Abstracts
OBJECTIVE: The objective of the present study was to evaluate cardiac tissue adaptations in rats submitted to aerobic training after nitric oxide (NO) synthesis blockade. METHODS: The animals (n=48) were divided into four groups: sedentary (CONTROL group); hypertensive after administration of NG-nitro-L-arginine methyl ester for 7 days (L-NAME Group); trained for 8 weeks through swimming exercises (TRAINED Group);trained and treated with L-NAME during the last week (L-NAME TRAINED Group). All the animals were submitted to the experiment procedures for blood pressure (BP) readings and cardiac morphometric evaluation. RESULTS: In comparison to the other groups, the L-NAME and L-NAME TRAINED groups were hypertensive (p<0.05); however, BP elevation in the L-NAME TRAINED group was significantly lower than the L-NAME group (p<0.05). The heart weight indexes for the TRAINED and L-NAME TRAINED groups were higher than the CONTROL and L-NAME groups (p<0.05). Also they had presented higher rates of macroscopic cardiac area and cardiac fibrosis in relation to the rest (p<0.05); comparisons revealed that the values for the L-NAME TRAINED group were significantly higher (p<0.05) than the others. CONCLUSION: Short term NO synthesis blockade in sedentary animals induced hypertension but did not cause cardiac hypertrophy. In the trained animals, the inhibition of NO synthesis attenuated hypertension, induced cardiac hypertrophy and significantly increased myocardial fibrosis, indicating that NO plays an important role in cardiac tissue adaptations caused by aerobic exercise.
Hypertension; exercise; nitric oxide; cardiomegaly; endomyocardial fibrosis
OBJETIVO: O presente estudo avaliou as adaptações teciduais cardíacas em ratos submetidos a treinamento aeróbio, após o bloqueio da síntese de óxido nítrico (NO). MÉTODOS: Os animais (n = 48) foram divididos em quatro grupos: sedentários (grupo CONTROLE), hipertensos após administração de Ng-nitro-L-arginina metil éster durante sete dias (grupo L-NAME), treinados por meio de natação durante oito semanas (grupo TREINADO) e treinados e tratados com L-NAME na última semana (grupo TREINADO L-NAME). Em todos os animais foi registrada a pressão arterial (PA) e realizada a avaliação morfométrica cardíaca. RESULTADOS: Os grupos L-NAME e TREINADO L-NAME apresentaram-se hipertensos em relação aos demais (p < 0,05), porém a elevação da PA no grupo TREINADO L-NAME foi significativamente menor em relação ao L-NAME (p < 0,05). Os grupos TREINADO e TREINADO L-NAME apresentaram índice de peso cardíaco maior que os grupos CONTROLE e L-NAME (p < 0,05). Também apresentaram maiores índices de área cardíaca macroscópica e de fibrose cardíaca em relação aos demais (p < 0,05) e, quando comparados, o grupo TREINADO L-NAME mostrou-se significativamente superior (p < 0,05). CONCLUSÃO: O bloqueio a curto prazo da síntese de NO, em animais sedentários, induziu hipertensão, sem no entanto causar hipertrofia cardíaca. Nos animais treinados, a inibição da síntese de NO atenuou a hipertensão e promoveu hipertrofia cardíaca com aumento expressivo da fibrose miocárdica, sugerindo importante papel do NO nas adaptações teciduais cardíacas induzidas pelo treinamento físico aeróbio.
Hipertensão; exercício aeróbico; óxido nítrico; cardiomegalia; fibrose endomiocárdica
ORIGINAL ARTICLE
Nitric oxide synthesis blockade increases hypertrophy and cardiac fibrosis in rats submitted to aerobic training
Hugo Celso Dutra de Souza; Daniel Martins Dias Penteado; Marli Cardoso Martin-Pinge; Octávio Barbosa Neto; Vicente de Paula Antunes Teixeira; João Henrique Dutra Blanco; Valdo José Dias da Silva
Faculdade de Medicina de Ribeirão Preto, Universidade Estadual de Londrina, Faculdade de Medicina do Triângulo Mineiro - Ribeirão Preto, SP, Londrina, PR, Uberaba, MG - Brazil
Mailing address Mailing address: Hugo Celso Dutra de Souza Rua Luís Basso, 130 14040-150 - Ribeirão Preto, SP - Brazil E-mail: hugocds@fmrp.usp.br
SUMMARY
OBJECTIVE: The objective of the present study was to evaluate cardiac tissue adaptations in rats submitted to aerobic training after nitric oxide (NO) synthesis blockade.
METHODS: The animals (n=48) were divided into four groups: sedentary (CONTROL group); hypertensive after administration of NG-nitro-L-arginine methyl ester for 7 days (L-NAME Group); trained for 8 weeks through swimming exercises (TRAINED Group);trained and treated with L-NAME during the last week (L-NAME TRAINED Group). All the animals were submitted to the experiment procedures for blood pressure (BP) readings and cardiac morphometric evaluation.
RESULTS: In comparison to the other groups, the L-NAME and L-NAME TRAINED groups were hypertensive (p<0.05); however, BP elevation in the L-NAME TRAINED group was significantly lower than the L-NAME group (p<0.05). The heart weight indexes for the TRAINED and L-NAME TRAINED groups were higher than the CONTROL and L-NAME groups (p<0.05). Also they had presented higher rates of macroscopic cardiac area and cardiac fibrosis in relation to the rest (p<0.05); comparisons revealed that the values for the L-NAME TRAINED group were significantly higher (p<0.05) than the others.
CONCLUSION: Short term NO synthesis blockade in sedentary animals induced hypertension but did not cause cardiac hypertrophy. In the trained animals, the inhibition of NO synthesis attenuated hypertension, induced cardiac hypertrophy and significantly increased myocardial fibrosis, indicating that NO plays an important role in cardiac tissue adaptations caused by aerobic exercise.
Key words: Hypertension; exercise; nitric oxide; cardiomegaly; endomyocardial fibrosis.
Introduction
The discovery of endothelial derived endogenous factors, particularly nitric oxide (NO)1, began a new chapter in the understanding of cardiovascular pathology mechanisms, as well as in methods of prevention and treatment.
NO, in addition to its potent vasodilator action, induces other important vascular, renal and cardiac effects including inhibiting platelet aggregation, modulation of the glomerular filtration rate and effect on vascular and cardiac remodeling2,3. In turn the diminished endogenous NO production is related to the reduction of endothelial-dependent vasodilatation in patients with hypertension, hypercholesterolemia, diabetes or arteriosclerosis4.
In relation to cardiac remodeling, NO seems to be an important endogenous factor in the modulation of hypertrophic growth, both indirectly through its hypotensive effect via peripheral artery dilatation, promoting afterload reduction and through via ditation of the venous system reducing the preload5,6. Additionally, other studies have shown that NO is also more directly involved in cardiac remodeling, since its reduction causes significant morphological alterations such as necrosis foci, increased fibrosis, apoptosis, reduction of cardiac angiogenesis and consequent pathological hypertrophy7-11. Nevertheless, literature on cardiac hypertrophy development and the effects of acute or chronic NO synthesis inhibition with L-arginine analogous agents, such as NG-monomethyl-L-arginine (L-NMMA) or NG-nitro-L-arginine methyl ester (L-NAME) that result in the consequent elevation of BP is controversial2,3,12-15.
On the other hand, aerobic exercise has been widely used as an anti-hypertensive therapy to attenuate the effects of cardiovascular risk factors. It has also been demonstrated that aerobic exercise, in addition to promoting significant alterations in cardiocirculatory autonomic control16-18, also induces tissue adaptations, mainly in the heart19-21. These adaptations promote excentric cardiac hypertrophy (addition of sarcomeres in series), which improves ejection function, promotes increased life expectancy and helps prevent cardiac events. Even though various mechanisms have been indicated to explain these adaptations, very few studies have been conducted on the possible participation of NO in this physiological myocardial hypertrophy modality as a result of physical exercise.
Thus, based on the abovementioned facts, the objective of the present study was to evaluate the effects of short term NO synthesis blockade on cardiac tissue adaptations in rats submitted to aerobic exercise.
Methods
All experimental procedures involved in this study were approved by the ethics committee for animal experiments of the University of São Paulo, Ribeirão Preto School of Medicine.
Animals - Male Wistar rats (120 150g) were separated in individual cages with controlled temperature (21ºC) and a 12 hour light/dark cycle. The animals were given animal feed and water ad libitum for 3 days before the start of the experimental procedure.
Experiment groups - The animals were divided into 4 experimental groups: 1) normotensive rats treated with animal feed and water ad libitum for 8 weeks (CONTROL group; n=12); 2) rats treated with animal feed and water ad libitum for 8 weeks, with L-NAME dissolved in the drinking water (70mg/kg) during the last week (L-NAME group; n=12); 3) rats submitted to swim training for 8 weeks (TRAINED group; n=12); 4) rats submitted to swim training for 8 weeks and given L-NAME during the last week (L-NAME TRAINED group; n=12).
Physical training - The 8-week swim training program was conducted in a glass aquarium (100 cm long, 80 cm wide and 80 cm high) with heated water (30ºC). Before beginning the training program, the animals were submitted to a two-week adaptation program, that is, the water exercise began with 10 minutes and was gradually increased to 45 minutes. After the adaptation program, the animals were further divided into subgroups of 4 animals and trained for 1 hour per day, 6 days per week. The animal groups that were not trained were submitted to water stress for 2 minutes per day during the training timeframe.
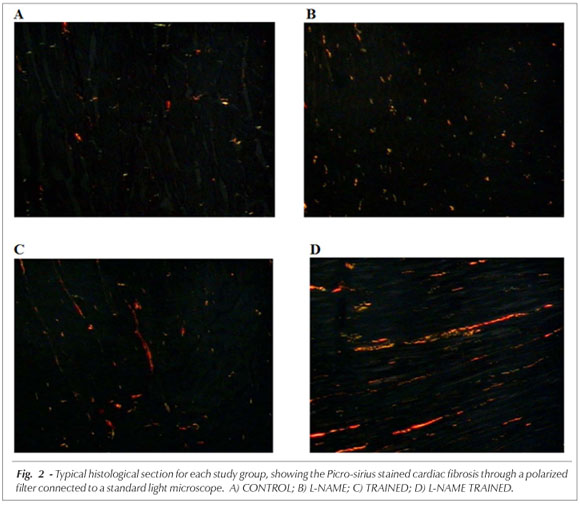
Experimental protocol - On the sixth day of the last week, the animals underwent surgery with tribromoethanol anesthesia (250 mg/kg, i.p.), to insert the femoral artery cannula for BP monitoring. The animals remained sedated with tribromoethanol for 24 hours after the surgery. When the animals came to, BP was recorded for 30 minutes using a pressure transducer (ADInstruments MLT0380) connected to the artery cannula that sent the signal to an amplifier (ADInstruments ML110) and then to a computerized data acquisition system (ADInstruments - PowerLab 8/30). After the BP measurements were recorded, the animals were sacrificed and their hearts were removed, photographed and fixed in formaldehyde (10%). The macroscopic morphometry was conducted using Image J (NIH, 2004) software as per the instructions in the online operation manual22. Next, 3mm thick cross sections of the heart were taken from two regions: one at the entry point of the inferior vena cava into the right atrium and the other at the mid third of the left ventricle. The two section fragments were then processed and embedded in paraffin. The histochemical methods used were: hematoxylineosin (HE) staining to measure left ventricle thickness, and Picro-sirius staining to quantify the cardiac fibrosis areas. After HE and Picro-sirius staining, the left ventricle thickness and myocardial fibrosis area were quantified using a video camera connected to a standard light microscope that sent the microscopy images to an image analysis system (KS 300 Kontron-Zeiss). This system is basically comprised of a standard light microscope connected to a color video camera, a high definition video monitor and a processing unit with a video card.
The left ventricle thickness of each case was estimated using four equally distributed measurements in the area visible on the video which covered the right, left, upper and lower sides of left ventricle cross section on the slide. The slide was positioned before the condenser and an x5 objective lens was used for these measurements. An average left ventricle thickness was calculated for each case using the measurements from the four distinct positions on the left ventricle cross section. Quantification of cardiac fibrosis was performed using the accumulated mean calculation method, estimating the number of representative fields. Eight fields were quantified for each case. Fibrosis was calculated using the arithmetic mean of the eight fields. A standard light microscope and polarizer filter were used for visualization of the Pico-sirius stained fibrosis areas; these areas were quantified morphometrically.
Statistical analysis - The results were presented as MEAN ± SEM. A computer spreadsheet was prepared for statistical analysis of the information using the software program Sigma-Stat, version 2.03. Analysis of normal homoscedastic variables was conducted using ANOVA variance analysis with the Tukey post-hoc test. Statistically significant difference was established as p less than 5% (p<0.05).
Results
Table 1 shows that the L-NAME and L-NAME TRAINED groups were hypertensive in relation to the CONTROL and TRAINED groups. Comparison of the hypertensive groups revealed that the L-NAME group was significantly more hypertensive than the L-NAME TRAINED group (p<0.05). In relation to heart rate (HR) the L-NAME group, in comparison to the other groups, was tachycardic (p<0.05). On the other hand, the TRAINED and L-NAME TRAINED groups were bradycardic in relation to the CONTROL group (p<0.001). When compared to the TRAINED group, the L-NAME TRAINED group presented more intense bradycardia (p<0.05). Figure 1 shows the morphometric cardiac parameters for all study groups. Figure 1A represents the heart weight index (mg/g) for all groups and shows increases for the TRAINED (3.57 ± 0.04 mg/g) and L-NAME TRAINED (3.79 ± 0.08 mg/g) groups in relation to the CONTROL (3.2 ± 0.04 mg/g) and L-NAME (3.29 ± 0.04 mg/g) groups (p<0.05). Figure 1A also shows that the L-NAME TRAINED group presented higher heart weight indexes than the TRAINED group (p<0.05).
Discussion
The NO synthesis blockade using L-NAME in the sedentary rats caused hypertension, however, no visible morphometric alterations were observed in the cardiac tissue when compared with the sedentary normotensive group. On the other hand, aerobic swim training alleviated the L-NAME induced hypertension during the last week of exercise. Additionally, the NO synthesis blockade during seven days in rats submitted to aerobic exercise induced significant morphometric cardiac alterations.
According to all indications, attenuation of hypertension in the TRAINED AND L-NAME TRAINED groups, demonstrated the preventative effect of physical exercise during the progression of this experimental hypertension model. Nevertheless, in this study we were not able to identify the mechanism responsible for hypertension attenuation. On the other hand, various studies have demonstrated the importance of physical activity as a preventative measure and treatment for hypertension indicate some mechanisms that could contribute to BP attenuation for both experimental animals and humans. Among the mechanisms indicated, it appears that the reduction of cardiac and vascular sympathetic activity associated with diminished peripheral vascular resistance is involved18,23,24. Other studies also demonstrated that physical training promotes BP reduction through the drop in cardiac output, reduction of serum catecholamine levels and oxidative stress25-27. Improvement in endothelial vasodilatation function and alterations in the renin-angiotensin-aldosterone system also appear to be involved28-32.
In relation to the absence of cardiac hypertrophy in the sedentary rats submitted to experimental L-NAME induced hypertension, it is possible that NO synthesis inhibition over a period of seven days was not sufficient to cause hypertrophy. Various authors that have demonstrated cardiac hypertrophy induced by chronic NO synthesis blockade, used treatment timeframes of more than four weeks even though the daily L-NAME doses were lower than those used in our study3,33. On the other hand, the purpose of the present study was to investigate blood pressure trends and cardiac morphometric alterations in animals submitted to an established aerobic exercise protocol with the main objective of evaluating the ability of the established training program to preventively attenuate these alterations. The relevant fact of this study was the observation that the hypertensive sedentary animals did not present cardiac hypertrophy. whereas the animals submitted to the 8-week aerobic exercise program did. It appears that the two groups presented excentric hypertrophysince, for all study groups, no difference was observed in ventricle thicknesses which coul be associated with an increase in the macro area of the trained groups. This type of cardiac hypertrophy, called physiologic, is a result of remodeling induced by exercise and in particular aerobic exercise. It is caused by mechanical adaptations such as ventricle wall dilation due to the volume overload induced by the increased venous return. This dilation causes the cardiomyocytes to add sarcomeres in series which increases contractile strength34. On the other hand, concentric cardiac hypertrophy occurs in pathological conditions such as hypertension. It is caused by the addition of sarcomeres in parallel, however, with a large proliferation of fibroblasts, which increases the extracellular matrix (fibrosis), apoptosis and necrotic tissue. As a consequence, this series of events leads to heart failure34,35.
Even though concentric hypertrophy, characteristic of the hypertension, was not observed in this study, the L-NAME TRAINED animals presented significant increases of fibrosis, demonstrating the important role of NO in exercise induced cardiac remodeling. The TRAINED group also presented increased cardiac fibrosis, although to a lesser degree, however, it is well known that this is common and is a result of physiological hypertrophy due to the proportional increase of all tissue components, or in other words, contractile tissue (cardiomyocyte) and non-contractile tissue (connective tissue and extracellular matrix)35.
The considerable increase of fibrosis presented by the L-NAME TRAINED group seems to reflect the important role of NO in exercise induced cardiac remodeling. Some authors have demonstrated that hypertension induced pathological hypertrophy is a result of the action of humoral factors, mainly angiotensin II36. The intracellular cascade of events induced by these factors appears to promote, among other effects, increased cardiac fibrosis, considerable cardiomyocyte apoptosis elevation, contractile depression and cardiac dysfunction in response to b-stimulation which gradually progresses to lethal cardiomyopathy37-39. The role of NO, in these situations is to prevent or attenuate myocardial hypertrophy, cardiac fibroblast proliferation and cardiomyocyte apoptosis. This effect appears to depend on the production and release of bradykinin via the AT2R angiotensin II receptor, leading to the formation of NO (bradykinin/NO-dependent), offsetting the pathological hypertrophy induced by the activation of the AT1 receptor11. On the other hand, even though the mechanism is unclear, NO is also involved in the angiogenesis process, which explains the increased cardiac fibrosis observed in the L-NAME TRAINED group40. Generally speaking, exercise induces angiogenesis in various tissues when they are stimulated. This is also true for the heart muscle and the increased demand induced by exercise promotes increased intramyocardial angiogenesis10. Since the NO production was blocked by L-NAME, it is possible that the fibrosis was caused by the reduction in the angiogenesis process as a result of the diminished cardiomyocyte vascularization. Nevertheless, further studies are required to clarify the NO mechanisms in the angiogenesis process in relation to pathological conditions and exercise.
In conclusion, the short term NO synthesis blockade with L-NAME caused severe hypertension but did not promote cardiac hypertrophy in the sedentary animals. In the rats submitted to the established aerobic exercise program, the inhibition of the NO synthesis resulted in a lower BP elevation; however, it was associated with a significant increase of cardiac hypertrophy and myocardial fibrosis. These findings, suggest the potentially significant role of NO in the cardiac remodeling observed in the physiological cardiac hypertrophy resulting from physical exercise.
Potential Conflict of Interest
No potential conflict of interest relevant to this article was reported.
Sources of Funding
This study was funded by FAPESP and FAEPA.
References
Manuscript received December 8, 2006; revised manuscript received January 23, 2007; accepted February 26, 2007
- 1. Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation in arterial smooth muscle by acetylcholine. Nature. 1980; 288: 373-6.
- 2. Bartunek J, Weinberg EO, Tajima M, Rohrbach S, Katz SE, Douglas PS, et al. Chronic NG-nitro-L-arginine methyl ester-induced hypertension. Circulation. 2000; 101: 423-9.
- 3. Hu CT, Chang HR, Hsu YH, Liu CJ, Chen HI. Ventricular hypertrophy and arterial hemodynamics following deprivation of nitric oxide in rats. Life Sci. 2005; 78: 164-73.
- 4. Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med. 1993; 329: 2002-12.
- 5. Matsuoka H, Nakata M, Kohno K, Koga Y, Nomura G, Toshima H, et al. Chronic L-arginine administration attenuates cardiac hypertrophy in spontaneously hypertensive rats. Hypertension. 1996; 27: 14-8.
- 6. Pereira LM, Mandarim-De-Lacerda CA. Stereology of cardiac hypertrophy induced by NO blockade in rats treated with enalapril and verapamil. Anal Quantitat Cytol Histol. 2001; 23: 330-8.
- 7. Akuzawa N, Nakamura T, Kurashina T, Saito Y, Hoshino J. Antihypertensive agents prevent nephrosclerosis and left ventricular hypertrophy induced in rats by prolonged inhibition of nitric oxide synthesis. Am J Hypertens. 1998; 11 (6 Pt 1): 697-707.
- 8. K-Laflamme A, Foucart S, Moreau P, Lambert C, Cardinal R, de Champlain J. Sympathetic functions in NG-nitro-L-arginine methyl ester-induced hypertension: modulation by the renin-angiotensin system. J Hypertens. 1998; 16: 63-76.
- 9. Luvara G, Pueyo ME, Phillippe M, Mondet C, Savoie F, Henrion D, et al. Chronic blocked of NO synthase activity induces a proinflammatory phenotype in the arterial wall: prevention by angiotensin II antagonism. Arterioscler Thromb Vasc Biol. 1998; 18: 1408-16.
- 10. Zorzi RLA, Pereira LMM, Mandarim-de-Lacerda CA. Beneficial effect of enalapril in spontaneously hypertensive rats cardiac remodeling with nitric oxide synthesis blockade. J Cell Mol Med. 2002; 6 (4): 599-608.
- 11. Kurisu S, Ozono R, Oshima T, Kambe M, Ishida T, Sugino H, et al. Cardiac angiotensin II type 2 receptor activates the kinin/NO system and inhibits fibrosis. Hypertension. 2003; 41 (1): 99-107.
- 12. Lantelme P, Lo M, Sassard J. Decreased cardiac baroreflex sensitivity is not due to cardiac hypertrophy in NG-nitro-L-arginine methyl ester-induced hypertension. J Hypertens. 1994; 12: 791-5.
- 13. Pechanova O, Bertanova I, Babal P. Structural alterations in the heart after long-term L-NAME and D-NAME treatment. Gen Physiol Biophys. 1999; 1: 6-9.
- 14. de Oliveira CF, Cintra KA, Teixeira SA, De Luca IMS, Antunes E, De Nucci G. Development of cardiomyocyte hypotrophy in rats under prolonged treatment with a low dose of a nitric oxide synthesis inhibitor. Eur J Pharmacol. 2000; 391: 121-6.
- 15. Simko F, Luptak I, Matuskova J, Krajcirovicova K, Sumbalova Z, Kucharska J, et al. L-arginine fails to protect against myocardial remodeling in L-NAME-induced hypertension. Eur J Clin Invest. 2005; 35: 362-8.
- 16. Arroll B, Beaglehole R. Does physical activity lower blood pressure: a critical review of the clinical trials. J Clin Epidemiol. 1992; 45 (5): 439-47.
- 17. Arakawa K. Antihypertensive mechanism of exercise. J Hypertens. 1993; 11 (3): 223-9.
- 18. Carter JB, Banister EW, Blaber AP. Effect of endurance exercise on autonomic control of heart rate. Sports Med. 2003; 33 (1): 33-46.
- 19. Perrault H, Turcotte RA. Exercise-induced cardiac hypertrophy. Fact or fallacy? Sports Med. 1994; 17 (5): 288-308.
- 20. Fagard RH. Effect of training on left ventricular structure and functioning of the normotensive and the hypertensive subject. Blood Press Monit. 1997; 2 (5): 241-5.
- 21. Ghorayeb N, Batlouni M, Pinto IM, Dioguardi GS. Left ventricular hypertrophy of athletes: adaptative physiologic response of the heart. Arq Bras Cardiol. 2005; 85 (3): 191-7.
- 22. Abdalla GK, Silveira MR, Lazo JE. Guia rápido de utilização do software de morfometria Image J. [acessado em 2006 abr 14]. Disponível em: <http://www.fmtm.br/instpub/fmtm/discbiologiacelular/guiarapidodeutilizacaodoimagej.pdf>.
- 23. Goldsmith RL, Bloomfield DM, Rosenwinkel ET. Exercise and autonomic function. Coron Artery Dis. 2000; 11 (2): 129-35.
- 24. Yamamoto K, Miyachi M, Saitoh T, Yoshioka A, Onodera S. Effects of endurance training on resting and post-exercise cardiac autonomic control. Med Sci Sports Exerc. 2001; 33 (9): 1496-502.
- 25. Chen HI, Chiang IP. Chronic exercise decreases adrenergic agonist-induced vasoconstriction in spontaneously hypertensive rats. Am J Physiol. 1996; 271 (3 Pt2): H977-H983.
- 26. Véras-Silva AS, Mattos KC, Gava NS, Brum PC, Negrão CE, Krieger EM. Low-intensity exercise training decreases cardiac output and hypertension in spontaneously hypertensive rats. Am J Physiol. 1997; 273 (42): H2627-H2631.
- 27. Oztasan N, Taysi S, Gumustekin K, Altinkaynak K, Aktas O, Timur H, et al. Endurance training attenuates exercise-induced oxidative stress in erythrocytes in rat. Eur J Appl Physiol. 2004; 91 (5-6): 622-7.
- 28. Yen MH, Yang JH, Sheu JR, Lee YM, Ding YA. Chronic exercise enhances endothelium-mediated dilation in spontaneously hypertensive rats. Life Sci. 1995; 57 (24): 2205-13.
- 29. Maiorana A, O'Driscoll G, Taylor R, Green D. Exercise and the nitric oxide vasodilator system. Sports Med. 2003; 33 (14): 1013-35.
- 30. Stewart KJ. Exercise training and the cardiovascular consequences of type 2 diabetes and hypertension: plausible mechanisms for improving cardiovascular health. JAMA. 2002; 288 (13): 1622-31.
- 31. Blanchard BE, Tsongalis GJ, Guidry MA, Labelle LA, Poulin M, Taylor AL, et al. RAAS polymorphisms alter the acute blood pressure response to aerobic exercise among men with hypertension. Eur J Appl Physiol. 2006; 97 (1): 26-33.
- 32. Ashley EA, Kardos A, Jack ES, Habenbacher W, Wheeler M, Kim YM, et al. Angiotensin-converting enzyme genotype predicts cardiac and autonomic responses to prolonged exercise. J Am Coll Cardiol. 2006; 48 (3): 523-31.
- 33. Raij L. Nitric oxide in hypertension: relationship with renal injury and left ventricular hypertrophy. Hypertension. 1998; 31 (1 Pt 2): 189-93.
- 34. Wakatsuki T, Schlessinger J, Elson EL. The biochemical response of the heart to hypertension and exercise. Trends Biochem Sci. 2004; 29 (11): 609-17.
- 35. Weber KT. Targeting pathological remodeling: concepts of cardioprotection and reparation. Circulation. 2000; 102 (12): 1342-5.
- 36. Weber KT, Brilla CG, Janicki JS. Signals for the remodeling of the cardiac interstitium in systemic hypertension. J Cardiovasc Pharmacol. 1991; 17 (Suppl 2): S14-9.
- 37. Adams JW, Sakata Y, Davis MG, Sah VP, Wang Y, Liggett SB, et al. Enhanced Galphaq signaling: a common pathway mediates cardiac hypertrophy and apoptotic heart failure. Proc Natl Acad Sci USA. 1998; 95 (17): 10140-5.
- 38. Wettschureck N, Rutten H, Zywietz A, Gehring D, Wilkie TM, Chen J, et al. Absence of pressure overload induced myocardial hypertrophy after conditional inactivation of Galphaq/Galpha11 in cardiomyocytes. Nat Med. 2001; 7 (11): 1236-40.
- 39. Liang Q, Molkentin JD. Redefining the roles of p38 and JNK signaling in cardiac hypertrophy: dichotomy between cultured myocytes and animal models. J Mol Cell Cardiol. 2003; 35 (12): 1385-94.
- 40. Sladek T, Gerova M, Znojil V, Devat L. Morphometric characteristics of cardiac hypertrophy induced by long-term inhibition of NO synthase. Physiol Res. 1996;45 (4): 335-8.
Publication Dates
-
Publication in this collection
05 Sept 2007 -
Date of issue
Aug 2007
History
-
Accepted
26 Feb 2007 -
Reviewed
23 Jan 2007 -
Received
08 Dec 2006