appelbaum L, Yigla M, Bendayan D, Reichart N, Fink G, Priel I, et al. Primary pulmonary hypertension in Israel: a national survey. Chest. 2001 Jun;119(6):1801-6..
Patients with PAH form a heterogeneous group, and it is necessary to distinguish the characteristics of PAH caused by schistosomiasis from other etiologies (Figure 1). Although there are some data regarding PAH-Sch, they primarily concern populations outside the known endemic zone. It remains unclear whether such findings can be extrapolated to populations living in endemic areas and manifesting the disease. Recent studies have indicated a possible improvement in survival using current therapies, alone or in combination, even in severe cases of PAH-Sch observed in endemic and non-endemic regions99. Roncal CGP, Mendes AA, Cartaxo MT, Oliveira AS, Neto LV, Filho DSC, et al. Shistosomiaiss-Associated Pulmonary Arterial Hypertension: Survival in Endemic Area in Brazil. J Am Coll Cardiol . 2014;63(12):A1494.,1010. Fernandes CJCS, Dias BA, Jardim CVP, Hovnanian A, Hoette S, Morinaga KL, et al. The Role of Target Therapies in Schistosomiasis-Associated Pulmonary Arterial Hypertension. Chest. 2012;141(4):923-928.. In the present study, we retrospectively assessed the clinical and hemodynamic characteristics of PAH-Sch carriers compared to the non-schistosomal PAH group (non-Sch PAH), considering the time period between symptoms and diagnosis in an endemic region of schistosomiasis in Northeastern Brazil.

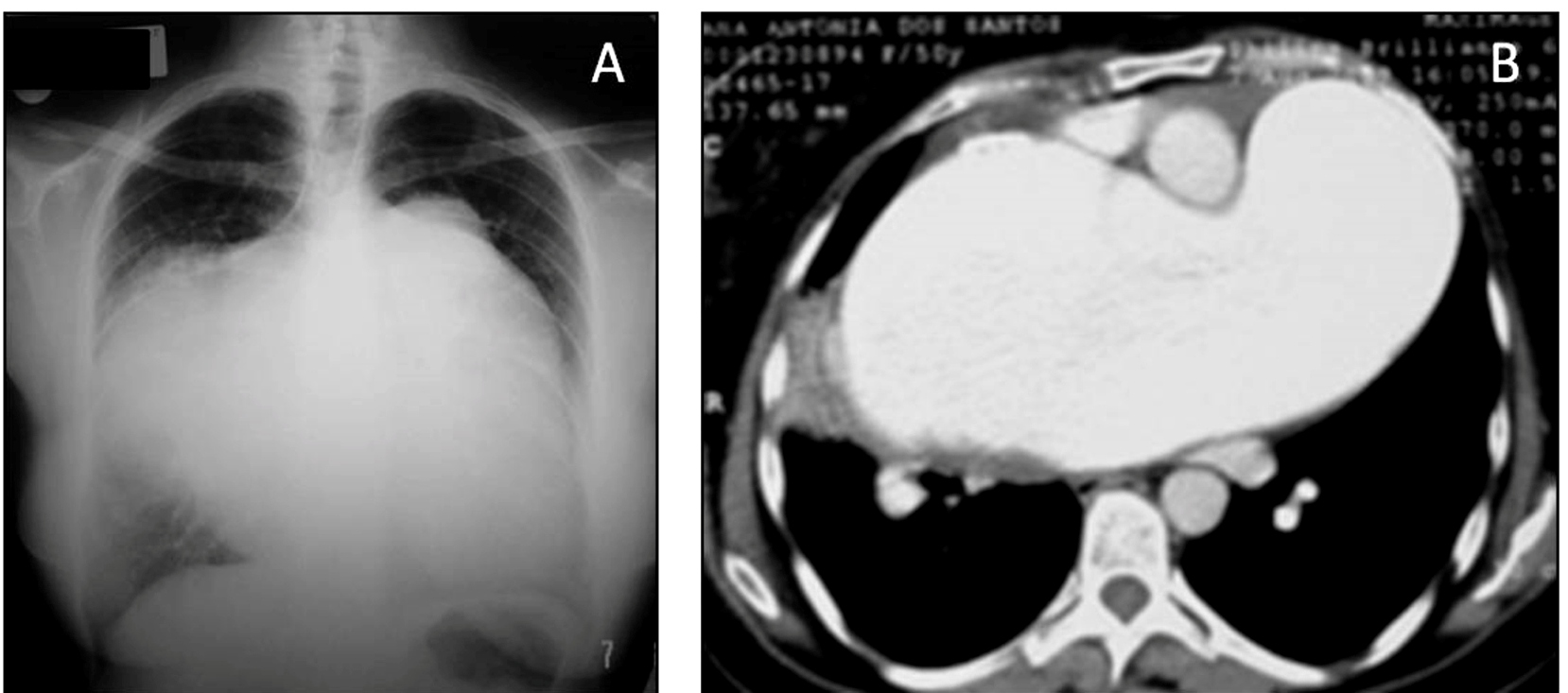
Chest X-ray (A) and computed tomography scan (B) of a patient with schistosomal pulmonary arterial hypertension and pulmonary artery aneurysm (“pulmonary artery ventricularization”).
METHODS
Patients enrolled in the Pulmonary Hypertension outpatient of the Pronto-Socorro Cardiológico de Pernambuco (PROCAPE/University of Pernambuco) from January 2001 to August 2009 were selected for the current study. We retrospectively reviewed patient charts and extracted the demographic, clinical, laboratory, echocardiographic, 6-minute walk test (6MWT), and hemodynamic variables. The diagnosis of PAH was established when the MPAP was >25 mmHg, pulmonary capillary pressure <15 mmHg, and PVR >3U Wood, as assessed by right cardiac catheterization22. Badesch DB, Champion HC, Gomez SMA, Hoeper MM, Loyd JE, Manes A, et al. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(1):S55-S66. with a nitric oxide vasoreactivity test. All included catheterizations were from the moment of diagnosis. Patients were classified according to guidelines proposed at the World Symposium on Pulmonary Hypertension held in Nice in 2013. The diagnosis of schistosomiasis was based on abdominal ultrasonography with the presence of periportal fibrosis associated with the origin (area endemic for schistosomiasis), which may or may not be associated with previous treatment or positive screening for Schistosoma eggs by routine methods. Patients with alcohol-related hepatic parenchymal disease, metabolic diseases, hepatitis C or B, as well as those with chronic pulmonary diseases, chronic pulmonary thromboembolism, and patients with evidence of pulmonary venous capillary hypertension (capillary pressure >15 mmHg) at the time of diagnosis, were excluded. The 6MWT was performed in all patients according to the American Thoracic Society protocol1111. American Thoracic Society. ATS statement: guidelines for the six-minute walk test. Am J Resp Crit Care Med. 2002;166(1):111-7.. Pulmonary arterial hypertension severity was assessed using the World Health Organization functional class (FC) adapted from the New York Heart Association1212. Hoper M, Ouzir R, Peacock A, Tapson VF, Haworth SG, Frost AE. End-points and clinical trial designs in pulmonary arterial hypertension: clinical and regulatory perspectives. J Am Coll Cardiol . 2004;43(12 Suppl S):48S-55S. . Laboratory tests were performed according to routine PAH protocols, as was the serum N-terminal-pro B-type natriuretic peptide (NT-proBNP) determination. We use transthoracic echocardiogram to estimate the pulmonary artery systolic pressure (PASP) by calculating the pressure gradient between the right ventricle and the atrium and applying the modified Bernoulli equation. A concomitant assessment of left ventricular function was performed to exclude the diagnosis of left ventricular failure1313. McQuillan BM, Picard HM, Leavitt M, Weyman AE. Clinical Correlates and Reference Intervals for Pulmonary Artery Systolic Pressure Among Echocardiographically Normal Subjects. Circulation. 2001;104(23):2797-802.. This study was approved by the Human Ethics and Research Committee of the University of Pernambuco (CAAE 0116.0.106.000-09).
Statistical analysis
Collected data were stored in an Excel database. Data were then exported to SPSS software version 19.0. To compare schistosomiasis and other etiologies (idiopathic, collagen, congenital, and human immunodeficiency virus [HIV]), the general characteristics, clinical complaints, physical characteristics, and hemodynamic variables were compared using the chi-square test or Fisher’s exact test (when necessary), in the case of categorical variables, and are presented as absolute (N) and relative (%) frequencies. For continuous variables, to determine whether the groups studied were homogeneous, the Student’s t-test was applied, and these variables are shown as absolute frequency (N) as well as mean ± standard deviation. A significance level of 5% was considered for all analyses.
RESULTS
The study included 98 patients (mean age 45 ± 14 years) registered at the PROCAPE/UPE reference center with a diagnosis of PAH between 2001 and 2009 (Table 1). The majority were women (69.4%; n = 68), and the age variation was not statistically significant. The age distribution analyses indicated that the PAH-Sch group predominantly included patients in the fifth decade of life (Table 1).
The most prevalent etiology was schistosomiasis in 56 patients (57.1%), followed by idiopathic in 19 (19.4%), collagen in seven (7.1%), congenital in ten (10.2%), and HIV in six (6.1%). Dyspnea was the most common symptom, reported by 93.8% (n = 92) of the study patients, and had been present for over two years in 43.3% of the study group. Table 2 presents descriptive statistics of the patient symptoms and the comparison between the patients with or without schistosomiasis. From physical examination, the most frequent finding was the presence of loud P2 (92.4%). Four patients (4.1%) were in FC I, 25 (25.5%) in FC II, 32 (32.7%) in FC III, and 37 (37.8%) in FC IV. There were no statistically significant differences between the PAH-Sch and non-Sch PAH groups (Table 1).
The average distance walked was 228.58 m (desvio padrão ± 125.38 m), and the median distance walked was 246.5 m. The analysis of variance test result indicated that that the mean 6MWT results varied according to the FC (p <0.001). At the time of diagnosis , the MPAP, measured by the hemodynamic study, and the PASP displayed a positive and significant correlation (r = 0.611, p <0.001).
The NT-proBNP results of FCs I and II compared to FCs III and IV in the general group presented a statistically significant difference (p = 0.001). These data indicated that natriuretic peptide values tended to be lower in FC I or II than in FC III or IV. There was no significant difference in serum NT-proBNP levels between patients in the PAH-Sch and non-Sch PAH groups (p = 0.081). The right cardiac catheterization was performed at the time of diagnosis in all 98 patients, and there was no significant difference between the means of hemodynamic parameters in PAH-Sch and non-Sch PAH groups (Table 1).
DISCUSSION
This is the first study comparing the clinical and hemodynamic characteristics of patients with different forms of PAH in an area endemic for schistosomiasis. Overall, the characteristics of patients with PAH-Sch were similar to those of patients with PAH resulting from other etiologies.
Among the 98 included patients with PAH, the prevalence of PAH-Sch was 57.1%, unlike other studies developed in a non-endemic region1414. Lapa MS, Ferreira MV, Jardim C, Martins BC, Arakaki JS, Rogerio S, et al. Características clínicas dos pacientes com hipertensão pulmonar em dois centros de referência. Rer Assoc Med Bras. 2006;52(3):139-43. that showed a prevalence of 30%. This difference can be attributed to the fact that the referral center in the current study is located in a region where disease control remains unsatisfactory, with high morbidity/mortality rates, increasing social and economic costs to the region, and worsening patient quality of life1515. Coura JR, Amaral RS. Epidemiological and Control Aspects of Schistosomiasis in Brazilian Endemic Areas. Mem Inst Oswaldo Cruz. 2004;99(5 Suppl 1):13-9.. In a United States registry of 578 patients, the prevalence of idiopathic PAH (IPAH) and connective tissue diseases (CTD) was 44% and 30%, respectively; in the French registry of 674 patients1717. Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habid G, Gressin V, et al. Pulmonary Arterial Hypertension in France: Results from a National Registry. Am J Respir Crit Care Med. 2006;173(9):1023-30., the prevalence of IPAH and CTD was 39% and 15%, respectively. However, in the present study, cases of IPAH and CTD comprised 19.4% and 10.2%, respectively, unlike other studies, which can be attributed to lack of recognition of the disease. In the French study1717. Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habid G, Gressin V, et al. Pulmonary Arterial Hypertension in France: Results from a National Registry. Am J Respir Crit Care Med. 2006;173(9):1023-30., the presence of PAH resulting from HIV was 6.2% versus 1.9% as shown by the REVEAL1818. Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey ChS, et al. Predicting Survival in Pulmonary Arterial Hypertension. Circulation. 2010;122(2):164-72. study. Six cases of HIV (6.1%) were identified in this study, although we believe that they are still underdiagnosed but similar to those previously reported in Brazil (4.5%)1919. Alves Jr JL, Gavilanes F, Jardim C, Fernandes CJC, Morinaga LTK, Dias B, et al. Pulmonary Hypertension in the Southern Hemisphere. Chest. 2015;147(2):495-501.. Mortality remains high even with specific therapies for PAH2020. Sitbon O, Lascoyx-Combe C, Delfraissy Jf, Yeni PG, Raffi F, De Zuttere, et al. Prevalence of HIV-related pulmonary arterial hypertension in the current antiretroviral therapy era. Am J Respir Crit Care Med. 2008;177(1):108-13.,2121. Mendes AA, Roncal CGP, Costa VLZ, Japyassu FAA, Oliveira FRA, Sepúlveda DL, et al. Hipertensão pulmonar associada à síndrome da imunodeficiência adquirida: apresentação de cinco casos e revisão da literatura. Rev Soc Bras Med Trop. 2009;42(4):452-457.. Further, women (69.4%) predominated, corroborating the higher incidence of PAH in the female sex, similar to the data from the French registry (65%) and the study by Rich et al. that reported a ratio of 1.7:1.0 between the sexes1717. Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habid G, Gressin V, et al. Pulmonary Arterial Hypertension in France: Results from a National Registry. Am J Respir Crit Care Med. 2006;173(9):1023-30.,2222. Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Primary pulmonary hypertension: a national prospective study. Ann Intern Med. 1987;107(2):216-230.. The mean age was 45 ± 14 years, while other studies indicated means of 48, 50, or 36 years1616. Thenappan T, Shah SJ, Rich S, Gonberg-Maitland M. A USA-based registry for pulmonary arterial hypertension: 1982-2006. Eur Resp J. 2007;30(6):1103-10.,1717. Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habid G, Gressin V, et al. Pulmonary Arterial Hypertension in France: Results from a National Registry. Am J Respir Crit Care Med. 2006;173(9):1023-30.,2222. Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Primary pulmonary hypertension: a national prospective study. Ann Intern Med. 1987;107(2):216-230.. Patients with PAH had a mean age of 56 years, suggesting late recognition of the disease in our region.
At the time of diagnosis, 70.5% of the patients were categorized as FC III or IV, similar to other studies that indicated rates of 80% and 75%, respectively1616. Thenappan T, Shah SJ, Rich S, Gonberg-Maitland M. A USA-based registry for pulmonary arterial hypertension: 1982-2006. Eur Resp J. 2007;30(6):1103-10.,1717. Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habid G, Gressin V, et al. Pulmonary Arterial Hypertension in France: Results from a National Registry. Am J Respir Crit Care Med. 2006;173(9):1023-30.. Dyspnea was the most common symptom, affecting 93.8% of patients (n = 92). In 43.3% of cases, dyspnea was present for over two years prior to diagnosis, demonstrating a long symptomatologic period occurring in advanced stages of the disease. Rich et al.2222. Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Primary pulmonary hypertension: a national prospective study. Ann Intern Med. 1987;107(2):216-230. reported that dyspnea was present as an initial symptom in 60% of patients. The importance of dyspnea at the time of early diagnosis of PAH is that this is an important prognostic factor77. Engels D, Chitsulo L, Montresor A, Savioli L. The global epidemiological situation of schistosomiasis and new approaches to control and research. Acta Trop. 2002;82(2):139-46., which makes it fundamental for identifying this pathology. In the REVEAL study1616. Thenappan T, Shah SJ, Rich S, Gonberg-Maitland M. A USA-based registry for pulmonary arterial hypertension: 1982-2006. Eur Resp J. 2007;30(6):1103-10., the recognition of PAH at more than two years after symptom onset was 39.3%, which was very different from the registry in the current study, suggesting that our population presents additional confounding factors such as difficulty accessing public health services, lower purchasing power, presence of comorbidities, and even a lack of knowledge of the disease, among other factors that make an early diagnosis by a general practitioner difficult. The aim of identifying early-stage PAH is a possibility of initiating specific therapy to prevent the development of right heart failure2323. Galié N, Rubin Lj, Hoeper M, Jansa P, Al-Hiti H, Meyer G, et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomized controlled trial. Lancet. 2008;371(9630):2093-100.. Two symptoms to be highlighted in this study are pre-syncope and syncope, which occurred in 22.7% and 25.8% of patients, respectively. These symptoms occur in PAH in advanced stages of the disease, with signs of right heart failure and low output, demonstrating the severity of the disease in this group. In another study of 187 patients, syncope was present in 13% at the time of diagnosis2222. Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Primary pulmonary hypertension: a national prospective study. Ann Intern Med. 1987;107(2):216-230..
Angina pectoris, another symptom of disease severity, was present in about 47% of patients with IPAH in a study of 187 patients; among these, etiology was not fully established in 22. Patients with PAH had few cardiovascular risk factors, since coronary atherosclerosis is unlikely as the cause of angina in these patients. Chest pain was reported by 31.8% of our patients. One of them presented spontaneous dissection of the marginal branch of the circumflex artery and was treated with a stent. Another with PAH-Sch presented with complete compression of the trunk of the left coronary artery, which was treated with a stent. The patient remained symptom free and showed good long-term outcomes, being the first case with this type of PAH-Sch treatment described in the literature2424. Mendes AA, Japyassu FA, Oliveira FR, Sepúlveda DL, Albuquerque ES, Roncal CG, et al. Tratamento por Stent em Tronco Artéria Coronária Esquerda por Compressão do Tronco Artéria Pulmonar em Paciente com Hipertensão Pulmonar Esquistossomótica. Rev Bras Cardiol Invas. 2010;42(4):89-94.. Although chest pain is present in diverse clinical situations, it is always recommended that coronary angiography be performed in those with this symptom or who have significant dilatation in the large pulmonary branches2525. Galié N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS). Eur Respir J. 2015;46:903-75.-2626. Galié N, Saia F, Palazzini M, Manes A, Russo V, Reggiani MLB, et al. Left Main Coronary Artery compression in Patients With Pulmonary Arterial Hypertension and Angina. J Am Coll Cardiol. 2017;69(23):2808-2817.. The physical examination of patients with PAH is essential for the clinical diagnosis since 92.4% of the patients in our study presented loud P2 , while the right ventricular gallop was present in 15.2%, reflecting an increase in right atrial pressure similar to a study by Rich et al.2222. Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Primary pulmonary hypertension: a national prospective study. Ann Intern Med. 1987;107(2):216-230..
The 6MWT is an important tool for the prognostic evaluation of PAH. In the present study, we observed a mean distance of 228.58 m with no statistically significant intergroup difference. The disease severity in these patients is evident compared to reports from the French registry1717. Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habid G, Gressin V, et al. Pulmonary Arterial Hypertension in France: Results from a National Registry. Am J Respir Crit Care Med. 2006;173(9):1023-30. (329 m) and Miyamoto et al.2727. Miaymoto S, Nagaya N, Satoh T, Kyotani S, Sakamaki F, Fujita M, et al. Clinical correlates and prognostic significance of six-minute walk test in patients with primary pulmonary hypertension: comparison with cardiopulmonary exercise testing. Am J Respir Crit Care Med. 2000;161(2 Pt 1):487-92. stating lower survival in patients who traveled less than 332 m. Transthoracic echocardiography is undoubtedly the method of choice of screening for the diagnosis of PAH, since it is noninvasive, safe, and carries a lower financial cost. In the studied patients, PASP levels were quite high (mean 97.28 mmHg for PAH-Sch and 88.76 mmHg for non-Sch PAH). There was a positive and significant association between PASP and MPAP measured in a hemodynamic study (p <0.001) in our patients, similar to the results of previous studies2828. Currie PJ, Seward JB, Chan KL, Fyfe DA, Hagler DJ, Mair DD, et al. Continuous wave Doppler determination of right ventricular pressure: a simultaneous Doppler catheterization study in 127 patients. J Am Coll Cardiol . 1985;6(4):750-6.
29. Chan KL, Currie PJ, Seward JB, Hagler DJ, Mair DD, Tajik AJ. Comparison or three Doppler ultrasound methods in the prediction of pulmonary artery pressure. J Am Coll Cardiol . 1987;9(3): 549-54.-3030. Denton CP, Cailes JB, Phillips GD, Welles UA, Black CM, Bois RM. Comparison of Doppler echocardiography and right heart catheterization to assess pulmonary hypertension in systemic sclerosis. Br J Rheumatol. 1997;36(2):239-43.. Transthoracic echocardiography presents parameters of great importance for the evaluation of patients with PAH, including the severity of right ventricular dysfunction, right atrial enlargement, and pericardial effusion3131. Hinderliter AL, Willis PW, Barst RJ. Effects of long-term infusion of prostacyclin (epoprostenol) on echocardiographic measures of right ventricular structure and function in primary pulmonary hypertension. Primary Pulmonary Hypertension Study Group. Circulation. 1997;95(6):1479-86.-3232. Raymond RJ, Hinderliter AL, Willis PW, Ralph D, Caldwell EJ, Williams W. Echocardiographic Predictors of Adverse Outcomes in Primary Pulmonary Hypertension. J Am Coll Cardiol . 2002;39(7):1214-9., in addition to being useful to exclude other causes of PAH. Investigating the role of natriuretic peptides, Nagaya et al.3333. Nagaya N, Nishikimi T, Uematsu M, Satoh T, Kyotani S, Sakamaki F, et al. Plasma brain natriuretic peptide as a prognostic indicator in patients with primary pulmonary hypertension. Circulation. 2000;102(8):865-70. evaluated a series of 60 patients with IPAH and demonstrated that BNP was correlated with HR, hemodynamic parameters of the right ventricle, and survival, functioning as an independent marker of mortality. Among the 43 patients with PAH-Sch in this study, the mean NT-proBNP was 1444.23 pg/mL, a value that was not significantly different from that of the non-Sch PAH group.
A study performed by Alonzo et al.3434. D’Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Survival in patients with primary pulmonary hypertension: results from a national prospective registry. Ann Intern Med . 1991;115(5):343-9. reported that the independent hemodynamic variables mean right atrium pressure (mRAP), MPAP, and cardiac index were associated with a reserved prognosis. Oxygen saturation in the pulmonary artery is another relevant survival predictor3535. Fuster V, Steele PM, Edwards WD, Gresh BJ, McGoon MD, Frye RL. Primary pulmonary hypertension: natural history and the importance of thrombosis. Circulation. 1984;70(4):580-7.. In our study, patients with PAH-Sch and non-Sch PAH with reduced cardiac index, and elevated mRAP , MPAP, and PVR demonstrated the severity of this population at the time of diagnosis, as suggested by other authors1717. Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habid G, Gressin V, et al. Pulmonary Arterial Hypertension in France: Results from a National Registry. Am J Respir Crit Care Med. 2006;173(9):1023-30.,1818. Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey ChS, et al. Predicting Survival in Pulmonary Arterial Hypertension. Circulation. 2010;122(2):164-72.. Patients with non-Sch PAH had values similar to those of the PAH-Sch group, but this difference was not statistically significant. Symptom severity, FC, 6MWT, NT-proBNP levels, and hemodynamic variables reflect the status of right ventricular failure, demonstrating the poor myocardial reserve of these patients and the disease severity. Thus, an early diagnosis is essential for the initiation of specific therapy and better patient prognosis3535. Fuster V, Steele PM, Edwards WD, Gresh BJ, McGoon MD, Frye RL. Primary pulmonary hypertension: natural history and the importance of thrombosis. Circulation. 1984;70(4):580-7..
This study demonstrates the significant prevalence of PAH-Sch in our country, as well as its severity at the time of diagnosis, in patients of productive age and provides important implications for the region’s economy. However, many questions remain open, such as the actual number of asymptomatic PAH patients or the best epidemiological, clinical, or laboratory methods for early disease detection. The authors’ experience with 98 patients with PAH in this registry allowed for the correlation of several clinical and hemodynamic variables for a better understanding of this pathology. As there is no curative therapy, perhaps a high index of suspicion and knowledge of the clinical and hemodynamic findings described in this article may contribute to an increased number of diagnoses in less advanced stages.
The present study has some limitations. First, it is retrospective and observational. There is also the possibility of selection bias since it uses data from a single center. In contrast, all clinical, diagnostic (echocardiographic, hemodynamic, and computed tomography), and laboratory investigations were always performed by the same group of investigators from the beginning, thus reducing possible bias. Pulmonary arterial hypertension is an important manifestation of schistosomiasis in endemic regions. The diagnosis is usually made late but should be considered in patients with chronic dyspnea, as the clinical, hemodynamic, and laboratory presentations are similar to those of PAH resulting from other etiologies.
ACKNOWLEDGMENTS
The authors thank Heloisa de Melo Rodrigues for the statistical analyses.
REFERENCES
-
1Rubin LJ. Primary pulmonary hypertension. N Engl J Med. 1997;336(2):111-7.
-
2Badesch DB, Champion HC, Gomez SMA, Hoeper MM, Loyd JE, Manes A, et al. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(1):S55-S66.
-
3Steinmann P, Keiser J, Bos R, Tanner M, Utzinger J. Schistosomiasis and water resources development: systematic review, meta-analysis, and estimates of people at risk. Lancet Infect Dis. 2006;6(7):411-25.
-
4Lapa M, Dias B, Jardim C, Fernandes CJC, Dourado PMM, Figueiredo M, et al. Cardiopulmonary manifestations of hepatosplenic schistosomiasis. Circulation 2009;119(11):1518-23.
-
5Ferreira RCS, Domingues ALC, Bandeira AP, Markman Filho B, Albuquerque Filho ES, Correia ACC, et al. Prevalence of pulmonary hypertension in patients with schistosomal liver fibrosis. Ann Trop Med Parasitol. 2009;103(2):129-43.
-
6Simonneau G, Gatzoulis MA, Adatia I, Celemajer D, Denton C, Ghofrani A, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol . 2013;62(25 Suppl):D34-41.
-
7Engels D, Chitsulo L, Montresor A, Savioli L. The global epidemiological situation of schistosomiasis and new approaches to control and research. Acta Trop. 2002;82(2):139-46.
-
8Appelbaum L, Yigla M, Bendayan D, Reichart N, Fink G, Priel I, et al. Primary pulmonary hypertension in Israel: a national survey. Chest. 2001 Jun;119(6):1801-6.
-
9Roncal CGP, Mendes AA, Cartaxo MT, Oliveira AS, Neto LV, Filho DSC, et al. Shistosomiaiss-Associated Pulmonary Arterial Hypertension: Survival in Endemic Area in Brazil. J Am Coll Cardiol . 2014;63(12):A1494.
-
10Fernandes CJCS, Dias BA, Jardim CVP, Hovnanian A, Hoette S, Morinaga KL, et al. The Role of Target Therapies in Schistosomiasis-Associated Pulmonary Arterial Hypertension. Chest. 2012;141(4):923-928.
-
11American Thoracic Society. ATS statement: guidelines for the six-minute walk test. Am J Resp Crit Care Med. 2002;166(1):111-7.
-
12Hoper M, Ouzir R, Peacock A, Tapson VF, Haworth SG, Frost AE. End-points and clinical trial designs in pulmonary arterial hypertension: clinical and regulatory perspectives. J Am Coll Cardiol . 2004;43(12 Suppl S):48S-55S.
-
13McQuillan BM, Picard HM, Leavitt M, Weyman AE. Clinical Correlates and Reference Intervals for Pulmonary Artery Systolic Pressure Among Echocardiographically Normal Subjects. Circulation. 2001;104(23):2797-802.
-
14Lapa MS, Ferreira MV, Jardim C, Martins BC, Arakaki JS, Rogerio S, et al. Características clínicas dos pacientes com hipertensão pulmonar em dois centros de referência. Rer Assoc Med Bras. 2006;52(3):139-43.
-
15Coura JR, Amaral RS. Epidemiological and Control Aspects of Schistosomiasis in Brazilian Endemic Areas. Mem Inst Oswaldo Cruz. 2004;99(5 Suppl 1):13-9.
-
16Thenappan T, Shah SJ, Rich S, Gonberg-Maitland M. A USA-based registry for pulmonary arterial hypertension: 1982-2006. Eur Resp J. 2007;30(6):1103-10.
-
17Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habid G, Gressin V, et al. Pulmonary Arterial Hypertension in France: Results from a National Registry. Am J Respir Crit Care Med. 2006;173(9):1023-30.
-
18Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey ChS, et al. Predicting Survival in Pulmonary Arterial Hypertension. Circulation. 2010;122(2):164-72.
-
19Alves Jr JL, Gavilanes F, Jardim C, Fernandes CJC, Morinaga LTK, Dias B, et al. Pulmonary Hypertension in the Southern Hemisphere. Chest. 2015;147(2):495-501.
-
20Sitbon O, Lascoyx-Combe C, Delfraissy Jf, Yeni PG, Raffi F, De Zuttere, et al. Prevalence of HIV-related pulmonary arterial hypertension in the current antiretroviral therapy era. Am J Respir Crit Care Med. 2008;177(1):108-13.
-
21Mendes AA, Roncal CGP, Costa VLZ, Japyassu FAA, Oliveira FRA, Sepúlveda DL, et al. Hipertensão pulmonar associada à síndrome da imunodeficiência adquirida: apresentação de cinco casos e revisão da literatura. Rev Soc Bras Med Trop. 2009;42(4):452-457.
-
22Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Primary pulmonary hypertension: a national prospective study. Ann Intern Med. 1987;107(2):216-230.
-
23Galié N, Rubin Lj, Hoeper M, Jansa P, Al-Hiti H, Meyer G, et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomized controlled trial. Lancet. 2008;371(9630):2093-100.
-
24Mendes AA, Japyassu FA, Oliveira FR, Sepúlveda DL, Albuquerque ES, Roncal CG, et al. Tratamento por Stent em Tronco Artéria Coronária Esquerda por Compressão do Tronco Artéria Pulmonar em Paciente com Hipertensão Pulmonar Esquistossomótica. Rev Bras Cardiol Invas. 2010;42(4):89-94.
-
25Galié N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS). Eur Respir J. 2015;46:903-75.
-
26Galié N, Saia F, Palazzini M, Manes A, Russo V, Reggiani MLB, et al. Left Main Coronary Artery compression in Patients With Pulmonary Arterial Hypertension and Angina. J Am Coll Cardiol. 2017;69(23):2808-2817.
-
27Miaymoto S, Nagaya N, Satoh T, Kyotani S, Sakamaki F, Fujita M, et al. Clinical correlates and prognostic significance of six-minute walk test in patients with primary pulmonary hypertension: comparison with cardiopulmonary exercise testing. Am J Respir Crit Care Med. 2000;161(2 Pt 1):487-92.
-
28Currie PJ, Seward JB, Chan KL, Fyfe DA, Hagler DJ, Mair DD, et al. Continuous wave Doppler determination of right ventricular pressure: a simultaneous Doppler catheterization study in 127 patients. J Am Coll Cardiol . 1985;6(4):750-6.
-
29Chan KL, Currie PJ, Seward JB, Hagler DJ, Mair DD, Tajik AJ. Comparison or three Doppler ultrasound methods in the prediction of pulmonary artery pressure. J Am Coll Cardiol . 1987;9(3): 549-54.
-
30Denton CP, Cailes JB, Phillips GD, Welles UA, Black CM, Bois RM. Comparison of Doppler echocardiography and right heart catheterization to assess pulmonary hypertension in systemic sclerosis. Br J Rheumatol. 1997;36(2):239-43.
-
31Hinderliter AL, Willis PW, Barst RJ. Effects of long-term infusion of prostacyclin (epoprostenol) on echocardiographic measures of right ventricular structure and function in primary pulmonary hypertension. Primary Pulmonary Hypertension Study Group. Circulation. 1997;95(6):1479-86.
-
32Raymond RJ, Hinderliter AL, Willis PW, Ralph D, Caldwell EJ, Williams W. Echocardiographic Predictors of Adverse Outcomes in Primary Pulmonary Hypertension. J Am Coll Cardiol . 2002;39(7):1214-9.
-
33Nagaya N, Nishikimi T, Uematsu M, Satoh T, Kyotani S, Sakamaki F, et al. Plasma brain natriuretic peptide as a prognostic indicator in patients with primary pulmonary hypertension. Circulation. 2000;102(8):865-70.
-
34D’Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Survival in patients with primary pulmonary hypertension: results from a national prospective registry. Ann Intern Med . 1991;115(5):343-9.
-
35Fuster V, Steele PM, Edwards WD, Gresh BJ, McGoon MD, Frye RL. Primary pulmonary hypertension: natural history and the importance of thrombosis. Circulation. 1984;70(4):580-7.
Publication Dates
-
Publication in this collection
07 Feb 2020 -
Date of issue
2020
History
-
Received
10 Oct 2019 -
Accepted
25 Nov 2019