Resumos
OBJETIVO: Avaliar as formas de apresentação da amilóidose cardíaca em hospital terciário. MÉTODOS: Nos últimos 15 anos, foram identificados 8 pacientes com amilóidose, sendo 5 mulheres , com idades entre 23 e 83 (média 62) anos. Após anamnese e exame físico foram submetidos a eletrocardiograma (ECG), ecocardiograma (ECO), estudo com pirofosfato de tecnécio e biópsia endomiocárdica, cujos resultados permitiram caracterizar suas formas clínicas. RESULTADOS: Sete pacientes apresentavam dispnéia aos esforços, 6 quadro de insuficiência cardíaca congestiva (ICC), 1 síncopes. O ECG identificou bloqueio atrioventricular total (BAVT) em 4 casos e área eletricamente inativa ântero-septal em outros 4. O ECO mostrou diâmetros normais em todos e fração de ejeção discretamente reduzida em 6. Hipertrofia do septo e parede posterior em todos, sendo em 7 com aspecto sugestivo de doença de depósito (aspecto granuloso). Os dados clínicos caracterizam dois grupos, um com BAVT e outro com cardiomiopatia restritiva. A evolução foi diferente com melhora clínica após o implante de marcapasso no primeiro grupo e má evolução no grupo com cardiomiopatia restritiva que evoluiu de maneira refratária, 3 falecendo em menos de 6 meses após diagnóstico. CONCLUSÃO: A presença de aumento da espessura das paredes ao ECO, discreta disfunção sistólica e aspecto de doença de depósito identificaram a quase totalidade dos casos. Cardiomiopatia restritiva e distúrbio de condução foram as formas de apresentação, sendo o prognóstico muito reservado nos pacientes com forma restritiva, evoluindo para ICC refratária.
amilóidose cardíaca; insuficiência cardíaca; bloqueio atrioventricular total
PURPOSE: To identify the principal forms of cardiac amiloydosis presentation in a terciary hospital. METHODS: Eight cases whith cardiac amyloidosis were identified. Five were woman, their ages ranged from 23 to 83 years (mean 62). After a medical history and clinical examination the patients, were submitted to complementary tests: electrocardiogram (EKG), echocardiogram (ECHO), scintigraphy with technecium pirophosfate and cardiac biopsy these results allowed the identification of their clinical situation. RESULTS: Seven patients refered dyspnea, 6 were in heart failure, 1 patient had syncope. The EKG identified complete atrioventricular (AV) block in 4 patients, and antero septal inactive area in the other 4. The ECHO showed normal cardiac diameter in all (mean left ventricular diastolic diameter of 46.8) and slight reduction of left ventricular ejection fraction; hypertrophy of the left ventricular septal and posterior walls in all cases, in 7 cases there was a hyper refractile granular sparkling ECHO. Two different groups were identified: one with complete AV block and the second with restrictive cardiomyopathy. The prognosis was different in these two groups. Those with complete AV block evolved better after pacemaker implantation and those with restrictive cardiomyopathy had refractary heart failure and 3 of them died. CONCLUSION: The increased free wall and septal thickness, the slight systolic dysfunction and the infiltration aspect at ECHO allow us to identify the great majority of the cases. Those patients with restrictive cardiomyopathy evolve with refractory heart failure and most of them die in a few months.
cardiac amyloidosis; heart failure; complete atrioventricular block
Artigo Original
Amilóidose Cardíaca. Uma Doença de Muitas Faces e Diferentes Prognósticos
Antonio Carlos Pereira Barretto, Dalton Precoma, João Batista Serro-Azul, Mauricio Wajngarten, Humberto Pierri, Ligia Pivotto, Amit Nussbacher, Otavio Celso Eluf Gebara, Giovanni Bellotti.
São Paulo, SP
OBJETIVO: Avaliar as formas de apresentação da amilóidose cardíaca em hospital terciário.
MÉTODOS: Nos últimos 15 anos, foram identificados 8 pacientes com amilóidose, sendo 5 mulheres , com idades entre 23 e 83 (média 62) anos. Após anamnese e exame físico foram submetidos a eletrocardiograma (ECG), ecocardiograma (ECO), estudo com pirofosfato de tecnécio e biópsia endomiocárdica, cujos resultados permitiram caracterizar suas formas clínicas.
RESULTADOS: Sete pacientes apresentavam dispnéia aos esforços, 6 quadro de insuficiência cardíaca congestiva (ICC), 1 síncopes. O ECG identificou bloqueio atrioventricular total (BAVT) em 4 casos e área eletricamente inativa ântero-septal em outros 4. O ECO mostrou diâmetros normais em todos e fração de ejeção discretamente reduzida em 6. Hipertrofia do septo e parede posterior em todos, sendo em 7 com aspecto sugestivo de doença de depósito (aspecto granuloso). Os dados clínicos caracterizam dois grupos, um com BAVT e outro com cardiomiopatia restritiva. A evolução foi diferente com melhora clínica após o implante de marcapasso no primeiro grupo e má evolução no grupo com cardiomiopatia restritiva que evoluiu de maneira refratária, 3 falecendo em menos de 6 meses após diagnóstico.
CONCLUSÃO: A presença de aumento da espessura das paredes ao ECO, discreta disfunção sistólica e aspecto de doença de depósito identificaram a quase totalidade dos casos. Cardiomiopatia restritiva e distúrbio de condução foram as formas de apresentação, sendo o prognóstico muito reservado nos pacientes com forma restritiva, evoluindo para ICC refratária.
Palavras-chave: amilóidose cardíaca, insuficiência cardíaca, bloqueio atrioventricular total
Cardiac Amyloidosis. A Disease with Many Faces and Different Prognoses
PURPOSE: To identify the principal forms of cardiac amiloydosis presentation in a terciary hospital.
METHODS: Eight cases whith cardiac amyloidosis were identified. Five were woman, their ages ranged from 23 to 83 years (mean 62). After a medical history and clinical examination the patients, were submitted to complementary tests: electrocardiogram (EKG), echocardiogram (ECHO), scintigraphy with technecium pirophosfate and cardiac biopsy these results allowed the identification of their clinical situation.
RESULTS: Seven patients refered dyspnea, 6 were in heart failure, 1 patient had syncope. The EKG identified complete atrioventricular (AV) block in 4 patients, and antero septal inactive area in the other 4. The ECHO showed normal cardiac diameter in all (mean left ventricular diastolic diameter of 46.8) and slight reduction of left ventricular ejection fraction; hypertrophy of the left ventricular septal and posterior walls in all cases, in 7 cases there was a hyper refractile granular sparkling ECHO. Two different groups were identified: one with complete AV block and the second with restrictive cardiomyopathy. The prognosis was different in these two groups. Those with complete AV block evolved better after pacemaker implantation and those with restrictive cardiomyopathy had refractary heart failure and 3 of them died.
CONCLUSION: The increased free wall and septal thickness, the slight systolic dysfunction and the infiltration aspect at ECHO allow us to identify the great majority of the cases. Those patients with restrictive cardiomyopathy evolve with refractory heart failure and most of them die in a few months.
Key-words: cardiac amyloidosis, heart failure, complete atrioventricular block
Amilóidose é uma doença complexa ocasionada por deposição de substância amilóide 1-3 . A substância amilóide pode ser encontrada em quase todos os órgãos, mas evidências clínicas da doença só surgem quando a deposição é extensa. Os órgãos mais comumente envolvidos são o sistema nervoso, coração e rim 3 .
Amilóidose cardíaca não é freqüentemente diagnosticada e a sua real incidência não é conhecida.
Várias formas de apresentação são descritas na amilóidose cardíaca 4 , manifestações clínicas são observadas em cerca de 1/3 dos casos e os portadores podem ser divididos em quatro grupos conforme as principais manifestações clínicas: cardiomiopatia restritiva, disfunção sistólica, hipotensão postural e distúrbio de condução 2 .
Apresentamos neste artigo nossa experiência com a doença, descrevendo as formas mais encontradas, métodos de diagnóstico, tratamento e suas implicações prognósticas.
Métodos
Foram internados em nossa Instituição, nestes últimos 15 anos, oito pacientes com a confirmação do diagnóstico de amilóidose cardíaca. Cinco eram do sexo feminino e três do masculino, variando as idades de 23 a 83 (x= 62,7) anos.
Os pacientes foram submetidos a consulta clínica e exame físico e, posteriormente, a exames complementares para confirmar o diagnóstico e avaliar a extensão da doença.
Na avaliação, todos fizeram eletrocardiograma (ECG) e ecocardiograma (ECO). Dois pacientes fizeram estudo cintilográfico com pirofosfato. Biópsia miocárdica foi realizada em quatro casos. Necropsia em três casos.
Os pacientes foram acompanhados em nosso ambulatório e, quando não retornavam a consulta, eram contactados por telefone para averiguar sua condição clínica.
Resultados
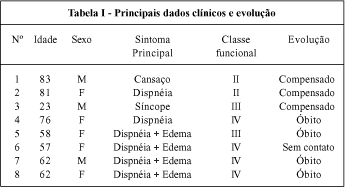
Na tabela I apresentamos os principais dados clínicos. Dois pacientes eram irmãos e apresentavam quadro clínico de insuficiência cardíaca congestiva (ICC) refratária, de aparecimento próximo aos 60 anos.
Na história clínica, a maioria apresentou sintomas de IC. Sete pacientes referiram dispnéia, sendo que em seis era aos mínimos esforços ou em repouso (CF III/IV). Três casos apresentavam edema importante e um procurou o hospital em decorrência de quadro sincopal.
Na tabela II são apresentados os principais achados eletro e ecocardiográficos. Ao ECG quatro apresentavam bloqueio atrioventricular total (BAVT), dois deles intermitente. Distúrbio de condução foi detectado em quatro, bloqueio completo de ramo direito (BCRD) em três, bloqueio da divisão ântero-septal (BDAS) do ramo esquerdo em um, bloqueio atrioventricular (BAV) de 1 o grau em um. Área inativa ântero-septal foi observada em quatro (fig. 1), alteração da repolarização ventricular em três e fibrilação atrial em um caso.
Ao ECO, sete dos oito casos apresentavam espessamento do septo ou parede livre e a espessura variou de 1,1 a 2,3 (x=1,4)cm. O ventrículo esquerdo (VE) apresentou diâmetro diastólico, variando de 3,5 a 5,3 (x=4,6)cm e o átrio esquerdo (AE) diâmetro de 3,5 a 5,0 (x= 4,3)cm. A fração de ejeção (FE) do VE variou de 0,44 a 0,73 (x=0,53). Aspecto sugestivo de doença de depósito (granulação na espessura do septo ou parede livre) foi descrito em sete casos (fig. 2).
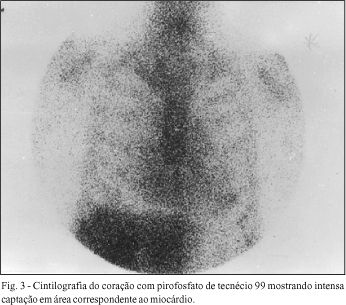
Os dois pacientes submetidos a cintilografia com pirofosfato de tecnécio apresentaram intensa captação difusa em área correspondente ao miocárdio (fig. 3).
Quatro casos foram submetidos a biópsia miocárdica, que identificou aspecto histológico compatível com deposição de substância amilóide (fig. 4) e quatro faleceram, sendo que três com diagnóstico da doença confirmado pela necropsia.
Com base nos resultados da avaliação clínica dos pacientes foi possível identificar duas formas principais de apresentação. Quatro pacientes apresentavam, predominantemente, distúrbio de condução e, na evolução, apresentaram BAVT, sendo em três necessário implante de marcapasso. No caso restante, o bloqueio surgiu em decorrência de intoxicação digitálica, revertido com a suspensão da droga. Um paciente deste grupo veio a falecer na evolução. Os outros quatro evoluíram com quadro de ICC de difícil controle, tendo três falecido com menos de seis meses de seguimento, Não dispomos de informações sobre o caso restante pois perdemos o contato.
O ECG foi diferente nos dois grupos. No grupo com distúrbio de condução, dois já se apresentaram com BAVT e todos tinham BCRD de base, e dois BCRD e BDAS. No grupo com ICC, todos tinham área eletricamente inativa no ECG, um apresentando também BCRD e outro BDAS.
A FE avaliada pelo ECO variou de 0,50 a 0,73 (x=0,59) no grupo com distúrbio de condução e de 0,44 a 0,52 (x=0,47) no grupo com ICC. O diâmetro do AE foi semelhante nos dois grupos e o diâmetro diastólico do VE foi maior no grupo com distúrbio de condução (51,5mm para 42,2mm).
Os dois estudos cintilográficos mostraram intensa captação miocárdica de pirofosfato de tecnécio.
A biópsia miocárdica identificou substância amilóide entre as fibras cardíacas, identificadas por diferentes colorações (hematoxilina/eosina, vermelho congo e por técnica imuno-histoquímica ao microscópio de luz polarizada).
A necropsia confirmou o diagnóstico nos três casos em que foi realizada e identificou lesão renal em dois e esplênica em um.
Discussão
Amilóidose cardíaca é pouco diagnosticada na prática clínica. É doença rara, mas provavelmente mais freqüente do que é usualmente reconhecida, uma vez que apenas os casos com manifestações clínicas mais evidentes são diagnosticados 1-3 .
Há muitos tipos de amilóidose em humanos, mas o envolvimento cardíaco é mais comum na amilóidose primária, onde há deposição de imunoglobulinas de cadeia leve produzida por plasmócitos no miocárdio, mais comumente decorrente de mieloma múltiplo. Amilóidose secundária é causada pela deposição de outras proteínas e pode ser familiar, senil ou devida a processos inflamatórios crônicos 1,2 . Seus depósitos são tipicamente pequenos e perivasculares, sendo raro envolvimento cardíaco expressivo.
Em gerontes é usualmente observada nas necropsias, com quadros de pequenos depósitos nos átrios, sem expressão clínica. Quando de origem familiar, somente cerca de 25% apresentam quadro cardíaco e, em geral, tardiamente 2 .
O nosso pequeno universo de oito casos confirma que a doença com significância clínica é realmente rara (pequena incidência com tantos anos de experiência clínica). A quase totalidade dos pacientes tinha mais de 50 anos e três deles mais de 70, à semelhança do que é discutido na literatura 2 . Na nossa experiência não houve predomínio de sexo, quando na mundial prevalece os homens 2 .
Dois dos nossos casos sugerem a possibilidade da etiologia familiar, pois eram irmãos com a mesma forma de doença (ICC), vindo a falecer com a mesma idade (62 anos). Como descrito na literatura, apesar de doença geneticamente determinada, suas manifestações clínicas são tardias e muito raras antes dos 35 anos 2 .
Do ponto de vista clínico, são descritas quatro formas de apresentação conforme predominância das manifestações clínicas: cardiomiopatia restritiva, disfunção sistólica, hipotensão postural e distúrbio de condução 2 .
Na nossa casuística, os pacientes apresentaram duas formas: insuficiência cardíaca e distúrbio da condução. Nos casos com falência cardíaca, o quadro foi de insuficiência cardíaca, com importante restrição diastólica e discreto comprometimento da função sistólica (tab. II). Os pacientes com distúrbio de condução em algum momento da evolução apresentaram BAVT, sendo que em três casos foi necessário implante de marcapasso para controle do quadro e no 4º, a retirada do digital reverteu o bloqueio.
Nos pacientes com distúrbio da condução observou-se associadamente insuficiência cardíaca e disfunção diastólica. Contudo esta disfunção era de menor importância clínica e sem a característica de refratariedade do outro grupo.
Os exames complementares na amilóidose representaram papel importante na caracterização dos grupos. Os exames auxiliaram a sua identificação e a previsão da evolução 5-8 . O ECG conforme descrito na literatura mostra área eletricamente inativa no grupo com insuficiência cardíaca (cardiomiopatia restritiva) e bloqueio de ramo direito que evoluiu para BAVT no grupo com distúrbio da condução 2-5 .
A ECO foi muito útil na caracterização da doença (fig. 2). As alterações ecocardiográficas sugestivas de formas avançadas da doença são aumento da espessura da parede dos ventrículos, pequenas câmaras ventriculares, dilatação atrial e espessamento do septo interatrial. O aspecto do aumento da espessura das paredes é pecu liar ao ECO bidimensional, onde se identifica textura granulosa 2,4,7 , achados presentes nos nossos casos. A maioria dos traçados (7 em 8) mostrou espessamento do septo e parede livre do VE com aspecto sugestivo de doença de depósito (granulação na espessura do septo). A alteração relativamente discreta da função sistólica em relação à intensidade das manifestações clínicas do quadro de IC foi outro dado útil para o diagnóstico.
Em muitos casos, o ECO foi o exame que fez a primeira suspeita do diagnóstico, pela identificação da disfunção diastólica e ausência de dilatação ventricular. Na nossa experiência, a maioria dos casos apresentava a disfunção diastólica e sistólica. Em somente dois casos a FE foi normal.
Outro método que auxilia no diagnóstico da doença é a cintilografia miocárdica com pirofosfato de tecnécio 99 . Os casos de amilóidose apresentam captação miocárdica proporcional à intensidade do depósito de substância amilóide. Não é alteração patognomônica, mas a presença de captação em portadores de insuficiência cardíaca, sem grande disfunção sistólica, é dado propedêutico que auxilia a caracterizar a doença. Conforme a literatura 2,8 , o método raramente apresenta resultado falso negativo. A positividade do exame é, em geral, encontrado na forma extensa da doença.
A biópsia é método que realmente confirma o diagnóstico, pois permite a caracterização histológica da substância amilóide. Coloração específica com vermelho congo ou através de coloração imuno-histoquímica ao microscópio com luz polarizada permite identificar os seus vários tipos. Na nossa experiência, entretanto, sua utilidade foi relativa, pois, nos três casos em que foi realizada havia forte suspeita clínica do diagnóstico e a biópsia apenas confirmou a suspeita, sem entretanto identificar casos insuspeitos.
Assim, na nossa experiência os dados clínicos e dos exames complementares permitiram caracterizar os casos diagnosticados.
O prognóstico da doença varia conforme a forma e sua intensidade. Pudemos observar que aqueles com predomínio do distúrbio de condução apresentaram melhor evolução que os com cardiomiopatia restritiva. Na forma com insuficiência cardíaca refratária, 3 dos 4 casos vieram a falecer com menos de seis meses de seguimento, apesar de tratamento intensivo. Resultado oposto foi observado na forma com distúrbio de condução quando a maioria estabilizou com a terapêutica proposta (marcapasso).
Para orientação terapêutica é fundamental o diagnóstico correto da doença, pois ela tem peculiaridades 2,3,10 . Os pacientes com amilóidose cardíaca apresentam maior sensibilidade aos digitálicos, sendo referido por Spyrove e Fsale 10 que nesses casos não devemos prescrever este medicamento. Os pacientes em uso do digital, com facilidade apresentam arritmias mais freqüentes, conforme observamos em um caso com BAVT, e por outro lado, não teriam benefício, pois, na doença, predomina a disfunção diastólica para qual o digital não tem indicação 9,10 . O controle da ICC é realizada basicamente com diuréticos. Antagonistas dos canais de cálcio, betabloqueadores e inibidores da enzima conversora podem não ser bem tolerados pela hipotensão que induzem e podem acentuar os distúrbios de condução freqüentes na doença 10 .
Nas formas menos avançada tem se indicado colchicina que parece reduzir a progressão da doença 9 . Transplante cardíaco não é indicado nas formas avançadas, pois a doença recidiva na maioria, em curto tempo de avaliação 11 .
Não tivemos no INCOR casos de hipotensão postural ou disfunção sistólica predominante, descritos na literatura como formas de apresentação da doença.
Na literatura, os autores descrevem marcadores prognósticos, que podem orientar o clínico. A espessura miocárdica da parede livre do VE avaliada ao ECO, não só é alteração diagnóstica, como pode-se dizer que quanto mais espessa pior a evolução. Pacientes com mais de 1,2 e menos que 1,5cm apresentam sobrevida de 1,3 anos e aqueles com mais de 1,5 de 0,4 anos 5,8 . Novos marcadores estão sendo desenvolvidos. A captação de componente P de substância amilóide (SAP) marcado com iodo 123 tem se mostrado útil no diagnóstico da doença e na avaliação do prognóstico 12 . Quanto maior a absorção nos vários órgãos pior a evolução.
Podemos concluir que amilóidose cardíaca com expressão clínica é doença rara. É mais freqüente a partir dos 50 anos, pode ser familiar e o seu prognóstico depende muito da sua forma de apresentação. A avaliação clínica complementada por ECG e ECO permite caracterizar bem a doença e a forma de manifestação: pacientes com distúrbio de condução predominante têm melhor evolução e para os pacientes com insuficiência cardíaca de difícil controle, a doença tem características malignas.
Instituto do Coração do Hospital das Clínicas - FMUSP
Correspondência: Antonio Carlos Pereira Barretto - InCor - Av. Dr. Enéas C. Aguiar, 44 - 05403-000 - São Paulo, SP
Recebido para publicação em 5/3/97
Aceito em 19/6/97
- 1. Kushwara SS, Fallon JT, Fuster V - Restrictive cardiomyopathy. N Engl J Med 1997; 336: 267-76.
- 2. Wynne J, Braunwald E - The cardiomyopathies and myocardities. In: Braunwald E - Heart Disease. A textbook of Cardiovascular Medicine. Philadelphia-London: WB Sanders, 1997; 1427-9.
- 3. Roberts WC, Waller BF - Cardiac amyloidosis causing cardiac dysfunctions: analysis of 54 necropsy patients. Am J Cardiol 1983; 52: 137-46.
- 4. Klein AL, Hatle LK, Burstow DJ et al - Doppler characterization of left ventricular diastolic functions in cardiac amyloidosis. J Am Coll Cardiol 1989;13:1017-26.
- 5. Parthenakis FI, Vardas PE, Ralidis L et al - QT interval in cardiac amyloidosis. Clin Cardiol 1996; 19: 51-4.
- 6. Brandt K, Cathcart ES, Cohen AS - A clinical analysis of the course and prognosis of forty two patients with amyloidosis. Am J Med 1968; 44: 955-69.
- 7. Cuesta-Cardia L, Reeder GS, Kyle RA et al - Echocardiographic findings in systemic amyloidosis: Spectrum cardiac involvement and relation to survival. J Am Coll Cardiol 1985; 6: 737-43.
- 8. Hartmann A, Frenkel J, Hopf R et al - Is technetium 99m pyrofhosphate scintigraphy valuable in the diagnosis of cardiac amyloidosis? Int J Card Imagins 1990; 5: 227-31.
- 9. Kyle RA - Amyloidosis. Circulation 1995; 91: 1269-75.
- 10. Spyrove N, Fsale R - Restrictive carrdiomyopathies. Curr Opin Cardiol 1994; 9: 344-64.
- 11. Hosenpude JD, De Marco T, Frazier OH et al - Progression of systemic disease and reduced long-term survival in patients with cardiac amyloidosis undergoing heart transplantation. Follow-up results of a multicenter survey. Circulation. 1991; 84: III-338.
- 12. Hachulla E, Maullin L, Deveaux M et al - Prospective and serial study of primary amyloidosis with serum amyloid p component scintigraphy. Form diagnosis to prognosis. Am J Med 1996; 101: 77-87.
Datas de Publicação
-
Publicação nesta coleção
14 Fev 2001 -
Data do Fascículo
Ago 1997
Histórico
-
Aceito
19 Jun 1997 -
Recebido
05 Mar 1997