1. Introduction
Significant advances in understanding cardiac amyloidosis (CA) have been made in recent years, leading to a thorough reformulation of its clinical significance. In addition to convincing evidence that CA is a relatively common cause of heart failure with preserved ejection fraction (HFpEF), we are witnessing the emergence of specific therapies that can change the course of the disease and prolong the survival of affected patients.
In parallel, relevant advances in cardiovascular imaging techniques have greatly contributed to earlier and more accurate identification of the disease. Cardiac scintigraphy with bone-seeking radiotracers has allowed non-invasive diagnosis of transthyretin (TTR) cardiac amyloidosis (ATTR-CA), eliminating the need for endomyocardial biopsy, which has greatly simplified the diagnostic flow.
Thus, we aim to present the most current recommendations for the diagnosis, prognostic staging, and treatment of CA based on a critical review of the current scientific evidence.
In this position paper, the recommendations and levels of evidence are classified according to the following parameters:
2. General Concepts
Systemic amyloidosis is caused by tissue deposition of fibrillar and insoluble protein aggregates in different organs, including the heart, which leads to organ dysfunction. 11. Sipe JD, Cohen AS. Review: history of the amyloid fibril. J Struct Biol. 2000;130(2-3):88-98. More than 30 types of amyloidogenic proteins have been described, 22. Benson MD, Buxbaum JN, Eisenberg DS, Merlini G, Saraiva MJM, Sekijima Y, et al. Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid. 2018;25(4):215-9. and five of them can affect the heart (immunoglobulin heavy and light chain (AL), TTR, amyloid A, and apolipoprotein A1), with the AL and ATTR types accounting for 95% of all CA cases, both in its wild type (ATTRwt) and hereditary/variant (ATTRv) forms. 33. Falk RH, Alexander KM, Liao R, Dorbala S. AL (Light-Chain) Cardiac Amyloidosis: A Review of Diagnosis and Therapy. J Am Coll Cardiol. 2016;68(12):1323-41. – 77. Gertz MA, Dispenzieri A, Sher T. Pathophysiology and treatment of cardiac amyloidosis. Nat Rev Cardiol. 2015;12(2):91-102.
TTR is a protein composed of four monomers, which circulate as a tetramer. 88. Koike H, Katsuno M. Ultrastructure in Transthyretin Amyloidosis: From Pathophysiology to Therapeutic Insights. Biomedicines. 2019;7(1):11. It acts as a thyroxine and retinol (vitamin A) transporter under physiological conditions. The limiting step in the amyloid fibril formation rate is the tetramer’s dissociation into monomers, which may involve proteolysis. Subsequently, partial denaturation of the monomer allows for incorrect assembly in various aggregate structures. Amyloidosis through mutation of the TTR gene (ATTRv) has an autosomal dominant character. This gene is located on chromosome 18, and more than 140 mutations of it have been described. By producing less stable TTR, aggressive and systemic amyloid deposition occurs. 99. Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019;73(22):2872-91.
In ATTRwt, the amino acid sequence is normal and the process by which the wild-type protein becomes unstable and aggregates into amyloid fibrils is not completely clear. However, aging appears to be involved in its pathophysiology. 88. Koike H, Katsuno M. Ultrastructure in Transthyretin Amyloidosis: From Pathophysiology to Therapeutic Insights. Biomedicines. 2019;7(1):11. , 99. Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019;73(22):2872-91.
In the AL form, amyloidogenic light chains originate from plasma cells or, less frequently, abnormal B lymphocytes. Thus, it is a clonal and neoplastic hematologic disease. In the heart, the deposition of amyloid fibrils causes structural damage by increasing cardiac and vascular rigidity, impairing cardiac contraction and relaxation and creating conduction disturbances. In parallel, circulating light chains are directly toxic to the myocardium through lysosomal dysfunction, defective autophagy, production of reactive oxygen species, cell and mitochondrial dysfunction, alterations in cardiomyocyte calcium homeostasis and, finally, cell death. 1010. Mankad AK, Sesay I, Shah KB. Light-chain cardiac amyloidosis. Curr Probl Cancer. 2017;41(2):144-56.
Figure 1 represents the physiopathogenesis of transthyretin (TTR) and light chain (AL) cardiac amyloidosis.

Different subtypes of amyloidosis can lead to overlapping clinical manifestations and, once diagnosed, it is essential to correctly characterize the precursor protein to determine a specific treatment. 1111. Wechalekar AD, Gillmore JD, Hawkins PN. Systemic amyloidosis. Lancet. 2016;387(10038):2641-54. – 1313. Muchtar E, Dispenzieri A, Magen H, Grogan M, Mauermann M, McPhail ED, et al. Systemic amyloidosis from A (AA) to T (ATTR): a review. J Intern Med. 2021;289(3):268-92.
Depending on the affected organs and degree of dysfunction, a wide spectrum of clinical manifestations can be observed, with a progressive and potentially fatal evolution. The main organs affected by systemic amyloidosis are the heart, kidneys, eyes, central and peripheral nervous system, and liver. Nonspecific clinical manifestations are frequently observed and include fatigue, weight loss, peripheral edema and orthostatic hypotension. For this reason, late diagnosis is common. Thus, knowledge of the disease and a high degree of clinical suspicion are necessary to complete the diagnosis.
In ATTRv, depending on the mutation, the clinical picture is dominated by neuropathy or heart disease. In ATTRwt, heart disease is the main clinical manifestation, occurring mainly in elderly men who develop HFpEF without previously known risk factors.
Some extracardiac alterations may precede CA by several years, especially bilateral carpal tunnel syndrome and spontaneous rupture of the biceps tendon. It is essential to recognize such signs as part of the clinical picture of amyloidosis, which could lead to earlier diagnosis and specific treatments that could prevent the progression of heart disease. 1414. Milandri A, Farioli A, Gagliardi C, Longhi S, Salvi F, Curti S, et al. Carpal tunnel syndrome in cardiac amyloidosis: implications for early diagnosis and prognostic role across the spectrum of aetiologies. Eur J Heart Fail. 2020;22(3):507-15. , 1515. Witteles RM, Bokhari S, Damy T, Elliott PM, Falk RH, Fine NM, et al. Screening for Transthyretin Amyloid Cardiomyopathy in Everyday Practice. JACC Heart Fail. 2019;7(8):709-16.
There is a higher incidence of ATTRwt in older patients, usually those over 70 years of age. However, since the clinical manifestations of ATTRv also usually occur in older adults, age should not be taken into account when differentiating between the two forms of ATTR. Regarding sex, there is a strong predominance (80% to 90%) of ATTRwt in men.
Regarding ATTRv, V30M is the most widespread mutation worldwide, being endemic in Portugal, Sweden, and Japan. It is probably the most common form in Brazil. Another common mutation is V122I, which is present in 3.4% of African-Americans and is related to the development of heart disease in patients over 60 years of age. 77. Gertz MA, Dispenzieri A, Sher T. Pathophysiology and treatment of cardiac amyloidosis. Nat Rev Cardiol. 2015;12(2):91-102. , 99. Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019;73(22):2872-91. , 1616. Jacobson D, Tagoe C, Schwartzbard A, Shah A, Koziol J, Buxbaum J. Relation of clinical, echocardiographic and electrocardiographic features of cardiac amyloidosis to the presence of the transthyretin V122I allele in older African-American men. Am J Cardiol. 2011;108(3):440-4. , 1717. Parman Y, Adams D, Obici L, Galan L, Guergueltcheva V, Suhr OB, et al. Sixty years of transthyretin familial amyloid polyneuropathy (TTR-FAP) in Europe: where are we now? A European network approach to defining the epidemiology and management patterns for TTR-FAP. Curr Opin Neurol. 2016;29 Suppl 1:S3-S13.
Table 1 summarizes the demographic and clinical characteristics of the AL, ATTRv, and ATTRwt subtypes.
Demographic and clinical presentation aspects, comparative between the forms AL, ATTRv and ATTRwt
The incidence of AL-CA is 6-10/million people/year and it is considered the main cause of CA. 1818. Kyle RA, Linos A, Beard CM, Linke RP, Gertz MA, O’Fallon WM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood. 1992;79(7):1817-22. With the development of less invasive CA diagnosis techniques for ATTR 1919. Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, et al. Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation. 2016;133(24):2404-12. and the prospect of more effective treatments, the number of diagnosed cases, especially of ATTRwt, has been increasing significantly. 2020. Lane T, Fontana M, Martinez-Naharro A, Quarta CC, Whelan CJ, Petrie A, et al. Natural History, Quality of Life, and Outcome in Cardiac Transthyretin Amyloidosis. Circulation. 2019;140(1):16-26. AL is currently the most common cause of CA. Studies have found ATTR deposits in the heart of 13% of HFpEF patients 2121. Gonzalez-Lopez E, Gallego-Delgado M, Guzzo-Merello G, de Haro-Del Moral FJ, Cobo-Marcos M, Robles C, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36(38):2585-94. and in 25% of autopsied older adults, 2222. Cornwell GG, 3rd, Murdoch WL, Kyle RA, Westermark P, Pitkanen P. Frequency and distribution of senile cardiovascular amyloid. A clinicopathologic correlation. Am J Med. 1983;75(4):618-23. , 2323. Tanskanen M, Peuralinna T, Polvikoski T, Notkola IL, Sulkava R, Hardy J, et al. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: a population-based autopsy study. Ann Med. 2008;40(3):232-9. mainly males. 2424. Cruz MW, Pinto MV, Pinto LF, Gervais R, Dias M, Perez C, et al. Baseline disease characteristics in Brazilian patients enrolled in Transthyretin Amyloidosis Outcome Survey (THAOS). Arq Neuropsiquiatr. 2019;77(2):96-100.
Thus, CA could be considered an underdiagnosed condition, rather than a rare disease. Recent data from the USA indicate a progressive increase in CA prevalence (from 18 to 55.2/100,000 person-years), 2525. Gilstrap LG, Dominici F, Wang Y, El-Sady MS, Singh A, Di Carli MF, et al. Epidemiology of Cardiac Amyloidosis-Associated Heart Failure Hospitalizations Among Fee-for-Service Medicare Beneficiaries in the United States. Circ Heart Fail. 2019;12(6):e005407. which supports this idea. The patient’s journey to diagnosis is long; it is estimated that there is a delay of more than 2 years from symptom onset to diagnosis, with the involvement of an average of five different professionals. 2626. Lousada I, Comenzo RL, Landau H, Guthrie S, Merlini G. Light Chain Amyloidosis: Patient Experience Survey from the Amyloidosis Research Consortium. Adv Ther. 2015;32(10):920-8. Thus, it is essential to disseminate knowledge about CA; clinicians and cardiologists must give greater consideration to this entity, aiming at earlier diagnosis and adequate therapeutic guidance, thus improving patient prognosis and survival.
Regarding prognosis, AL affects multiple organs and is more aggressive than other subtypes. Late diagnosis is associated with high early mortality in the first 6 to 12 months due to advanced heart disease complications. 77. Gertz MA, Dispenzieri A, Sher T. Pathophysiology and treatment of cardiac amyloidosis. Nat Rev Cardiol. 2015;12(2):91-102. , 88. Koike H, Katsuno M. Ultrastructure in Transthyretin Amyloidosis: From Pathophysiology to Therapeutic Insights. Biomedicines. 2019;7(1):11. The median estimated survival for ATTRwt is 3.6 years, while the prognosis for ATTRv depends on the mutation. In cases of neurological phenotype, the progression of neuropathy leads to sensorimotor disability, although mortality is more associated with cardiac impairment. 1212. Gertz MA, Dispenzieri A. Systemic Amyloidosis Recognition, Prognosis, and Therapy: A Systematic Review. JAMA. 2020;324(1):79-89. , 1313. Muchtar E, Dispenzieri A, Magen H, Grogan M, Mauermann M, McPhail ED, et al. Systemic amyloidosis from A (AA) to T (ATTR): a review. J Intern Med. 2021;289(3):268-92.
3. Neurological Manifestations
Mutations in the TTR gene are associated with a wide variety of clinical manifestations, which reflect the deposition of the variant protein in different types of tissues. Cardiac and peripheral nervous system tissue are the most frequent; the former is particularly associated with the V122I mutation, and the latter with the V30M mutation. 2727. Adams D, Polydefkis M, Gonzalez-Duarte A, Wixner J, Kristen AV, Schmidt HH, et al. Long-term safety and efficacy of patisiran for hereditary transthyretin-mediated amyloidosis with polyneuropathy: 12-month results of an open-label extension study. Lancet Neurol. 2021;20(1):49-59.
In this chapter, we will describe the main neurological manifestations suggestive of ATTRv.
Neurological manifestations in ATTRv can be divided into peripheral neuropathy, ie, late manifestations of central nervous system involvement linked to amyloid angiopathy, and central nervous system manifestations associated with oculoleptomeningeal infiltration.
3.1. Peripheral Neuropathy
The involvement of the peripheral nerves in ATTRv is typically mixed sensory and motor length-dependent axonal neuropathy, ie, it initially affects more distal segments of the limbs, especially the lower ones, progressing to the proximal segments and upper limbs. 2828. Plante-Bordeneuve V, Said G. Familial amyloid polyneuropathy. Lancet Neurol. 2011;10(12):1086-97. , 2929. Sekijima Y. Transthyretin (ATTR) amyloidosis: clinical spectrum, molecular pathogenesis and disease-modifying treatments. J Neurol Neurosurg Psychiatry. 2015;86(9):1036-43.
In its early onset form (< 50 years of age), it is usually associated with the ATTRv V30M (V50M) mutation, and thin fibers with little or no myelination (autonomic, heat, cold, and pain) are initially affected. This is followed, in the degree that the disease progresses, by thick fibers, which are very myelinated and responsible for vibratory, postural-kinetic, and motor sensitivity. The initial symptoms are erectile dysfunction, early satiety, nausea, vomiting, diarrhea, constipation, alternating diarrhea with constipation, orthostatic hypotension, syncope, arrhythmias, altered atrioventricular conduction, dry eye, urinary retention or incontinence, neuropathic pain, lost sensitivity to heat and cold, and significant weight loss. The initial phase can include painless lesions, plantar perforating ulcers and their repercussions, such as localized infections, cellulitis, osteomyelitis, and even septicemia. After a few years, gait instability and muscle atrophy appear, always evolving from distal to proximal segments. 2828. Plante-Bordeneuve V, Said G. Familial amyloid polyneuropathy. Lancet Neurol. 2011;10(12):1086-97. , 2929. Sekijima Y. Transthyretin (ATTR) amyloidosis: clinical spectrum, molecular pathogenesis and disease-modifying treatments. J Neurol Neurosurg Psychiatry. 2015;86(9):1036-43.
In its late forms, neuropathy compromises all types of fibers, although dysautonomia is not as important, at least in the initial phase. These forms can be associated with V30M or a number of other mutations, and the evolution is usually more aggressive. In a Brazilian study, 26% of ATTRv patients with V30M had late onset. 3030. Pinto MV, Pinto LF, Dias M, Rosa RS, Mundayat R, Pedrosa RC, et al. Late-onset hereditary ATTR V30M amyloidosis with polyneuropathy: Characterization of Brazilian subjects from the THAOS registry. J Neurol Sci. 2019;403:1-6.
Bilateral carpal tunnel syndrome is a frequent manifestation in ATTRv and may be the initial manifestation. Although it can be associated with any mutation, it is particularly important in some of them, including TTR V122I. This mutation appears to be frequent in Brazil and is associated with heart disease, which it can precede by several years. 3131. Rapezzi C, Quarta CC, Riva L, Longhi S, Gallelli I, Lorenzini M, et al. Transthyretin-related amyloidoses and the heart: a clinical overview. Nat Rev Cardiol. 2010;7(7):398-408.
3.2. Central Nervous System Manifestations
Prolonging survival, which was initially associated with liver transplantation and is currently possible with new drugs, has enabled the appearance of previously uncommon manifestations. The long-term production of TTR by the choroid plexus (only 2% of the total) is associated with both amyloid angiopathy and meningeal infiltration. Amyloid angiopathy manifests as focal stroke-like, transient ischemic attack-like, or aura-like episodes, as well as an irritative epileptic type. In more severe cases, ischemia or even intracranial hemorrhaging can occur. The following neurological manifestations stand out: hearing impairment, migraine, dementia, cerebellar syndrome, myelopathy, and radiculopathy.
Some rare mutations have a predilection for oculocerebral involvement and lead to oculoleptomeningeal amyloidosis (ATTR Y69H: oculocerebral; Val30Gly: oculoleptomeningeal). 3232. Kapoor M, Rossor AM, Laura M, Reilly MM. Clinical Presentation, Diagnosis and Treatment of TTR Amyloidosis. J Neuromuscul Dis. 2019;6(2):189-99.
Stages of neuropathy: The Coutinho stages for ATTRv are classified according to polyneuropathy. In stage 1 sensorimotor polyneuropathy affects gait, but walking support is not necessary. In stage 2, one or more support devices are needed for walking. In stage 3, the patient is confined to bed or a wheelchair.
3.3. Genetic Analysis
ATTRv is a disease of autosomal dominant inheritance but variable penetrance. It is mutation- and age-dependent, as well as regionally influenced. 3333. Lahuerta-Pueyo CL, Tubar Arregui MA, Gracia- Gutierrez AG, Buena Juana E, Guillén SM. Estimating the prevalence of allelic variants in the transthyretin gene by analysing large-scale sequencing data. Eur J Hum Genet. 2019;27(5):783-91. The V30Met mutation, for example, has an 80% and 91% penetrance in Portugal in individuals aged 50 and 70 years, respectively. On the other hand, in Sweden, the values are 11% and 36%, respectively, for the same age groups.
At least 140 different mutations have been described to date, but not all are pathogenic. Some polymorphisms are well-defined, while the significance of others is still indeterminate. 3434. Park GY, Jamerlan A, Shim KH, An SSA. Diagnostic and Treatment Approaches Involving Transthyretin in Amyloidogenic Diseases. Int J Mol Sci. 2019;20(12):2982. This suggests that the diagnostic test must always include complete TTR gene sequencing and that care must be taken regarding the interpretation of rare or undescribed variants. Among pathogenic mutations, some predominantly cause neuropathy (V30Met), heart disease (V122I), or both (Leu58Hist) ( Figure 2 ). 3535. Maurer MS, Bokhari S, Damy T, Dorbala S, Drachman BM, Fontana M, et al. Expert Consensus Recommendations for the Suspicion and Diagnosis of Transthyretin Cardiac Amyloidosis. Circ Heart Fail. 2019;12(9):e006075. It should be considered, however, that the genotypic/phenotypic correlation is not strict.

Distribution of hereditary transthyretin amyloidosis mutations associated with neurological, cardiological, and mixed phenotypes.
A special topic among genetic aspects is pre-symptomatic testing, ie, testing the relatives of individuals known to be affected. Unlike diagnostic testing, it must be performed by specialized personnel and there must be a support team, including a psychologist. It must include a preparation phase, pre-diagnosis, genetic testing and a post-result support phase. Such testing should not be performed on children and should only be applied to individuals who expressly state that it is their wish and are considered psychologically prepared for the results. 3636. Obici L, Kuks JB, Buades J, Adams D, Suhr OB, Coelho T, et al. Recommendations for presymptomatic genetic testing and management of individuals at risk for hereditary transthyretin amyloidosis. Curr Opin Neurol. 2016;29 Suppl 1:S27-35.
4. Cardiovascular Manifestations
CA progresses as the cardiac extracellular matrix is infiltrated by amyloid fibrils, resulting in a progressive increase in the thickness of the ventricular wall and a marked increase in chamber stiffness, resulting in impaired diastolic function, which leads to heart failure with restrictive physiology. 3737. Maurer MS, Elliott P, Comenzo R, Semigran M, Rapezzi C. Addressing Common Questions Encountered in the Diagnosis and Management of Cardiac Amyloidosis. Circulation. 2017;135(14):1357-77. Systolic function is also compromised and is usually indicated by abnormal longitudinal tension despite normal ejection fraction, which may be preserved until the late stages of the disease. 3838. Castano A, Drachman BM, Judge D, Maurer MS. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20(2):163-78. – 4040. Mohty D, Damy T, Cosnay P, Echahidi N, Casset-Senon D, Virot P, et al. Cardiac amyloidosis: updates in diagnosis and management. Arch Cardiovasc Dis. 2013;106(10):528-40. Atrial amyloid infiltration is frequent, leading to contractile dysfunction. Deposits can also occur in the heart valves, usually without causing major dysfunctions, as well as in the perivascular region. 33. Falk RH, Alexander KM, Liao R, Dorbala S. AL (Light-Chain) Cardiac Amyloidosis: A Review of Diagnosis and Therapy. J Am Coll Cardiol. 2016;68(12):1323-41.
Thus, the most frequent clinical manifestation is heart failure syndrome, most commonly with preserved ejection fraction (HFpEF), although a drop in ejection fraction can occur in more advanced stages of the disease. The clinical syndrome may present predominant left HF symptoms with pulmonary congestion (dyspnea, orthopnea, paroxysmal nocturnal dyspnea), right HF symptoms (edema, ascites, hepatomegaly, increased abdominal volume, early satiety, severe fatigue), or both sets of symptoms. Cardiac amyloidosis should be considered in the differential diagnosis of HFpEF etiology in older men, 4141. Mohammed SF, Mirzoyev SA, Edwards WD, Dogan A, Grogan DR, Dunlay SM, et al. Left ventricular amyloid deposition in patients with heart failure and preserved ejection fraction. JACC Heart Fail. 2014;2(2):113-22. particularly when there is no apparent history of systemic arterial hypertension or the thickness of the interventricular septum increases ≥ 12 mm, which raises the possibility of infiltrative cardiomyopathy. 2121. Gonzalez-Lopez E, Gallego-Delgado M, Guzzo-Merello G, de Haro-Del Moral FJ, Cobo-Marcos M, Robles C, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36(38):2585-94.
Syncope and orthostatic hypotension are common symptoms and indicate the presence of dysautonomia. One typical clinical aspect that may raise the suspicion of amyloidosis is the need for dose reduction or discontinuation of antihypertensive drugs in patients previously diagnosed with systemic arterial hypertension, especially beta-blockers and angiotensin-converting enzyme inhibitors/angiotensin receptor blockers. 1515. Witteles RM, Bokhari S, Damy T, Elliott PM, Falk RH, Fine NM, et al. Screening for Transthyretin Amyloid Cardiomyopathy in Everyday Practice. JACC Heart Fail. 2019;7(8):709-16.
Amyloid infiltration can also occur, causing cardiac conduction system disease from the early stages, with variable degrees of atrioventricular block, which results in high-risk bradycardia in some cases, requiring pacemaker implantation. Another important change is the hardening of the atrial walls, which leads to high rates of atrial arrhythmias, including atrial fibrillation, as well as atrial thrombi, with cardioembolic stroke being a common clinical manifestation, even in individuals with sinus rhythm. Complex ventricular arrhythmias seem to be frequent in advanced stages of the disease, an aspect that is best documented in AL amyloidosis.
4.1. Increased Suspicion of Cardiac Amyloidosis
CA, particularly the ATTR type, is often underdiagnosed due to factors associated with the medical evaluation, in addition to the characteristics of the disease itself, including: fragmented knowledge among different specialties and subspecialties, a scarcity of centers and specialists dedicated to the management of this disease, the mistaken belief that CA is a rare and incurable disease, and the phenotypic and genotypic heterogeneity of ATTR-CA. 4242. Rapezzi C, Lorenzini M, Longhi S, Milandri A, Gagliardi C, Bartolomei I, et al. Cardiac amyloidosis: the great pretender. Heart Fail Rev. 2015;20(2):117-24. It should be pointed out that early CA diagnosis is critical, since the prognosis rapidly worsens with continued amyloid protein deposition and increasing organ dysfunction.
Thus, recognizing “red flags” can help diagnose CA in HF patients, 3535. Maurer MS, Bokhari S, Damy T, Dorbala S, Drachman BM, Fontana M, et al. Expert Consensus Recommendations for the Suspicion and Diagnosis of Transthyretin Cardiac Amyloidosis. Circ Heart Fail. 2019;12(9):e006075. , 4343. Yilmaz A, Bauersachs J, Bengel F, Buchel R, Kindermann I, Klingel K, et al. Diagnosis and treatment of cardiac amyloidosis: position statement of the German Cardiac Society (DGK). Clin Res Cardiol. 2021;110(4):479-506. the most relevant of which are summarized in Table 2 .
Clinical clues that should raise suspicion of cardiac amyloidosis in patients with manifestations of heart failure
Bilateral carpal tunnel syndrome, often one of the first indicators of ATTR-CA, is the most common non-cardiac manifestation and can precede symptoms of HF by several years. A recent study found that approximately 50% of individuals with ATTRwt had carpal tunnel syndrome 5 to 7 years prior to diagnosis. 4444. Nakagawa M, Sekijima Y, Yazaki M, Tojo K, Yoshinaga T, Doden T, et al. Carpal tunnel syndrome: a common initial symptom of systemic wild-type ATTR (ATTRwt) amyloidosis. Amyloid. 2016;23(1):58-63. Lumbar stenosis and atraumatic rupture of the biceps tendon have also been identified as clinical manifestations of extracardiac deposition in ATTRwt. Biceps tendon rupture may occur in up to 33% of ATTRwt cases. 4545. Geller HI, Singh A, Alexander KM, Mirto TM, Falk RH. Association Between Ruptured Distal Biceps Tendon and Wild-Type Transthyretin Cardiac Amyloidosis. JAMA. 2017;318(10):962-3. On the other hand, macroglossia and periorbital purpura are highly specific to AL-CA, although they occur in only 15% of cases. 4646. Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349(6):583-96. Sensorimotor polyneuropathy or dysautonomia in HF patients should raise suspicion of CA. 4747. Conceicao I, Gonzalez-Duarte A, Obici L, Schmidt HH, Simoneau D, Ong ML, et al. “Red-flag” symptom clusters in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst. 2016;21(1):5-9. , 4848. Sekijima Y, Ueda M, Koike H, Misawa S, Ishii T, Ando Y. Diagnosis and management of transthyretin familial amyloid polyneuropathy in Japan: red-flag symptom clusters and treatment algorithm. Orphanet J Rare Dis. 2018;13(1):6.
Other warning signs may emerge from typical changes in routine complementary cardiac examinations, which are covered as specific topics in this document.
It should also be pointed out that CA can often simulate other heart diseases. Amyloidosis should be considered one possible etiology for patients with a hypertrophic cardiomyopathy phenotype, particularly if it developed after 60 years of age. The asymmetrical pattern of myocardial hypertrophy in ATTR-CA patients differs from that of AL-CA patients, which is usually symmetrical. In a study that compared 263 confirmed ATTR-CA patients with 50 AL-CA patients, among the ATTR cases, asymmetric hypertrophy was present in 79%, symmetric hypertrophy in 18%, and no myocardial hypertrophy in 3%. 4949. Martinez-Naharro A, Treibel TA, Abdel-Gadir A, Bulluck H, Zumbo G, Knight DS, et al. Magnetic Resonance in Transthyretin Cardiac Amyloidosis. J Am Coll Cardiol. 2017;70(4):466-77.
Older patients with severe low-flow/low-gradient aortic stenosis may have CA in 10% to 15% of cases, with an unfavorable prognosis. 5050. Pibarot P, Lancellotti P, Narula J. Concomitant Cardiac Amyloidosis in Severe Aortic Stenosis: The Trojan Horse? J Am Coll Cardiol. 2021;77(2):140-3.
5. Additional Diagnostic Examinations
5.1. Electrocardiogram
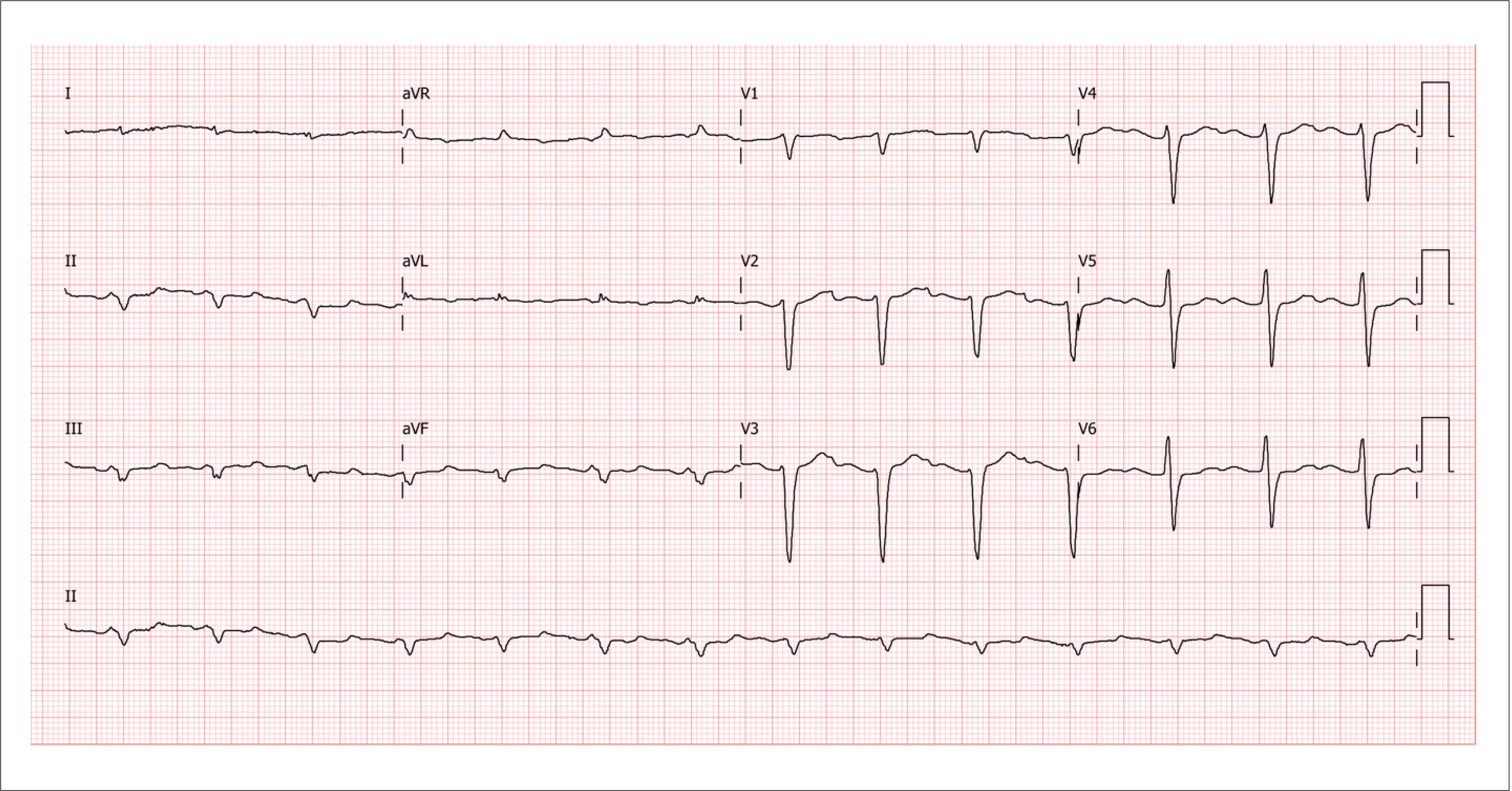
The electrocardiogram (ECG) is an essential test for diagnostic assessment and therapeutic planning, and its interpretation in conjunction with clinical and echocardiographic information is important. Although low-voltage ECG has great specificity in diagnosing myocardial infiltration secondary to CA, this is not the most prevalent finding. A lack of R-wave progression in precordial leads, which simulates an electrically inactive anteroseptal zone (pseudoinfarction pattern) is a much more frequent finding, with a prevalence of 60% to 70% in confirmed CA diagnoses, regardless of the type ( Figure 3 ).

Sample electrocardiogram image from a patient with wild-type transthyretin cardiac amyloidosis, showing low voltage in peripheral leads, no R-wave progression in precordial leads V1 to V3 (pseudoinfarction pattern) and first degree atrioventricular block. (Image from the authors’ personal archive)
In ATTR-CA, less than 40% of patients with a biopsy-confirmed diagnosis have low-voltage ECG. 5151. Cyrille NB, Goldsmith J, Alvarez J, Maurer MS. Prevalence and prognostic significance of low QRS voltage among the three main types of cardiac amyloidosis. Am J Cardiol. 2014;114(7):1089-93.
Thus, without low-voltage ECG criteria or signs of left ventricle (LV) overload, diagnostic suspicion of CA should not be ruled out, especially ATTR-CA. Disproportionate voltage in relation to myocardial thickness is also an important warning sign, reaching a prevalence of 73% to 80% in CA patients, regardless of type. 5252. Quarta CC, Solomon SD, Uraizee I, Kruger J, Longhi S, Ferlito M, et al. Left ventricular structure and function in transthyretin-related versus light-chain cardiac amyloidosis. Circulation. 2014;129(18):1840-9. , 5353. Carroll JD, Gaasch WH, McAdam KP. Amyloid cardiomyopathy: characterization by a distinctive voltage/mass relation. Am J Cardiol. 1982;49(1):9-13.
Among cardiac rhythm alterations, atrial fibrillation is more prevalent in ATTR patients, as well as atrioventricular blocks.
5.2. Echocardiogram
Echocardiography should be performed in all patients with clinical suspicion of the disease. Classic CA findings are usually present at an advanced stage of the disease and characterize a restrictive, infiltrative-type cardiomyopathy. The dimensions of the LV are not increased, volumes are normal or reduced, and there is a thickening of the ventricular walls. Increased atrial dimensions are common, reflecting early and progressive diastolic dysfunction, with increased filling pressures. Atrioventricular valves may be thickened and mitral and tricuspid regurgitations are functional. There may also be signs suggestive of infiltration of the interatrial septum, as well as an increase in the pulmonary artery systolic pressure. The right ventricle (RV) may also be affected. Pleural and pericardial effusions are very common and, in cases involving intense tissue infiltration, a granular sparkling appearance can be observed ( Figure 4 ) 3535. Maurer MS, Bokhari S, Damy T, Dorbala S, Drachman BM, Fontana M, et al. Expert Consensus Recommendations for the Suspicion and Diagnosis of Transthyretin Cardiac Amyloidosis. Circ Heart Fail. 2019;12(9):e006075. , 5454. Kittleson MM, Maurer MS, Ambardekar AV, Bullock-Palmer RP, Chang PP, Eisen HJ, et al. Cardiac Amyloidosis: Evolving Diagnosis and Management: A Scientific Statement From the American Heart Association. Circulation. 2020;142(1):e7-e22.

Classic echocardiographic presentation of cardiac amyloidosis. In A, the longitudinal parasternal projection shows a normal-sized left ventricle (LV) with increased wall thickness and a granular aspect in the interventricular septum (green arrow). There are also signs of left atrium (LA) enlargement and mild pericardial effusion (yellow arrow). In B, the apical projection shows large atria, normal-sized ventricles, and increased wall thickness, as well as a granular aspect in the interventricular septum (green arrow). The mitral and tricuspid valves are slightly thickened. LV: left ventricle; RV: right ventricle; LA: left atrium. (Images from the authors’ personal archive).
The LV ejection fraction is normally preserved until the more advanced stages of CA, although the longitudinal contractile function is reduced early. 5555. Mitchell C, Rahko PS, Blauwet LA, Canaday B, Finstuen JA, Foster MC, et al. Guidelines for Performing a Comprehensive Transthoracic Echocardiographic Examination in Adults: Recommendations from the American Society of Echocardiography. J Am Soc Echocardiogr. 2019;32(1):1-64. Quantitatively, systolic function can be assessed in 2D mode by calculating LV stroke volume and ejection fraction, as well as by Doppler-derived techniques such as estimating left ventricular dP/dt. 5656. Kolias TJ, Aaronson KD, Armstrong WF. Doppler-derived dP/dt and -dP/dt predict survival in congestive heart failure. J Am Coll Cardiol. 2000;36(5):1594-9.
Assessing the ventricular diastolic filling pattern is essential and often demonstrates some degree of diastolic dysfunction. In the initial phases, alterations compatible with type I diastolic dysfunction can be observed (inversion of the mitral inflow E/A wave ratio, prolongation of the isovolumetric relaxation time, and early diastolic deceleration. In tissue Doppler recording of myocardial velocities, E’ wave deceleration may be observed ( Figure 5 ).

Tissue Doppler images of the mitral annulus. There is E’ wave(*) deceleration in both the medial (a) and lateral (b) mitral annulus. Both speeds are below 4 cm/s (right ventricle > 8 cm/s). (Images from the authors’ personal archive).
As the disease progresses, a pseudonormal pattern of diastolic dysfunction (normal E/A wave ratio and normal deceleration time) may develop as a result of increased left atrial pressure.
In more advanced phases of the disease, there is a restrictive pattern of ventricular filling (E/A wave ratio > 2, decreased relaxation time, and an increased deceleration slope of the E-wave).
Analyzing myocardial deformation allows the early identification of signs of myocardial dysfunction in relation to LV ejection fraction. 5757. Voigt JU, Pedrizzetti G, Lysyansky P, Marwick TH, Houle H, Baumann R, et al. Definitions for a common standard for 2D speckle tracking echocardiography: consensus document of the EACVI/ASE/Industry Task Force to standardize deformation imaging. J Am Soc Echocardiogr. 2015;28(2):183-93. – 5959. Pagourelias ED, Vassilikos VP, Voigt JU. Left Ventricular Pressure Strain-Derived Myocardial Work at Rest and during Exercise in Patients with Cardiac Amyloidosis. J Am Soc Echocardiogr. 2020;33(10):1295-6. Global longitudinal strain – a function predominantly performed by the endocardium – is reduced early. 6060. Di Bella G, Pizzino F. Myocardial Deformation Analysis and Late-Gadolinium Enhancement: Important Markers of Cardiac Amyloidosis Involvement That Can Masquerade as a False-Negative Diagnosis. Circ J. 2018;82(10):2687. Regional myocardial deformation also frequently presents an apical sparing (“cherry on top”) pattern. 6161. Bravo PE, Fujikura K, Kijewski MF, Jerosch-Herold M, Jacob S, El-Sady MS, et al. Relative Apical Sparing of Myocardial Longitudinal Strain Is Explained by Regional Differences in Total Amyloid Mass Rather Than the Proportion of Amyloid Deposits. JACC Cardiovasc Imaging. 2019;12(7 Pt 1):1165-73. , 6262. Rapezzi C, Fontana M. Relative Left Ventricular Apical Sparing of Longitudinal Strain in Cardiac Amyloidosis: Is it Just Amyloid Infiltration? JACC Cardiovasc Imaging. 2019;12(7 Pt 1):1174-6. This aspect is best visualized in a parametric (“bulls eye”) image of the LV ( Figure 6 ).

Analyses of global and regional LV myocardial deformation in a patient with cardiac amyloidosis, showing the parametric graphical representation (bulls-eye), apical segments in dark red (preserved values), and basal segments in light red (reduced values) in the “cherry-on-top” pattern. (Image from the authors’ personal archive)
Other analyses of myocardial deformation, such as RV systolic function, 6363. Monivas Palomero V, Durante-Lopez A, Sanabria MT, Cubero JS, Gonzalez-Mirelis J, Lopez-Ibor JV, et al. Role of Right Ventricular Strain Measured by Two-Dimensional Echocardiography in the Diagnosis of Cardiac Amyloidosis. J Am Soc Echocardiogr. 2019;32(7):845-53 e1. atrial function, 6565. Syed IS, Glockner JF, Feng D, Araoz PA, Martinez MW, Edwards WD, et al. Role of cardiac magnetic resonance imaging in the detection of cardiac amyloidosis. JACC Cardiovascular imaging. 2010;3(2):155-64. and estimating myocardial work, 5959. Pagourelias ED, Vassilikos VP, Voigt JU. Left Ventricular Pressure Strain-Derived Myocardial Work at Rest and during Exercise in Patients with Cardiac Amyloidosis. J Am Soc Echocardiogr. 2020;33(10):1295-6. have been applied to CA patients and have shown good diagnostic accuracy.
5.3. Cardiac Magnetic Resonance
Cardiovascular magnetic resonance (CMR) allows accurate assessment of myocardial tissue changes in CA. 6565. Syed IS, Glockner JF, Feng D, Araoz PA, Martinez MW, Edwards WD, et al. Role of cardiac magnetic resonance imaging in the detection of cardiac amyloidosis. JACC Cardiovascular imaging. 2010;3(2):155-64. Classically, the deposition of myofibrils leads to an increase in the thickness of the LV myocardial wall and the interatrial septum, which can be visualized by the morphological techniques of CMR. 6666. Kwong RY, Falk RH. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2005;111(2):122-4. , 6767. Maceira AM, Joshi J, Prasad SK, Moon JC, Perugini E, Harding I, et al. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2005;111(2):186-93. Another tissue change is increased total water content in the myocardium, which may be derived from increased extracellular volume, a direct result of protein deposition and the water it attracts through osmosis, as well as from the increased intracellular water in myocytes suffering the cytotoxic effects of the deposition or even from decreased myocardial perfusion (increased distance from the capillaries and/or obstruction by the deposition).
The global increase in myocardial tissue water content leads to an increase in mean hydrogen relaxation time, whether T1 (longitudinal) or T2 (transverse). 6868. Kotecha T, Martinez-Naharro A, Treibel TA, Francis R, Nordin S, Abdel-Gadir A, et al. Myocardial Edema and Prognosis in Amyloidosis. Journal of the American College of Cardiology. 2018;71(25):2919-31. However, the most dramatic and relevant myocardial tissue change in CA is the extreme increase in myocardial extracellular volume, which occurs in the clinical stages of the disease, not only due to the deposition of amyloid fibrils, but also to myocardial fibrosis repair.
Thus, the combination of myofibril deposition and interstitial fibrosis can be detected easily and precisely, and can even be quantified by late enhancement (LE) techniques 6767. Maceira AM, Joshi J, Prasad SK, Moon JC, Perugini E, Harding I, et al. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2005;111(2):186-93. , 6969. Fontana M, Corovic A, Scully P, Moon JC. Myocardial Amyloidosis: The Exemplar Interstitial Disease. JACC Cardiovascular imaging. 2019;12(11 Pt 2):2345-56. and by calculating the extracellular myocardial volume. 7070. Mongeon FP, Jerosch-Herold M, Coelho-Filho OR, Blankstein R, Falk RH, Kwong RY. Quantification of extracellular matrix expansion by CMR in infiltrative heart disease. JACC Cardiovascular imaging. 2012;5(9):897-907. – 7272. Martinez-Naharro A, Kotecha T, Norrington K, Boldrini M, Rezk T, Quarta C, et al. Native T1 and Extracellular Volume in Transthyretin Amyloidosis. JACC Cardiovascular imaging. 2019;12(5):810-9. As an example, normal values for myocardial extracellular space are approximately 25%, while in CA they can reach 60% (mainly in ATTR-CA). 7272. Martinez-Naharro A, Kotecha T, Norrington K, Boldrini M, Rezk T, Quarta C, et al. Native T1 and Extracellular Volume in Transthyretin Amyloidosis. JACC Cardiovascular imaging. 2019;12(5):810-9. , 7373. Fontana M, Banypersad SM, Treibel TA, Maestrini V, Sado DM, White SK, et al. Native T1 mapping in transthyretin amyloidosis. JACC Cardiovascular imaging. 2014;7(2):157-65.
The CMR contrast medium is based on gadolinium, which is bound to a macromolecular chelator that does not allow it to pass through the entire cell membrane. Thus it is distributed exclusively in the myocardial extracellular volume. The distribution pattern in the LE image can raise suspicion of CA (global subendocardial, apical non-involvement of the LV, and distribution outside the coronary vascular territory). 6767. Maceira AM, Joshi J, Prasad SK, Moon JC, Perugini E, Harding I, et al. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2005;111(2):186-93.
5.3.1. Assessment of Cardiac Morphology and Function
CA can modify the appearance of all cardiac chambers. 7474. Fontana M, Chung R, Hawkins PN, Moon JC. Cardiovascular magnetic resonance for amyloidosis. Heart Fail Rev. 2015;20(2):133-44. , 7575. Baggiano A, Boldrini M, Martinez-Naharro A, Kotecha T, Petrie A, Rezk T, et al. Noncontrast Magnetic Resonance for the Diagnosis of Cardiac Amyloidosis. JACC Cardiovasc Imaging. 2020;13(1 Pt 1):69-80. Changes in the atrial plane can be observed the initial stages, including dilation and apparent thickening of the interatrial septum, which, in most cases, consists of fat. 7474. Fontana M, Chung R, Hawkins PN, Moon JC. Cardiovascular magnetic resonance for amyloidosis. Heart Fail Rev. 2015;20(2):133-44. In later stages of cardiac impairment, when atrial function decreases, signs of slow flow and thrombi in the left atrial appendage can be seen, which may not appear in cardiovascular magnetic resonance imaging if there are artifacts related to rhythm or if specific series are not used for this purpose.
CA is also commonly associated with increased myocardial thickness, which is, in most cases, more expressive than in cases secondary to hypertension. The thickness of the cardiac muscle is generally greater in ATTR-CA than AL-CA. 5252. Quarta CC, Solomon SD, Uraizee I, Kruger J, Longhi S, Ferlito M, et al. Left ventricular structure and function in transthyretin-related versus light-chain cardiac amyloidosis. Circulation. 2014;129(18):1840-9. , 7676. Pozo E, Kanwar A, Deochand R, Castellano JM, Naib T, Pazos-Lopez P, et al. Cardiac magnetic resonance evaluation of left ventricular remodelling distribution in cardiac amyloidosis. Heart. 2014;100(21):1688-95. The increased thickness may be concentric or eccentric, 7474. Fontana M, Chung R, Hawkins PN, Moon JC. Cardiovascular magnetic resonance for amyloidosis. Heart Fail Rev. 2015;20(2):133-44. , 7575. Baggiano A, Boldrini M, Martinez-Naharro A, Kotecha T, Petrie A, Rezk T, et al. Noncontrast Magnetic Resonance for the Diagnosis of Cardiac Amyloidosis. JACC Cardiovasc Imaging. 2020;13(1 Pt 1):69-80. and may involve the RV. 7474. Fontana M, Chung R, Hawkins PN, Moon JC. Cardiovascular magnetic resonance for amyloidosis. Heart Fail Rev. 2015;20(2):133-44. , 7575. Baggiano A, Boldrini M, Martinez-Naharro A, Kotecha T, Petrie A, Rezk T, et al. Noncontrast Magnetic Resonance for the Diagnosis of Cardiac Amyloidosis. JACC Cardiovasc Imaging. 2020;13(1 Pt 1):69-80. The ejection fraction may be preserved for a long time, and the earliest changes in ventricular function include diastolic restriction and changes in ventricular strain. 7575. Baggiano A, Boldrini M, Martinez-Naharro A, Kotecha T, Petrie A, Rezk T, et al. Noncontrast Magnetic Resonance for the Diagnosis of Cardiac Amyloidosis. JACC Cardiovasc Imaging. 2020;13(1 Pt 1):69-80. , 7676. Pozo E, Kanwar A, Deochand R, Castellano JM, Naib T, Pazos-Lopez P, et al. Cardiac magnetic resonance evaluation of left ventricular remodelling distribution in cardiac amyloidosis. Heart. 2014;100(21):1688-95.
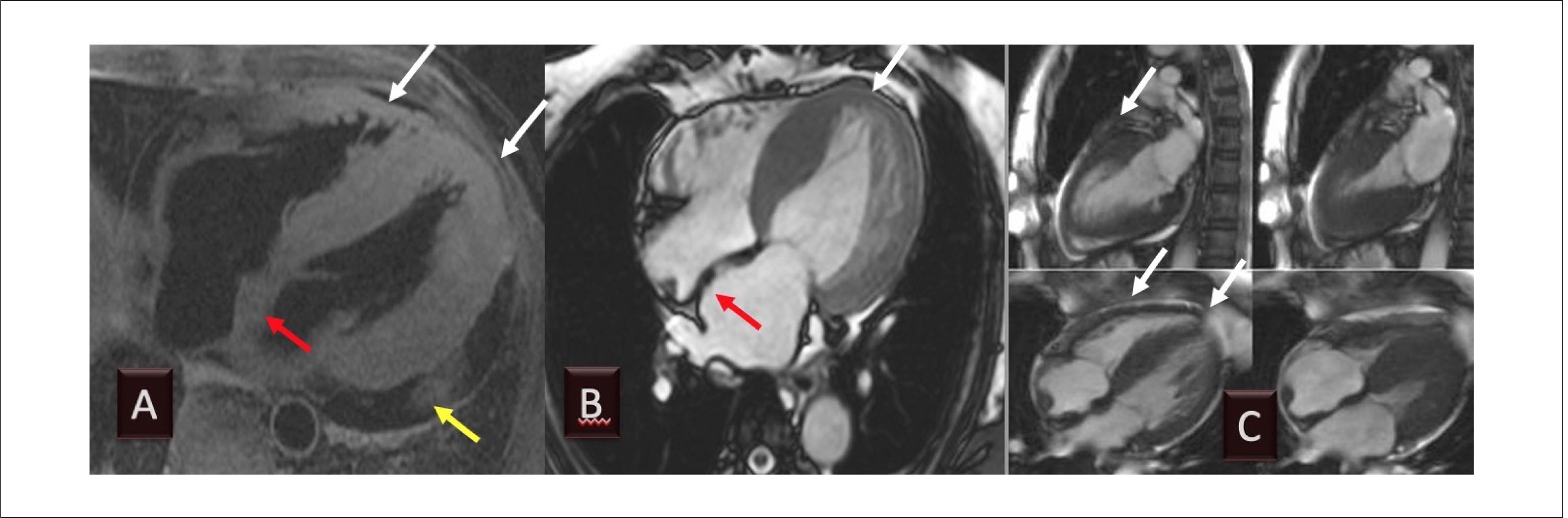
As a result of diastolic dysfunction, it is not uncommon to observe pericardial or pleural effusions. 7474. Fontana M, Chung R, Hawkins PN, Moon JC. Cardiovascular magnetic resonance for amyloidosis. Heart Fail Rev. 2015;20(2):133-44. Assessing morphological characteristics with cardiovascular magnetic resonance imaging ( Figure 7 ) is helpful and can suggest the diagnosis. However, tissue characterization is usually performed with LE and T1 mapping techniques, which we will discuss below in other sections.

Example of cardiovascular magnetic resonance imaging of transthyretin amyloidosis, showing pericardial effusion (A, yellow arrow and C), increased atrial dimensions (A and B), and increased interatrial septum (A and B, red arrows) and ventricular wall thickness (A to C, white arrows). Diastolic function is reduced, but contractility may be preserved until the later stages of the disease. (Image from the authors’ personal archive).
5.3.2. Assessment with Late Enhancement
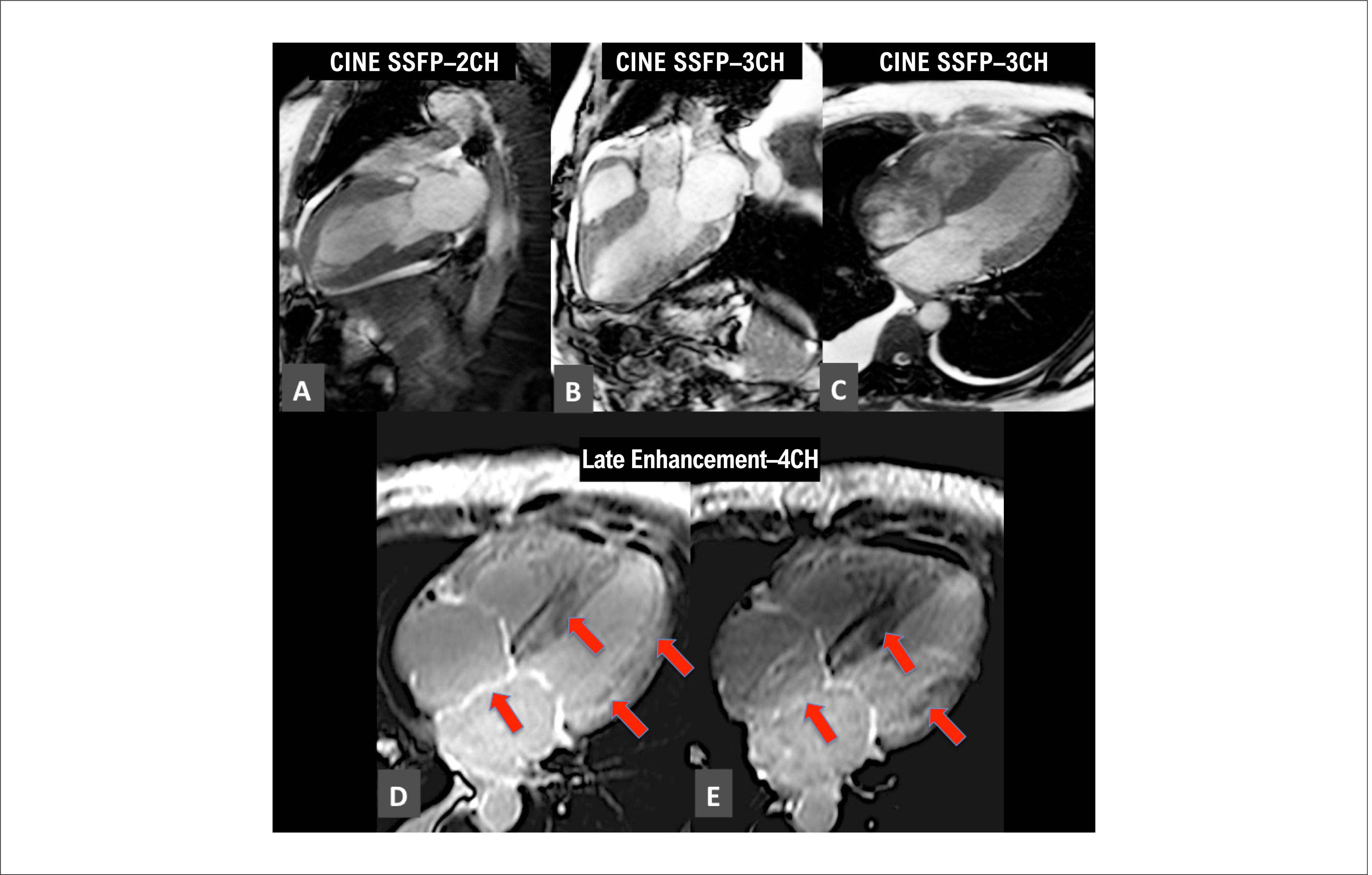
The LE technique after gadolinium contrast injection has been widely recognized as a pillar of CA imaging diagnosis. 7777. Jurcut R, Onciul S, Adam R, Stan C, Coriu D, Rapezzi C, et al. Multimodality imaging in cardiac amyloidosis: a primer for cardiologists. European heart journal cardiovascular Imaging. 2020;21(8):833-44. , 7878. Dorbala S, Ando Y, Bokhari S, Dispenzieri A, Falk RH, Ferrari VA, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI Expert Consensus Recommendations for Multimodality Imaging in Cardiac Amyloidosis: Part 1 of 2-Evidence Base and Standardized Methods of Imaging. J Card Fail.2019;25(11):e 1-e39. When crossing the interstitial space in normal tissue, gadolinium contrast medium is not delayed and the agent quickly diffuses, leaving normal tissue dark. When there is amyloid deposition in the interstitial space, it begins to slow the transit of the gadolinium-based contrast agent, and the myocardium “shines” in the dedicated sequences, for which an anatomopathological correlation has been demonstrated. 6565. Syed IS, Glockner JF, Feng D, Araoz PA, Martinez MW, Edwards WD, et al. Role of cardiac magnetic resonance imaging in the detection of cardiac amyloidosis. JACC Cardiovascular imaging. 2010;3(2):155-64. Subendocardial, transmural and focal patterns of amyloid infiltration have been described, the latter being less frequent ( Figure 8 ). 6767. Maceira AM, Joshi J, Prasad SK, Moon JC, Perugini E, Harding I, et al. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2005;111(2):186-93. A recent meta-analysis found that LE had a sensitivity and specificity of 85% and 92%, respectively, for amyloidosis. 7979. Zhao L, Tian Z, Fang Q. Diagnostic accuracy of cardiovascular magnetic resonance for patients with suspected cardiac amyloidosis: a systematic review and meta-analysis. BMC Cardiovasc Dis. 2016;16:129.

Example of a patient with ATTR transthyretin with concentric hypertrophy of the left ventricle and enlargement of both atria in cine images (A to C). Late enhancement shows a predominantly diffuse transmural pattern (red arrows, D and E). (Image from the authors’ personal archive).
In a series of 250 patients with different forms of amyloidosis, LE with a transmural pattern was associated with a 5.4 times greater risk of death (CI: 2.1-13.7; p < 0.0001). 8080. Fontana M, Pica S, Reant P, Abdel-Gadir A, Treibel TA, Banypersad SM, et al. Prognostic Value of Late Gadolinium Enhancement Cardiovascular Magnetic Resonance in Cardiac Amyloidosis. Circulation. 2015;132(16):1570-9. In addition, LE has an incremental prognostic effect for cardiac markers in AL-CA 8181. Boynton SJ, Geske JB, Dispenzieri A, Syed IS, Hanson TJ, Grogan M, et al. LGE Provides Incremental Prognostic Information Over Serum Biomarkers in AL Cardiac Amyloidosis. JACC Cardiovascular imaging. 2016;9(6):680-6. or in isolation. 8282. Lin L, Li X, Feng J, Shen KN, Tian Z, Sun J, et al. The prognostic value of T1 mapping and late gadolinium enhancement cardiovascular magnetic resonance imaging in patients with light chain amyloidosis. Journal of cardiovascular magnetic resonance . J Cardiovasc Magn Reson. 2018;20(1):2.
5.3.3. T1 Mapping
Different groups have investigated the utility of cardiovascular magnetic resonance-derived T1 maps to improve diagnostic and prognostic performance in CA. 7070. Mongeon FP, Jerosch-Herold M, Coelho-Filho OR, Blankstein R, Falk RH, Kwong RY. Quantification of extracellular matrix expansion by CMR in infiltrative heart disease. JACC Cardiovascular imaging. 2012;5(9):897-907. , 7171. Banypersad SM, Sado DM, Flett AS, Gibbs SD, Pinney JH, Maestrini V, et al. Quantification of myocardial extracellular volume fraction in systemic AL amyloidosis: an equilibrium contrast cardiovascular magnetic resonance study. Circulation Cardiovascular imaging. 2013;6(1):34-9. , 7373. Fontana M, Banypersad SM, Treibel TA, Maestrini V, Sado DM, White SK, et al. Native T1 mapping in transthyretin amyloidosis. JACC Cardiovascular imaging. 2014;7(2):157-65. , 8383. Fontana M, White SK, Banypersad SM, Sado DM, Maestrini V, Flett AS, et al. Comparison of T1 mapping techniques for ECV quantification. Histological validation and reproducibility of ShMOLLI versus multibreath-hold T1 quantification equilibrium contrast CMR. Journal of cardiovascular magnetic resonance : J Cardiovasc Magn Reson. 2012;14:88. , 8484. White JA, Kim HW, Shah D, Fine N, Kim KY, Wendell DC, et al. CMR imaging with rapid visual T1 assessment predicts mortality in patients suspected of cardiac amyloidosis. JACC Cardiovasc Imaging. 2014;7(2):143-56. Both native T1 data, which do not require a contrast agent, as well as extracellular volume data have effectively identified patients with CA, who show markedly higher native T1 and extracellular volume values than healthy controls. 7070. Mongeon FP, Jerosch-Herold M, Coelho-Filho OR, Blankstein R, Falk RH, Kwong RY. Quantification of extracellular matrix expansion by CMR in infiltrative heart disease. JACC Cardiovascular imaging. 2012;5(9):897-907. , 7373. Fontana M, Banypersad SM, Treibel TA, Maestrini V, Sado DM, White SK, et al. Native T1 mapping in transthyretin amyloidosis. JACC Cardiovascular imaging. 2014;7(2):157-65. , 8383. Fontana M, White SK, Banypersad SM, Sado DM, Maestrini V, Flett AS, et al. Comparison of T1 mapping techniques for ECV quantification. Histological validation and reproducibility of ShMOLLI versus multibreath-hold T1 quantification equilibrium contrast CMR. Journal of cardiovascular magnetic resonance : J Cardiovasc Magn Reson. 2012;14:88. These changes can be detected before LE. 7070. Mongeon FP, Jerosch-Herold M, Coelho-Filho OR, Blankstein R, Falk RH, Kwong RY. Quantification of extracellular matrix expansion by CMR in infiltrative heart disease. JACC Cardiovascular imaging. 2012;5(9):897-907. , 7272. Martinez-Naharro A, Kotecha T, Norrington K, Boldrini M, Rezk T, Quarta C, et al. Native T1 and Extracellular Volume in Transthyretin Amyloidosis. JACC Cardiovascular imaging. 2019;12(5):810-9. , 7373. Fontana M, Banypersad SM, Treibel TA, Maestrini V, Sado DM, White SK, et al. Native T1 mapping in transthyretin amyloidosis. JACC Cardiovascular imaging. 2014;7(2):157-65. Therefore, combining native T1 mapping and extracellular volume measurements can help determine the amyloid burden and confirm the diagnosis of CA. 8585. Dorbala S, Cuddy S, Falk RH. How to Image Cardiac Amyloidosis: A Practical Approach. JACC Cardiovascular imaging. 2020;13(6):1299-310. , 8686. Pan JA, Kerwin MJ, Salerno M. Native T1 Mapping, Extracellular Volume Mapping, and Late Gadolinium Enhancement in Cardiac Amyloidosis: A Meta-Analysis. JACC Cardiovascular imaging. 2020;13(6):1299-310.
5.4. Cardiac Scintigraphy with Bone-seeking Radiotracers
Technetium-99m-labeled bisphosphonate-derived radiotracers, originally developed for bone imaging, have found a new role as a non-invasive diagnostic tool for ATTR-CA. 11. Sipe JD, Cohen AS. Review: history of the amyloid fibril. J Struct Biol. 2000;130(2-3):88-98. – 77. Gertz MA, Dispenzieri A, Sher T. Pathophysiology and treatment of cardiac amyloidosis. Nat Rev Cardiol. 2015;12(2):91-102. Bone radiotracers safely allow non-invasive diagnosis of ATTR-CA once the presence of monoclonal gammopathy is excluded. 9191. Hanna M, Ruberg FL, Maurer MS, Dispenzieri A, Dorbala S, Falk RH, et al. Cardiac Scintigraphy With Technetium-99m-Labeled Bone-Seeking Tracers for Suspected Amyloidosis: JACC Review Topic of the Week. J Am Coll Cardiol. 2020;75(22):2851-62.
The main99mTc-labeled bone radiotracers used in ATTR-CA diagnosis are99mTc-pyrophosphate,99mTc-DPD (3,3-diphosphono-1,2-propanedicarboxylic acid), and99mTc-HMDP (99mTc-labeled hydroxymethylene diphosphonate). 11. Sipe JD, Cohen AS. Review: history of the amyloid fibril. J Struct Biol. 2000;130(2-3):88-98. – 77. Gertz MA, Dispenzieri A, Sher T. Pathophysiology and treatment of cardiac amyloidosis. Nat Rev Cardiol. 2015;12(2):91-102. 99mTc-pyrophosphate is the only one of these available in Brazil. It should be pointed out that although99mTc-MDP (99mTc-labeled methylene diphosphonate) has proven efficient for bone scintigraphy, it has low sensitivity for diagnosing ATTR-CA and should not be used for this purpose. 8989. Perugini E, Guidalotti PL, Salvi F, Cooke RM, Pettinato C, Riva L, et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol. 2005;46(6):1076-84.
Although the structural component of the amyloid deposition to which99mTc-pyrophosphate binds in the heart is unknown, it is widely accepted that a calcium-dependent uptake mechanism is involved. 9393. Pepys MB, Dyck RF, de Beer FC, Skinner M, Cohen AS. Binding of serum amyloid P-component (SAP) by amyloid fibrils. Clin Exp Immunol. 1979;38(2):284-93. Several binding sites have been found in animals: microcalcifications, calcium deposits, intracellular pyrophosphate, and intracellular macromolecules. The mechanism of99mTc-pyrophosphate uptake in the myocardium is probably related to the presence of microcalcifications. 8888. Stats MA, Stone JR. Varying levels of small microcalcifications and macrophages in ATTR and AL cardiac amyloidosis: implications for utilizing nuclear medicine studies to subtype amyloidosis. Cardiovasc Pathol. 2016;25(5):413-7. ATTR-CA involves more microcalcifications than AL-CA and has greater99mTc-pyrophosphate uptake, while AL-CA has little or no affinity for bone radiotracers. In addition, ATTR-CA has a more indolent evolution, providing more microcalcifications and, consequently, greater radiotracer accumulation.
Although the echocardiogram and cardiac magnetic resonance findings may be indicative of CA, they cannot differentiate ATTR-CA from AL-CA. This is the main advantage of99mTc-pyrophosphate cardiac scintigraphy: it is a simple, easy, widely available method with low dosimetry that can differentiate ATTR-CA from AL-CA noninvasively and with high specificity, and thus guide treatment. This differentiation is useful, since AL-CA and ATTR-CA have completely different prognostic and therapeutic implications.
The role of bone radiotracers in ATTR diagnosis was recently re-evaluated by an international group of several centers with expertise in CA: in a cumulative analysis of 1,217 patients, 867 with biopsy-confirmed amyloidosis and 360 with non-amyloid cardiomyopathy, scintigraphy was highly sensitive (99%) and specific (86%) for ATTR-CA. 1919. Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, et al. Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation. 2016;133(24):2404-12. This study further demonstrated that the combined finding of positive bone radiotracer scintigraphy in patients with no evidence of detectable monoclonal protein in urine or serum (serum free light chain analysis and electrophoresis with immunofixation) was 100% specific for ATTR-CA, leading the authors to conclude that scintigraphy allows for accurate detection without the need for cardiac biopsy. 1919. Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, et al. Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation. 2016;133(24):2404-12. Another recent study with pooled cases from three U.S. centers showed that, among a total of 171 patients (121 ATTR, 34 AL, and 16 non-amyloid HFpEF),99mTc-pyrophosphate had an 88% sensitivity and 88% specificity for ATTR when only visual assessment was used (score ≥ 2). 9494. Castano A, Haq M, Narotsky DL, Goldsmith J, Weinberg RL, Morgenstern R, et al. Multicenter Study of Planar Technetium 99m Pyrophosphate Cardiac Imaging: Predicting Survival for Patients With ATTR Cardiac Amyloidosis. JAMA Cardiol. 2016;1(8):880-9. When semiquantitative analysis was used (heart/contralateral ratio > 1.6), its sensitivity and specificity were 91% and 92%, respectively, for detecting ATTR. Furthermore, when considering all variables, a heart/contralateral ratio ≥ 1.6 was predictive of worse survival in ATTR-CA patients. 9494. Castano A, Haq M, Narotsky DL, Goldsmith J, Weinberg RL, Morgenstern R, et al. Multicenter Study of Planar Technetium 99m Pyrophosphate Cardiac Imaging: Predicting Survival for Patients With ATTR Cardiac Amyloidosis. JAMA Cardiol. 2016;1(8):880-9. Vraniam et al. 9595. Vranian MN, Sperry BW, Hanna M, Hachamovitch R, Ikram A, Brunken RC, et al. Technetium pyrophosphate uptake in transthyretin cardiac amyloidosis: Associations with echocardiographic disease severity and outcomes. J Nucl Cardiol. 2018;25(4):1247-56. also demonstrated that, in patients with suspected CA, the intensity of cardiac99mTc-pyrophosphate uptake was predictive of overall mortality and hospitalization for HF. In these studies, combined assessment of the intensity of radiotracer uptake in the myocardium (heart/contralateral ratio) with anatomical, functional and biomarker variables improved risk stratification.
5.4.1. Recommended Technical Aspects for Image Acquisition
No preparation required. Images are obtained after intravenous administration of 10 to 25 mCi (370 to 925 Mbq) of99mTc-pyrophosphate (dosimetry: 3.2 mSv for the whole body for 15 mCi). Flat and single photon emission computed tomography (SPECT) images of the chest are taken 1 and 3 hours after administration of the radiopharmaceutical.
-
Planar chest images: can be obtained in the anterior, left anterior oblique, and left lateral projections using99mTc photopic (140 keV, 15% window), low energy/high resolution collimator and a 256 × 256 matrix, 500,000-750,000 counts.
-
SPECT chest images: obtained with a 128 × 128 matrix (64 × 64 is acceptable), 180-degree rotation from right anterior oblique to left posterior oblique (360-degree is acceptable), 1 image every 3 to 6 degrees. If available, SPECT/CT images (SPECT combined with computed tomography) provide greater confidence for interpreting the images.
-
Minimum recommended images: 1 h SPECT and 1 h and 3 h flat images in the anterior projection. The use of SPECT images is recommended to differentiate diffuse uptake of the radiopharmaceutical in the myocardium from its persistence in the blood pool, focal uptake by myocardial infarction, and bone overlay. In addition, 1 h and 3 h flat images are useful for quantifying and monitoring the blood pool washout, which is variable, eg, much slower in patients with renal failure.
5.4.2. Image Analysis
-
Semiquantitative analysis (1 h planar image): For99mTc-pyrophosphate, the semiquantitative analysis was defined as the ratio between uptake in the cardiac projection and uptake in the contralateral hemithorax, measured in a planar 1 h image in the anterior view. To do this, a circular (or elliptical) area of interest is drawn over the cardiac projection without including the sternum, avoiding the inclusion of both the adjacent lung and areas of focal uptake in the costal arches. An identical (“mirror”) area of interest is placed in the contralateral hemithorax following the same procedure. The count within the cardiac area of interest is divided by the count in the contralateral area of interest, thus obtaining the heart/contralateral (H/CL) ratio. H/CL ≥ 1.5 at 1 h identifies ATTR-CA with high accuracy if systemic AL-CA has been excluded 9090. Dorbala S, Ando Y, Bokhari S, Dispenzieri A, Falk RH, Ferrari VA, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: Part 1 of 2-evidence base and standardized methods of imaging. J Nucl Cardiol. 2019;26(6):2065-123. , 9191. Hanna M, Ruberg FL, Maurer MS, Dispenzieri A, Dorbala S, Falk RH, et al. Cardiac Scintigraphy With Technetium-99m-Labeled Bone-Seeking Tracers for Suspected Amyloidosis: JACC Review Topic of the Week. J Am Coll Cardiol. 2020;75(22):2851-62. ( Figure 9 ).
-
Visual graduation (3 h image): visual grading is performed by comparing cardiac uptake with physiological uptake in adjacent costal arch, and can be performed with planar images in the anterior projection, in SPECT images, or even in whole-body images obtained 3 h after injecting the radiopharmaceutical. Uptake intensity is defined as grade 0 (no myocardial uptake), grade 1 (myocardial uptake lower than that of adjacent costal arch), grade 2 (uptake similar to that of costal arch), and grade 3 (greater than that of the costal arch). Grades 2 or 3 are strongly suggestive of ATTR if monoclonal gammopathy has been excluded ( Figure 9 ).

99mTc-pyrophosphate cardiac scintigraphy in patients with suspected cardiac amyloidosis (flat images in anterior chest view taken 3 h after administration) showing negative and positive transthyretin (ATTR) amyloidosis cases. Visual analysis: images on the left: negative ATTR cases (grades 0 and 1); images on the right: positive ATTR cases (grades 2 and 3). Semi-quantitative analysis: on the left, negative ATTR case (heart/contralateral ratio = 1.2 – in this case, the SPECT images showed activity in the blood pool and not in the heart walls); on the right, positive ATTR case (heart/contralateral ratio = 1.6 –SPECT imaging confirmed uptake in the left ventricle walls).
5.4.3. False-Positive ATTR
The operational characteristics of bone scintigraphy are very favorable for use in clinical diagnosis, with a specificity of 100% for ATTR when the uptake is grade 2 or 3 and there is no monoclonal gammopathy. However, it must be pointed out that failure to exclude monoclonal gammopathy (either due to inappropriate use or to misinterpretation of laboratory tests) entails a risk of inaccurate diagnosis. The most common cause of ATTR misdiagnosis is AL-CA. Recent studies indicate that up to 22% of patients with AL-CA may have grade 2 or 3 uptake in99mTc-pyrophosphate scintigraphy. 66. Buxbaum JN, Tagoe C, Gallo G, Walker JR, Kurian S, Salomon DR. Why are some amyloidoses systemic? Does hepatic “chaperoning at a distance” prevent cardiac deposition in a transgenic model of human senile systemic (transthyretin) amyloidosis? FASEB J. 2012;26(6):2283-93. It should also be pointed out that SPECT imaging is crucial to differentiate abnormal myocardial uptake from residual uptake in the blood pool ( Figure 10 ). Table 3 lists the main causes of false-positive diagnosis with99mTc-pyrophosphate scintigraphy.
Causes of false-positive99mTc-pyrophosphate scintigraphy (myocardial99mTc-pyrophosphate uptake that is not associated with amyloid transthyretin)

Illustrative images of a 76-year-old male patient with ATTRv (VAL30MET) cardiac amyloidosis. Cardiac scintigraphy with99mTc-pyrophosphate (flat images A and B) showed intense radiotracer uptake in the 1 h and 3 h images. Visual analysis (A) showed grade 3 uptake in both 1 h and 3 h images (myocardial uptake intensity greater than that of the costal margins). The semiquantitative analysis (B) showed a 2.1 heart/contralateral ratio at 1 h 2.0 and at 3 h. Single photon emission computed tomography (SPECT) showed radiopharmaceutical uptake in all left ventricle walls and confirmed right ventricle involvement, as shown in the flat images (arrows). The99mTc-pyrophosphate cardiac scintigraphy findings are highly suggestive of ATTR-CA. (Images from the authors’ personal files).
Finally, scintigraphy is not recommended for patient follow-up, since there is no current evidence of a correlation between a change in image pattern and disease progression or response to treatment. 8787. Singh V, Falk R, Di Carli MF, Kijewski M, Rapezzi C, Dorbala S. State-of-the-art radionuclide imaging in cardiac transthyretin amyloidosis. J Nucl Cardiol. 2019;26(1):158-73. , 9090. Dorbala S, Ando Y, Bokhari S, Dispenzieri A, Falk RH, Ferrari VA, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: Part 1 of 2-evidence base and standardized methods of imaging. J Nucl Cardiol. 2019;26(6):2065-123.
Table 4 summarizes the main findings and practical guidelines for image acquisition and analysis in CA diagnosis.
Summary of the main findings suggestive of cardiac amyloidosis in complementary examinations
5.5. Biomarkers
No specific laboratory marker can be used to diagnose CA.
Troponin and natriuretic peptides have been found useful for assessing cardiac damage due to amyloidosis and are non-invasive, accessible, and relatively low-cost diagnostic aids. 9696. Kyriakou P, Mouselimis D, Tsarouchas A, Rigopoulos A, Bakogiannis C, Noutsias M, et al. Diagnosis of cardiac amyloidosis: a systematic review on the role of imaging and biomarkers. BMC Cardiovasc Disord. 2018;18(1):221. When these biomarkers detect persistent changes, it is a warning sign of cardiac damage due to amyloidosis.
5.5.1. Natriuretic Peptides
Analysis of data from the THAOS (Transthyretin Amyloidosis Outcomes Survey) registry showed that natriuretic peptide levels can be used as a diagnostic aid, with higher values observed in mutations associated with amyloid cardiomyopathy, such as V122I and late-onset V30M, than in those predominantly associated with neurologic manifestations, such as early-onset V30M. Higher levels of biomarkers were also observed in ATTRwt than ATTRv. However, it was observed that even in patients with a predominantly neurological phenotype, 45% to 90% had altered levels of these biomarkers, indicating some degree of subclinical myocardial involvement. 9797. Kristen AV, Maurer MS, Rapezzi C, Mundayat R, Suhr OB, Damy T, et al. Impact of genotype and phenotype on cardiac biomarkers in patients with transthyretin amyloidosis - Report from the Transthyretin Amyloidosis Outcome Survey (THAOS). PLoS One. 2017;12(4):e0173086.
Higher NT-proBNP values have been observed in AL-CA than ATTR-CA. This is because amyloidogenic light chains modulate p38 mitogen-activated protein kinase, which directly promotes NT-proBNP expression. Thus, despite the same degree of hemodynamic changes in both forms of amyloidosis, serum levels of NT-proBNP may be higher in AL-CA. 9898. Perfetto F, Bergesio F, Grifoni E, Fabbri A, Ciuti G, Frusconi S, et al. Different NT-proBNP circulating levels for different types of cardiac amyloidosis. J Cardiovasc Med (Hagerstown). 2016;17(11):810-7.
Of note, patients with HFpEF due to ATTR have disproportionately high NT-ProBNP values with respect to HF severity compared to patients with non-amyloid HFpEF. 9999. Hahn VS, Yanek LR, Vaishnav J, Ying W, Vaidya D, Lee YZJ, et al. Endomyocardial Biopsy Characterization of Heart Failure With Preserved Ejection Fraction and Prevalence of Cardiac Amyloidosis. JACC Heart Fail. 2020;8(9):712-24.
5.5.2. Troponins
Mild and persistent elevation of troponin levels is frequently observed and suggests subclinical myocardial damage in several non-ischemic cardiomyopathies. 100100. Kociol RD, Pang PS, Gheorghiade M, Fonarow GC, O’Connor CM, Felker GM. Troponin elevation in heart failure prevalence, mechanisms, and clinical implications. J Am Coll Cardiol. 2010;56(14):1071-8. However, it has been reported that levels are higher in CA than in other forms of cardiomyopathy. 2121. Gonzalez-Lopez E, Gallego-Delgado M, Guzzo-Merello G, de Haro-Del Moral FJ, Cobo-Marcos M, Robles C, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36(38):2585-94. A study of patients with hypertrophic cardiomyopathy who underwent endomyocardial biopsy identified markedly higher troponin levels in CA patients than in those with amyloid-free heart disease and had high diagnostic sensitivity. 101101. Takashio S, Yamamuro M, Izumiya Y, Hirakawa K, Marume K, Yamamoto M, et al. Diagnostic utility of cardiac troponin T level in patients with cardiac amyloidosis. ESC Heart Fail. 2018;5(1):27-35.
Several mechanisms have been postulated to explain the elevated troponin levels in these patients: myocardial ischemia, increased wall stress, direct myocyte damage by inflammatory cytokines and/or oxidative stress, neurohormonal activation, and microvascular dysfunction in heart failure. Microvascular dysfunction in amyloidosis is presumably caused by interstitial and perivascular deposition, increased ventricular filling pressure, and endothelial dysfunction due to immunoglobulin-induced toxicity in AL-CA. In addition, it has also been found that light chains have a direct cardiotoxic effect, regardless of extracellular deposition of fibrils, which might explain the higher troponin levels in AL-CA than in ATTR-CA, in which mild and persistent elevation is usually observed. 101101. Takashio S, Yamamuro M, Izumiya Y, Hirakawa K, Marume K, Yamamoto M, et al. Diagnostic utility of cardiac troponin T level in patients with cardiac amyloidosis. ESC Heart Fail. 2018;5(1):27-35.
It is important to point out that many ATTR patients have comorbidities, such as ischemic cardiomyopathy, which can cause altered troponin values. Therefore, troponin levels should not be used to rule out or confirm cardiac involvement. Rather, they should serve as a potential warning sign for the disease, which can be better evaluated through more specific tests.
5.5.3New Biomarkers
A number of other biomarkers have been studied, some with high specificity for amyloidosis subtypes, such as retinol-binding protein 4 for hereditary amyloidosis through the Val142Ile mutation. However, further studies are necessary, as is the commercial availability of tests for routine analysis. 102102. Hafeez AS, Bavry AA. Diagnosis of Transthyretin Amyloid Cardiomyopathy. Cardiol Ther. 2020;9(1):85-95.
6. Rational Diagnostic Approach to Cardiac Amyloidosis
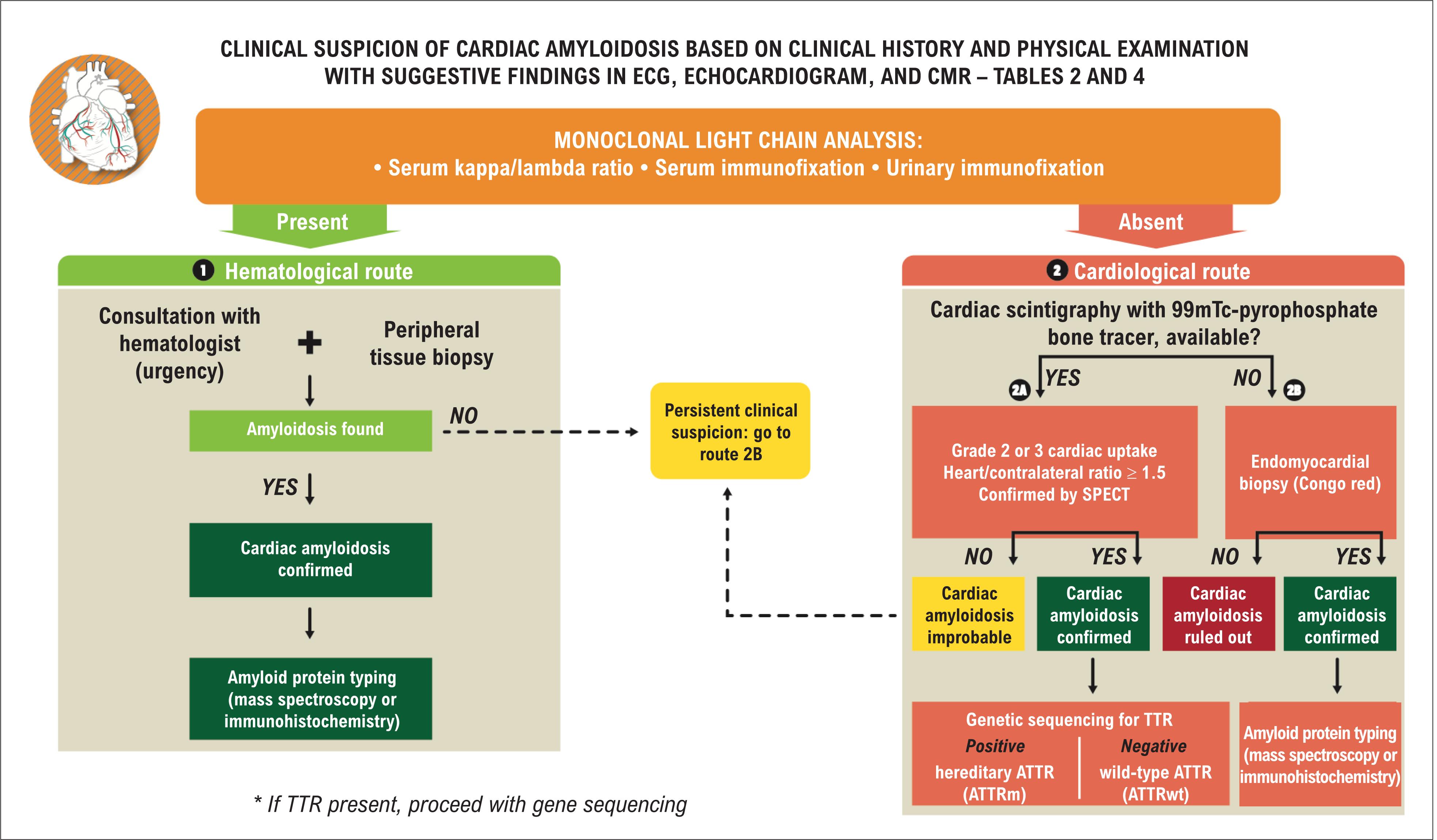
Recent evidence indicates that CA, particularly the ATTRwt form, is more prevalent than previously estimated, and this is due in part to widespread underdiagnosis and the fact that this disease mimics other heart diseases, such as hypertrophic cardiomyopathy, non-amyloid HFpEF, and low-flow/low-gradient aortic stenosis. 4242. Rapezzi C, Lorenzini M, Longhi S, Milandri A, Gagliardi C, Bartolomei I, et al. Cardiac amyloidosis: the great pretender. Heart Fail Rev. 2015;20(2):117-24. These factors indicate that high suspicion of the disease in different clinical scenarios is needed for a more rational diagnostic process. 5454. Kittleson MM, Maurer MS, Ambardekar AV, Bullock-Palmer RP, Chang PP, Eisen HJ, et al. Cardiac Amyloidosis: Evolving Diagnosis and Management: A Scientific Statement From the American Heart Association. Circulation. 2020;142(1):e7-e22. Figure 11 shows a diagnostic algorithm for CA, whose steps are discussed below. The main recommendations and classes of evidence for diagnosis are listed in Table 5 .

The first and most important step is clinical suspicion, which is based on clinical history, a physical examination (see Table 2 ) with findings suggestive of CA in ECG, echocardiogram, and CMR (see Table 4 ).
Thus, in cases with high clinical suspicion, an investigation of immunoglobulin monoclonal light chains should be performed for effective screening of AL-CA, given that an AL-CA diagnosis is a medical emergency, and treatment delay should be avoided, since it is associated with a markedly worse prognosis.
Protein electrophoresis is not an adequate screening test since this method may not detect the monoclonal component in blood and/or urine. Thus, it is important to carry out immunofixation in blood and urine, which increases the detection sensitivity for clonal light chains to around 90%. 103103. Rajkumar SV. Multiple myeloma: 2011 update on diagnosis, risk-stratification, and management. Am J Hematol. 2011;86(1):57-65. Adding the serum free light chain ratio, which detects an abnormal relationship between kappa/lambda chains (> 1.65 or < 0.26), increases the detection sensitivity to > 99%. 103103. Rajkumar SV. Multiple myeloma: 2011 update on diagnosis, risk-stratification, and management. Am J Hematol. 2011;86(1):57-65. , 104104. Comenzo RL, Reece D, Palladini G, Seldin D, Sanchorawala V, Landau H, et al. Consensus guidelines for the conduct and reporting of clinical trials in systemic light-chain amyloidosis. Leukemia. 2012;26(11):2317-25. Therefore, electrophoresis with immunofixation in blood and urine associated with free light chain ratio analysis represents the best non-invasive method for detecting clonal light chains, which indicate the presence of AL.
The patients with positive results for monoclonal light chains must be referred to a hematologist (following the hematological route on the algorithm), and a tissue biopsy must be performed, which is fundamental for confirming amyloid protein deposition and definition of the therapeutic strategy.
6.1. Hematological Route
6.1.1Hematologist Participation and Peripheral Tissue Biopsy
An AL-CA diagnosis must be confirmed by biopsy. Abdominal fat biopsy, a simple and safe method, is preferred initially. 105105. Duston MA, Skinner M, Meenan RF, Cohen AS. Sensitivity, specificity, and predictive value of abdominal fat aspiration for the diagnosis of amyloidosis. Arthritis Rheum. 1989;32(1):82-5.
Congo red staining with birefringence under polarized light is used to determine tissue amyloid protein. In cases of systemic amyloidosis, ie, affecting numerous organs or tissues, abdominal fat biopsy with this stain has a sensitivity of 60-80% and a specificity of 90-100% for diagnosing amyloidosis, 106106. van G, II, Hazenberg BP, Bijzet J, van Rijswijk MH. Diagnostic accuracy of subcutaneous abdominal fat tissue aspiration for detecting systemic amyloidosis and its utility in clinical practice. Arthritis Rheum. 2006;54(6):2015-21. as well as a strong association with the total body load of amyloid deposition. 3737. Maurer MS, Elliott P, Comenzo R, Semigran M, Rapezzi C. Addressing Common Questions Encountered in the Diagnosis and Management of Cardiac Amyloidosis. Circulation. 2017;135(14):1357-77. However, in cases of localized amyloidosis, ie, restricted to one organ or tissue, a subcutaneous fat tissue biopsy is seldom positive. 107107. Andrews TR, Colon-Otero G, Calamia KT, Menke DM, Boylan KB, Kyle RA. Utility of subcutaneous fat aspiration for diagnosing amyloidosis in patients with isolated peripheral neuropathy. Mayo Clin Proc. 2002;77(12):1287-90. In general, there is a higher chance of positive extracardiac biopsy results when the site is abdominal fat and the amyloidosis is AL. AL is followed by ATTRv and ATTRwt in this regard. 3737. Maurer MS, Elliott P, Comenzo R, Semigran M, Rapezzi C. Addressing Common Questions Encountered in the Diagnosis and Management of Cardiac Amyloidosis. Circulation. 2017;135(14):1357-77. In a series of 131 patients whose ATTR-CA diagnosis was confirmed by endomyocardial biopsy, the abdominal fat biopsy was positive in 67% of ATTRv patients but only in 14% of ATTRwt patients. 88. Koike H, Katsuno M. Ultrastructure in Transthyretin Amyloidosis: From Pathophysiology to Therapeutic Insights. Biomedicines. 2019;7(1):11. Therefore, although abdominal fat is the preferred initial site for extracardiac biopsies, a negative result should not exclude the diagnosis, and an endomyocardial biopsy should be performed. 3737. Maurer MS, Elliott P, Comenzo R, Semigran M, Rapezzi C. Addressing Common Questions Encountered in the Diagnosis and Management of Cardiac Amyloidosis. Circulation. 2017;135(14):1357-77. In these cases, biopsy of the affected organ has 100% sensitivity and specificity.
It should be pointed out that although Congo red staining can confirm amyloid infiltration in tissue, it cannot identify the type of precursor protein. In addition, 40% of ATTR patients may have a monoclonal gammopathy of undetermined significance, presenting positive results in a monoclonal light chain analysis. 109109. Phull P, Sanchorawala V, Connors LH, Doros G, Ruberg FL, Berk JL, et al. Monoclonal gammopathy of undetermined significance in systemic transthyretin amyloidosis (ATTR). Amyloid. 2018;25(1):62-7.
In light of these aspects, since identifying the amyloid deposition type is fundamental for appropriate treatment, immunohistochemistry or, preferably, laser microdissection and mass spectrometry of the amyloid biopsy material must be performed. Immunohistochemistry remains the most widely available method for identifying the deposition type. However, when the amyloidosis is light chain, the results are not always conclusive; they may be positive for more than one type of antiserum, usually TTR and kappa or lambda chain. Thus, mass spectrometry has become the new gold standard for identifying amyloid deposition type. 3737. Maurer MS, Elliott P, Comenzo R, Semigran M, Rapezzi C. Addressing Common Questions Encountered in the Diagnosis and Management of Cardiac Amyloidosis. Circulation. 2017;135(14):1357-77. , 5454. Kittleson MM, Maurer MS, Ambardekar AV, Bullock-Palmer RP, Chang PP, Eisen HJ, et al. Cardiac Amyloidosis: Evolving Diagnosis and Management: A Scientific Statement From the American Heart Association. Circulation. 2020;142(1):e7-e22. , 110110. Vrana JA, Gamez JD, Madden BJ, Theis JD, Bergen HR, 3rd, Dogan A. Classification of amyloidosis by laser microdissection and mass spectrometry-based proteomic analysis in clinical biopsy specimens. Blood. 2009;114(24):4957-9.
For negative monoclonal light chain results , ie, when ATTR is more likely, although other rare forms of CA may also be diagnosed, the investigation should follow the cardiological route , taking two sub-routes according to the availability of scintigraphy with bone markers.
6.2. Cardiological Route
When scintigraphy with bone tracers (sub-route 2A) is available and monoclonal light chains are absent, grades 2 or 3 cardiac uptake (equivalent to or greater than that of the costal archs) and a ≥ 1.5 H/CL uptake ratio, with SPECT imaging showing that the increased uptake is in the ventricular walls, confirms ATTR-CA without the need for endomyocardial biopsy. TTR gene sequencing should then be performed to determine whether the ATTR is hereditary or wild type.
Differentiating between hereditary and wild-type ATTR has prognostic and therapeutic implications and is also important for family screening and genetic counseling.
When cardiac scintigraphy with bone tracers is negative and is associated with an absence of monoclonal light chains, CA is unlikely. However, when clinical suspicion persists, based mainly on the results of other imaging methods highly suggestive of amyloidosis, endomyocardial biopsy can have a relevant diagnostic role and should be performed. Such cases could indicate ATTRv involving mutations and amyloid deposits, to which bone tracers do not adhere, such as early-onset V30M and P64I, in addition to other unusual types of amyloidosis. 111111. Musumeci MB, Cappelli F, Russo D, Tini G, Canepa M, Milandri A, et al. Low Sensitivity of Bone Scintigraphy in Detecting Phe64Leu Mutation-Related Transthyretin Cardiac Amyloidosis. JACC Cardiovasc Imaging. 2020;13(6):1314-21.
When bone scintigraphy is unavailable (sub-route 2B) , endomyocardial biopsy is recommended to clarify the diagnosis.
7. Prognosis and Staging
7.1. AL-CA
AL-CA patients have faster-forming and more toxic amyloid deposits than ATTR-CA patients. As a consequence, monthly increases of 1.45 to 2.16 mm can be observed in myocardial thickness, which are associated with higher elevations of biomarkers, such as troponin and BNP, 112112. Binder C, Duca F, Stelzer PD, Nitsche C, Rettl R, Aschauer S, et al. Mechanisms of heart failure in transthyretin vs. light chain amyloidosis. Eur Heart J Cardiovasc Imaging. 2019;20(5):512-24. as well as the development of HF symptoms and death within an average of 6 months after diagnosis. 113113. Podduturi V, Armstrong DR, Hitchcock MA, Roberts WC, Guileyardo JM. Isolated atrial amyloidosis and the importance of molecular classification. Proc (Bayl Univ Med Cent). 2013;26(4):387-9.
Recent technological and laboratory advances indicate that the main prognostic determinant in AL-CA is the extent of amyloid deposition in the heart. 114114. Gertz MA. Immunoglobulin light chain amyloidosis: 2018 Update on diagnosis, prognosis, and treatment. Am J Hematol. 2018;93(9):1169-80. The most widely used staging criteria were developed by the Mayo Clinic (shown in Table 6 ), which consider individuals to be at higher risk when troponin T ≥ 0.025 ng/mL, NT-ProBNP ≥ 1,800 pg/ml, and the difference between the light chains ≥ 18 mg/dL. 115115. Kumar S, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Colby C, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. 2012;30(9):989-95.
Prognostic staging of light-chain amyloidosis according to revised Mayo Clinic criteria 115115. Kumar S, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Colby C, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. 2012;30(9):989-95.
Additionally, echocardiographic findings such as a marked increase in wall thickness, diastolic dysfunction, LV dysfunction, valve thickening, and reduction in LV global longitudinal strain, combined with BNP and troponin biomarkers and hematological status, are predictors of higher mortality. 116116. Koyama J, Falk RH. Prognostic significance of strain Doppler imaging in light-chain amyloidosis. JACC Cardiovasc Imaging. 2010;3(4):333-42. Myocardial elastography, a new non-invasive myocardial assessment technique that assesses heart stiffness, has shown a good correlation with ventricular mass, myocardial thickness, biomarkers such as BNP, filling pressures, worsening functional class, and diastolic dysfunction. 117117. Pislaru C, Ionescu F, Alashry M, Petrescu I, Pellikka PA, Grogan M, et al. Myocardial Stiffness by Intrinsic Cardiac Elastography in Patients with Amyloidosis: Comparison with Chamber Stiffness and Global Longitudinal Strain. J Am Soc Echocardiogr. 2019;32(8):958-68 e4.
CMR also provides data that correlates with survival in CA patients. 8484. White JA, Kim HW, Shah D, Fine N, Kim KY, Wendell DC, et al. CMR imaging with rapid visual T1 assessment predicts mortality in patients suspected of cardiac amyloidosis. JACC Cardiovasc Imaging. 2014;7(2):143-56. , 119119. Raina S, Lensing SY, Nairooz RS, Pothineni NV, Hakeem A, Bhatti S, et al. Prognostic Value of Late Gadolinium Enhancement CMR in Systemic Amyloidosis. JACC Cardiovasc Imaging. 2016;9(11):1267-77. – 121121. Rapezzi C, Merlini G, Quarta CC, Riva L, Longhi S, Leone O, et al. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation. 2009;120(13):1203-12. The presence and extent of myocardial fibrosis, detected by late gadolinium enhancement, indicate poor prognosis. 118118. Kuruvilla S, Adenaw N, Katwal AB, Lipinski MJ, Kramer CM, Salerno M. Late gadolinium enhancement on cardiac magnetic resonance predicts adverse cardiovascular outcomes in nonischemic cardiomyopathy: a systematic review and meta-analysis. Circ Cardiovasc Imaging. 2014;7(2):250-8. – 119119. Raina S, Lensing SY, Nairooz RS, Pothineni NV, Hakeem A, Bhatti S, et al. Prognostic Value of Late Gadolinium Enhancement CMR in Systemic Amyloidosis. JACC Cardiovasc Imaging. 2016;9(11):1267-77. A recent study found that T1 mapping measurements > 1,044 ms and extracellular volume measurements (calculated in the equilibrium phase) > 0.45 are associated with 5.84 and 3.48 times higher cardiovascular mortality, respectively. 99. Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019;73(22):2872-91. , 1010. Mankad AK, Sesay I, Shah KB. Light-chain cardiac amyloidosis. Curr Probl Cancer. 2017;41(2):144-56.
Biomarkers not only play a role in staging these patients, but also enable treatment response assessment, which is always a challenge in clinical practice, especially for AL-CA, since chemotherapy can be an additional factor in myocardial damage. What has been well established in the literature is the agreement between lower NT-proBNP levels and hematologic response to treatment, as well as improvement in New York Heart Association (NYHA) functional class. The reverse is also true, such as increased levels of NT-proBNP and troponin and reduced ejection fraction. (see Table 9 regarding AL-CA treatment)
7.2. ATTR-CA
ATTR-CA has a more benign profile and is associated with longer survival than AL-CA. 121121. Rapezzi C, Merlini G, Quarta CC, Riva L, Longhi S, Leone O, et al. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation. 2009;120(13):1203-12. In addition to the specific type of amyloid deposition, other factors such as genotype, clinical data, laboratory biomarkers, and radiological findings can guide therapeutic planning and determine prognosis. 1313. Muchtar E, Dispenzieri A, Magen H, Grogan M, Mauermann M, McPhail ED, et al. Systemic amyloidosis from A (AA) to T (ATTR): a review. J Intern Med. 2021;289(3):268-92.
In cases of hereditary ATTR, the genetic profile determines the clinical phenotype and evolution of the disease, although the gene’s distribution may vary in different populations. Thus, the survival time for pathogenic variants with predominantly myocardial involvement, such as V122I, is shorter than for ATTRwt and mixed or predominantly neuropathic variants such as V30M. 2020. Lane T, Fontana M, Martinez-Naharro A, Quarta CC, Whelan CJ, Petrie A, et al. Natural History, Quality of Life, and Outcome in Cardiac Transthyretin Amyloidosis. Circulation. 2019;140(1):16-26. , 122122. Chacko L, Martone R, Bandera F, Lane T, Martinez-Naharro A, Boldrini M, et al. Echocardiographic phenotype and prognosis in transthyretin cardiac amyloidosis. Eur Heart J. 2020;41(14):1439-47. , 123123. Siddiqi OK, Ruberg FL. Cardiac amyloidosis: An update on pathophysiology, diagnosis, and treatment. Trends Cardiovasc Med. 2018;28(1):10-21.
From a clinical point of view, the onset and duration of HF symptoms have prognostic value. 124124. Sugiura A, Kitahara H, Iwahana T, Suzuki N, Okada S, Miyauchi H, et al. Association of heart failure duration with clinical prognosis in advanced heart failure. Clin Res Cardiol. 2020;109(3):350-7. It has been determined that the longer the clinical decompensation and the more advanced the NYHA functional class, the lower the survival. 125125. Kristen AV, Perz JB, Schonland SO, Hegenbart U, Schnabel PA, Kristen JH, et al. Non-invasive predictors of survival in cardiac amyloidosis. Eur J Heart Fail. 2007;9(6-7):617-24. , 126126. Pinney JH, Whelan CJ, Petrie A, Dungu J, Banypersad SM, Sattianayagam P, et al. Senile systemic amyloidosis: clinical features at presentation and outcome. J Am Heart Assoc. 2013;2(2):e000098. The median survival observed for each functional class is: I = 4.6 years, II = 4.1 years, III = 2.1 years and IV = 1.3 years. Although sex, age at diagnosis, and comorbidities such as aortic valve disease or tachyarrhythmias do not have a direct effect on mortality, a greater association with unfavorable cardiac outcomes has been observed in older men with associated diseases. 127127. Maurer MS, Hanna M, Grogan M, Dispenzieri A, Witteles R, Drachman B, et al. Genotype and Phenotype of Transthyretin Cardiac Amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J Am Coll Cardiol. 2016;68(2):161-72.
The results of inexpensive, quick, and easy-to-interpret laboratory tests have been used in different proposals for staging the disease, being closely correlated with life expectancy. ( Table 7 ). 128128. Grogan M, Scott CG, Kyle RA, Zeldenrust SR, Gertz MA, Lin G, et al. Natural History of Wild-Type Transthyretin Cardiac Amyloidosis and Risk Stratification Using a Novel Staging System. J Am Coll Cardiol. 2016;68(10):1014-20. – 130130. Gillmore JD, Damy T, Fontana M, Hutchinson M, Lachmann HJ, Martinez-Naharro A, et al. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J. 2018;39(30):2799-806. Serum levels of troponins T and I, NT-proBNP, and glomerular filtration rate reflect the toxicity of amyloid deposition in target organs by direct action, as well as oxidative stress and inflammation processes. 131131. Cappelli F, Martone R, Gabriele M, Taborchi G, Morini S, Vignini E, et al. Biomarkers and Prediction of Prognosis in Transthyretin-Related Cardiac Amyloidosis: Direct Comparison of Two Staging Systems. Can J Cardiol. 2020;36(3):424-31. Recommendations for CA risk stratification/staging and treatment response monitoring are shown in Table 8 .
Recommendations for risk stratification/staging of cardiac amyloidosis and monitoring treatment response and/or disease progression
Imaging findings, such as echocardiography, cardiac scintigraphy with technetium-pyrophosphate (99mTc-PYP), and CMR, provide complementary data that facilitate prognostic stratification. 99. Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019;73(22):2872-91. , 8080. Fontana M, Pica S, Reant P, Abdel-Gadir A, Treibel TA, Banypersad SM, et al. Prognostic Value of Late Gadolinium Enhancement Cardiovascular Magnetic Resonance in Cardiac Amyloidosis. Circulation. 2015;132(16):1570-9. , 9494. Castano A, Haq M, Narotsky DL, Goldsmith J, Weinberg RL, Morgenstern R, et al. Multicenter Study of Planar Technetium 99m Pyrophosphate Cardiac Imaging: Predicting Survival for Patients With ATTR Cardiac Amyloidosis. JAMA Cardiol. 2016;1(8):880-9. , 132132. Rubin J, Steidley DE, Carlsson M, Ong ML, Maurer MS. Myocardial Contraction Fraction by M-Mode Echocardiography Is Superior to Ejection Fraction in Predicting Mortality in Transthyretin Amyloidosis. J Card Fail. 2018;24(8):504-11. – 134134. Knight DS, Zumbo G, Barcella W, Steeden JA, Muthurangu V, Martinez-Naharro A, et al. Cardiac Structural and Functional Consequences of Amyloid Deposition by Cardiac Magnetic Resonance and Echocardiography and Their Prognostic Roles. JACC Cardiovasc Imaging. 2019;12(5):823-33. ( Table 9 ). The increasing quality of the images, associated with greater quantitative detailing of amyloid deposits, has reduced the need for myocardial biopsy as a predictor of disease. 99. Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019;73(22):2872-91.
8. Treatment
CA treatment consists of specific measures aimed at reducing or preventing the progression of amyloid fibril deposition. These measures will be addressed separately for AL-CA and ATTR-CA. General measures are also needed to manage the resultant clinical and hemodynamic abnormalities, including heart failure and cardiac rhythm disturbances, which apply to both AL-CA and ATTR-CA.
8.1. Specific Therapy for AL-CA
AL-CA is caused by the aberrant production of kappa or lambda immunoglobulin light chains ( kappa or lambda ). Thus, specific treatment is based on eliminating this light chains production by eradicating plasma cell clones in bone marrow. The rapid reduction and, ideally, normalization of light chain levels (hematologic response) is a primary goal in AL-CA treatment. The reversal of damage to tissues and organs affected by amyloid deposits (organ response) is the second major objective of treatment. Further goals include improved quality of life and overall survival. 135135. Palladini G, Milani P, Merlini G. Management of AL amyloidosis in 2020. Blood. 2020;136(23):2620-7.
After the diagnosis of systemic AL-CA has been confirmed, the therapeutic plan must be defined by the hematologist. However, the identification and management of organ dysfunction is essential, as is multidisciplinary work, including joint follow-up with a cardiologist and other specialists (see Table 12 ). Cardiac involvement is the main prognostic factor in AL-CA, determining not only survival, but also tolerance to cytotoxic treatment. The impact of cardiac impairment on survival has been well established in the literature, and validated staging systems can help with patient risk stratification. These systems are used together with other parameters to determine therapeutic strategies. 135135. Palladini G, Milani P, Merlini G. Management of AL amyloidosis in 2020. Blood. 2020;136(23):2620-7.
Survival in AL-CA is related to the production of amyloidogenic light chains and target organ damage, especially to the heart. Thus, staging includes biomarkers related to these factors, 104104. Comenzo RL, Reece D, Palladini G, Seldin D, Sanchorawala V, Landau H, et al. Consensus guidelines for the conduct and reporting of clinical trials in systemic light-chain amyloidosis. Leukemia. 2012;26(11):2317-25. , 115115. Kumar S, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Colby C, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. 2012;30(9):989-95. , 136136. Palladini G, Dispenzieri A, Gertz MA, Kumar S, Wechalekar A, Hawkins PN, et al. New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. J Clin Oncol. 2012;30(36):4541-9. as described above (see Table 5 ).
The value of this prognostic stratification has been confirmed in individuals treated with autologous hematopoietic stem cell transplantation (HSCT), as well as in those treated without this procedure. The median survival for stage I, II, III, and IV patients is 55, 19, 12 and 5 months, respectively, in patients who did not receive or failed HSCT, and 97, 58 and 22 months, respectively for stages II, III and IV, in patients who underwent HSCT. 115115. Kumar S, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Colby C, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. 2012;30(9):989-95.
Low-risk patients are candidates for high-dose chemotherapy followed by autologous HSCT ( Table 12 ). This is considered the most effective current strategy to eradicate plasma cell clones. 135135. Palladini G, Milani P, Merlini G. Management of AL amyloidosis in 2020. Blood. 2020;136(23):2620-7. Intermediate-risk patients usually receive chemotherapy at conventional doses and, if they show clinical and laboratory improvement, they can become candidates for autologous HSCT. Finally, older or frail patients with multiple affected organs or patients with advanced cardiomyopathy are treated with chemotherapy at adjusted doses and have a poor prognosis, since they generally cannot tolerate the treatment. 135135. Palladini G, Milani P, Merlini G. Management of AL amyloidosis in 2020. Blood. 2020;136(23):2620-7.
Three months after the end of treatment, the hematological and organ responses must be evaluated with specific tests for monoclonal gammopathy and each affected organ. The hematological and organ response criteria are summarized in Tables 10 and 11 . 136136. Palladini G, Dispenzieri A, Gertz MA, Kumar S, Wechalekar A, Hawkins PN, et al. New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. J Clin Oncol. 2012;30(36):4541-9. , 137137. Mahmood S, Palladini G, Sanchorawala V, Wechalekar A. Update on treatment of light chain amyloidosis. Haematologica. 2014;99(2):209-21.
Even when a complete hematological response occurs, dysfunction improves more slowly, and may take months or even years to appear after the light chain ratio has been normalized. 1919. Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, et al. Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation. 2016;133(24):2404-12.
8.1.1. Treating Patients Eligible for Autologous Hematopoietic Stem Cell Transplantation
Only 20% of patients diagnosed with AL amyloidosis are eligible for HSCT, although this number could increase with organ response after induction regimens with effective anti-cancer therapies (bortezomib and daratumumab). 135135. Palladini G, Milani P, Merlini G. Management of AL amyloidosis in 2020. Blood. 2020;136(23):2620-7. , 139139. Palladini G, Kastritis E, Maurer MS, Zonder J, Minnema MC, Wechalekar AD, et al. Daratumumab plus CyBorD for patients with newly diagnosed AL amyloidosis: safety run-in results of ANDROMEDA. Blood. 2020;136(1):71-80. , 140140. Cornell RF, Zhong X, Arce-Lara C, Atallah E, Blust L, Drobyski WR, et al. Bortezomib-based induction for transplant ineligible AL amyloidosis and feasibility of later transplantation. Bone Marrow Transplant. 2015;50(7):914-7.
HSCT eligibility assessment is central to therapeutic success in this strategy. A single prospective randomized study compared HSCT with conventional chemotherapy, finding no overall survival benefit compared to a less intensive strategy. However, in this study the high mortality rate associated with HSCT (24%) was related to the inclusion criteria, the inexperience of the transplant center and the use of a subtherapeutic dose of melphalan. 141141. Jaccard A, Moreau P, Leblond V, Leleu X, Benboubker L, Hermine O, et al. High-dose melphalan versus melphalan plus dexamethasone for AL amyloidosis. N Engl J Med. 2007;357(11):1083-93.
Since the literature provides no precise well-established prospectively validated HSCT eligibility criteria, each center establishes its own. Although subjective assessment is relevant, HSCT can be guided by certain factors: “physiological age” ≤ 70 years, creatinine clearance > 30 mL/min (except for patients on chronic dialysis), troponin T < 0.06 ng/mL, Eastern Cooperative Oncology Group performance scale ≤ 2, NYHA functional class I or II, and systolic blood pressure > 90 mmHg. Adequate pre-treatment risk stratification, concomitant with the development of better supportive conditions, such as antibiotic therapy and intensive care, has led to a reduction in HSCT-associated mortality in recent decades, with rates of 2.4% to 3.4% in retrospective analyses. 142142. Moreau P, Leblond V, Bourquelot P, Facon T, Huynh A, Caillot D, et al. Prognostic factors for survival and response after high-dose therapy and autologous stem cell transplantation in systemic AL amyloidosis: a report on 21 patients. Br J Haematol. 1998;101(4):766-9. – 148148. Gertz MA, Lacy MQ, Dispenzieri A, Kumar SK, Dingli D, Leung N, et al. Refinement in patient selection to reduce treatment-related mortality from autologous stem cell transplantation in amyloidosis. Bone Marrow Transplant. 2013;48(4):557-61.
Therefore, HSCT should be recommended as the first-line therapy in eligible patients ( Table 12 ). This recommendation is due to the achievement of high hematological response rates, leading to reduced production and potential reabsorption of amyloid fibrils, with consequent improvement in organ dysfunction and performance status, as well as increased survival and quality of life. As an example, in a series of 672 AL-CA patients undergoing HSCT, 84% had a hematologic response, 39% of which were a complete response, and survival was > 50% at 15 years. 146146. Sanchorawala V, Sun F, Quillen K, Sloan JM, Berk JL, Seldin DC. Long-term outcome of patients with AL amyloidosis treated with high-dose melphalan and stem cell transplantation: 20-year experience. Blood. 2015;126(20):2345-7. , 147147. Sidiqi MH, Aljama MA, Buadi FK, Warsame RM, Lacy MQ, Dispenzieri A, et al. Stem Cell Transplantation for Light Chain Amyloidosis: Decreased Early Mortality Over Time. J Clin Oncol. 2018;36(13):1323-9. , 149149. Gertz MA, Lacy MQ, Dispenzieri A, Hayman SR, Kumar SK, Leung N, et al. Effect of hematologic response on outcome of patients undergoing transplantation for primary amyloidosis: importance of achieving a complete response. Haematologica. 2007;92(10):1415-8.
An upfront HSCT can be performed soon after diagnosis in patients with < 10% clonal plasma cells in the bone marrow or when preceded by induction therapy with regimens containing bortezomib and/or anti-CD38 monoclonal antibody. The latter should be considered when there is an association with multiple myeloma, a predictable delay in HSCT, poor performance that may improve with induction therapy, and > 10% infiltration of the bone marrow by clonal plasma cells, which is associated with worse prognosis. 135135. Palladini G, Milani P, Merlini G. Management of AL amyloidosis in 2020. Blood. 2020;136(23):2620-7. , 150150. Kourelis TV, Kumar SK, Gertz MA, Lacy MQ, Buadi FK, Hayman SR, et al. Coexistent multiple myeloma or increased bone marrow plasma cells define equally high-risk populations in patients with immunoglobulin light chain amyloidosis. J Clin Oncol. 2013;31(34):4319-24.
The main regimens used as pre-transplant therapy are cyclophosphamide, bortezomib and dexamethasone (CyBorD) and daratumumab, bortezomib, cyclophosphamide and dexamethasone (Dara-CyBorD). 139139. Palladini G, Kastritis E, Maurer MS, Zonder J, Minnema MC, Wechalekar AD, et al. Daratumumab plus CyBorD for patients with newly diagnosed AL amyloidosis: safety run-in results of ANDROMEDA. Blood. 2020;136(1):71-80.
Given that new therapies for treating AL amyloidosis are emerging and the scarcity of randomized studies on transplantation, further studies will be needed to determine whether HSCT will remain the most effective therapy in coming decades.
8.1.2. Treating Patients Ineligible for HSCT/Conventional Chemotherapy
Most patients diagnosed with AL-CA are not eligible for higher intensity autologous HSCT therapy due to comorbidities, such as advanced heart disease, renal failure, the involvement of more than two organs, or advanced age. Thus, in this group of patients, chemotherapy with anti-cancer medications is the basis of treatment ( Table 12 ). The regimens are similar to those for multiple myeloma.
The CyBorD regimen was found to be highly effective (81% to 94%), well tolerated, and capable of producing a rapid hematological response (within 3 months) in two retrospective studies with small samples (n = 17 and 43). Two-year overall survival reached 92% in these studies. 151151. Venner CP, Lane T, Foard D, Rannigan L, Gibbs SD, Pinney JH, et al. Cyclophosphamide, bortezomib, and dexamethasone therapy in AL amyloidosis is associated with high clonal response rates and prolonged progression-free survival. Blood. 2012;119(19):4387-90. , 152152. Mikhael JR, Schuster SR, Jimenez-Zepeda VH, Bello N, Spong J, Reeder CB, et al. Cyclophosphamide-bortezomib-dexamethasone (CyBorD) produces rapid and complete hematologic response in patients with AL amyloidosis. Blood. 2012;119(19):4391-4. The largest sample to have been tested with this regimen included 230 patients, 60% of whom had a hematologic response, and 23% of whom had a complete response. However, cardiac and renal organ responses were observed in only 17% and 25% of patients, respectively. A lower hematologic response rate was observed among patients with advanced heart disease (42% global and 14% complete), with a median overall survival of 7 months for this group 153153. Palladini G, Sachchithanantham S, Milani P, Gillmore J, Foli A, Lachmann H, et al. A European collaborative study of cyclophosphamide, bortezomib, and dexamethasone in upfront treatment of systemic AL amyloidosis. Blood. 2015;126(5):612-5. Since the CyBorD regimen may make patients who are initially ineligible for autologous HSCT eligible (after hematologic response and clinical improvement), it should be considered when developing the therapeutic plan. 153153. Palladini G, Sachchithanantham S, Milani P, Gillmore J, Foli A, Lachmann H, et al. A European collaborative study of cyclophosphamide, bortezomib, and dexamethasone in upfront treatment of systemic AL amyloidosis. Blood. 2015;126(5):612-5. – 155155. Kastritis E, Roussou M, Gavriatopoulou M, Migkou M, Kalapanida D, Pamboucas C, et al. Long-term outcomes of primary systemic light chain (AL) amyloidosis in patients treated upfront with bortezomib or lenalidomide and the importance of risk adapted strategies. Am J Hematol. 2015;90(4):E60-5.
Two recent randomized studies tested chemotherapy combinations as new treatments of choice in AL-CA patients who are ineligible for HSCT. The first compared a regimen of bortezomib, melphalan and dexamethasone to melphalan and dexamethasone, finding that adding bortezomib resulted in a higher overall hematologic response rate after 3 months (79% vs. 52%), a greater cardiac organ response (38% vs. 28%), and improved overall survival, with a 2-fold reduction in mortality. 156156. Kastritis E, Leleu X, Arnulf B, Zamagni E, Cibeira MT, Kwok F, et al. Bortezomib, Melphalan, and Dexamethasone for Light-Chain Amyloidosis. J Clin Oncol. 2020;38(28):3252-60. The largest randomized trial for AL-CA (ANDROMEDA) associated an anti-CD38 monoclonal antibody (daratumumab) with the CyBorD regimen, finding promising results. 139139. Palladini G, Kastritis E, Maurer MS, Zonder J, Minnema MC, Wechalekar AD, et al. Daratumumab plus CyBorD for patients with newly diagnosed AL amyloidosis: safety run-in results of ANDROMEDA. Blood. 2020;136(1):71-80. The Dara-CyBorD group had an overall hematologic response of 92%, compared to 77% in the CyBorD group, with an OR of 53% vs. 18%, which took a median of 2 months to achieve. Better organ function was also observed in the group that received the monoclonal antibody, with improved progression-free survival. Importantly, advanced heart disease patients (classified as stage IIIb) were excluded from both of these randomized studies, and treatment for this group remains a challenge in clinical practice.
8.2. ATTR-specific Therapies
Several steps in the pathophysiological process of amyloid fibril formation and deposition in cardiac tissue are potential therapeutic targets in ATTR-CA, as illustrated in Figure 12 , including: 1) liver transplantation; 2) TTR tetramer stabilizers; 3) hepatic TTR synthesis inhibitors; and 4) the degradation and resorption of deposited amyloid fibrils.

The pathophysiological process of amyloid fibril deposition in ATTR and recognized therapeutic targets.
8.2.1. Liver Transplantation
In the past, liver transplantation has been proposed as a treatment for patients with ATTRv-associated polyneuropathy. 157157. Holmgren G, Steen L, Ekstedt J, Groth CG, Ericzon BG, Eriksson S, et al. Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP-met30). Clin Genet. 1991;40(3):242-6. Liver transplantation, which removes the source of mutated TTR molecules, is associated with increased survival, with a reported 20-year survival rate of 55.3%. However, TTR deposition may continue after liver transplantation and is associated with heart disease progression, probably because the accumulated amyloid fibrils in the myocardium promote additional deposition of wild-type TTR over time. 158158. Liepnieks JJ, Zhang LQ, Benson MD. Progression of transthyretin amyloid neuropathy after liver transplantation. Neurology. 2010;75(4):324-7. , 159159. Okamoto S, Zhao Y, Lindqvist P, Backman C, Ericzon BG, Wijayatunga P, et al. Development of cardiomyopathy after liver transplantation in Swedish hereditary transthyretin amyloidosis (ATTR) patients. Amyloid. 2011;18(4):200-5. Thus, heart and liver transplantation may be possible and appears to be associated with better prognosis than the transplantation of either organ alone. 160160. Sack FU, Kristen A, Goldschmidt H, Schnabel PA, Dengler T, Koch A, et al. Treatment options for severe cardiac amyloidosis: heart transplantation combined with chemotherapy and stem cell transplantation for patients with AL-amyloidosis and heart and liver transplantation for patients with ATTR-amyloidosis. Eur J Cardiothorac Surg. 2008;33(2):257-62. Considering the reduced availability of organs and transplant centers, in addition to the risks posed by lifelong immunosuppression, the development of new therapies capable of blocking hepatic TTR synthesis should replace liver transplantation as a means of suppressing TTR production.
8.2.2. Selective Transthyretin Kinetic Stabilizers
8.2.2.1. Tafamidis
Tafamidis is a small molecule that selectively inhibits the dissociation of TTR tetramers by binding to thyroxine binding sites, thereby effectively inhibiting the cascade that results in amyloid fibril formation. 161161. Bulawa CE, Connelly S, Devit M, Wang L, Weigel C, Fleming JA, et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci U S A. 2012;109(24):9629-34. When tested in a phase 3 clinical trial, tafamidis was effectively reduced the progression of neurological manifestations in patients with early-stage ATTRv polyneuropathy. 162162. Coelho T, Maia LF, Martins da Silva A, Waddington Cruz M, Plante-Bordeneuve V, Lozeron P, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79(8):785-92.
Tafamidis is the only drug to have been specifically tested in CA patients in a prospective, randomized, placebo-controlled, multicenter clinical trial (ATTR-ACT). 163163. Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington-Cruz M, et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N Engl J Med. 2018;379(11):1007-16. This phase 3 trial included 441 patients (18 to 90 years of age) diagnosed with hereditary or wild-type ATTR cardiomyopathy who were characterized by a history of heart failure, interventricular septal thickness > 12 mm on echocardiography, TTR amyloid deposits (confirmed by biopsy or positive bone marker scintigraphy), NT-Pro-BNP > 600 pg/mL, and > 100 meters walked in the 6-minute walk test. The main exclusion criteria were: NYHA functional class IV, AL-CA, and GFR < 25 mL/min/1.73 m2. The patients were randomized to receive tafamidis 80 mg/day, tafamidis 20 mg/day, or placebo in a 2:1:2 proportion. The study’s primary endpoint was hierarchically assessed all-cause mortality, followed by the frequency of cardiovascular hospitalization over 30 months of follow-up. The main secondary outcomes were change in 6-minute walk test results and quality of life scores according to the Kansas City Cardiomyopathy Questionnaire. The tafamidis 80 and 20 mg groups were merged for the statistical analysis, and the results indicated that these patients (n = 264) had a 30% lower relative risk of all-cause mortality (RR = 0.70 [95% CI: 0 .51-0.96]), 32% fewer cardiovascular hospitalizations (RR = 0.68 [95% CI: 0.56-0.81]), a lower rate of decline in 6-minute walk test results (p < 0.001), and a lower rate of decline in Kansas City Cardiomyopathy Questionnaire scores (p < 0.001) than the placebo group (n = 177). The Kaplan-Meier survival curves showed that tafamidis resulted in lower all-cause mortality, with the curves diverging after approximately 18 months of treatment, a result that agrees with the concept that tafamidis modifies the natural history of the disease. Tafamidis was well tolerated, with a similar incidence of adverse effects in the treatment and placebo groups. In the patient subgroup analysis, tafamidis was associated with lower all-cause mortality than placebo independently of NYHA functional class or ATTR genotype (hereditary or wild-type). However, patients in NYHA functional class III at the time of inclusion who were allocated to the tafamidis group had higher rates of hospitalization than the placebo group, a result likely explained by longer survival during a more severe disease phase. This subgroup analysis demonstrates the need for a trial with a large enough sample size to specifically assess the effect of tafamidis in ATTR patients with more advanced symptoms of heart failure.
More recently, an open-label extension study of ATTR-ACT found that 80 mg/day of tafamidis resulted in significantly higher survival than 20 mg/day (RR = 0.70 [95%CI: 0.50 - 0.979], p = 0.0374). 164164. Damy T, Garcia-Pavia P, Hanna M, Judge DP, Merlini G, Gundapaneni B, et al. Efficacy and safety of tafamidis doses in the Tafamidis in Transthyretin Cardiomyopathy Clinical Trial (ATTR-ACT) and long-term extension study. Eur J Heart Fail. 2020.
Based on this evidence, tafamidis 80 mg/day is recommended for patients with ATTRv or ATTRwt, with NYHA I to III HF, without severe renal dysfunction, and who are beginning therapy at the earliest stages of the disease ( Table 12 ). In Brazil, tafamidis 80 mg/day was approved by the Brazilian Health Regulatory Agency for treating ATTR-CA.
8.2.2.2. AG10
AG10 is a selective TTR tetramer stabilizer designed to mimic the structural influence of a super-stabilizing mutation (T119M), which significantly reduces the dissociation rate of tetramers. 165165. Hammarstrom P, Jiang X, Hurshman AR, Powers ET, Kelly JW. Sequence-dependent denaturation energetics: A major determinant in amyloid disease diversity. Proc Natl Acad Sci U S A. 2002;99 Suppl 4:16427-32. This agent was evaluated in a multicenter, phase 2, randomized, double-blind, placebo-controlled trial that included 49 patients with symptomatic ATTR cardiomyopathy and NYHA functional class II or III. The treatment was well tolerated and resulted in almost complete TTR stabilization. 166166. Judge DP, Heitner SB, Falk RH, Maurer MS, Shah SJ, Witteles RM, et al. Transthyretin Stabilization by AG10 in Symptomatic Transthyretin Amyloid Cardiomyopathy. J Am Coll Cardiol. 2019;74(3):285-95. There is an ongoing phase 3 multicenter clinical trial testing the effects of AG10 in CA patients. (ClinicalTrial.gov – Identifier: NCT03860935).
8.2.3. Inhibitors of Hepatic TTR Synthesis
Therapies based on silencing the expression of genes that encode hepatic TTR production are very promising, including RNA interference (patisiran) and antisense oligonucleotide (inotersen) strategies. Both drugs have been tested in multicenter phase 3 trials in patients with ATTRv polyneuropathy and were shown to be effective in reducing the progression of neurological manifestations. 167167. Adams D, Gonzalez-Duarte A, O’Riordan WD, Yang CC, Ueda M, Kristen AV, et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med. 2018;379(1):11-21. , 168168. Benson MD, Waddington-Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N Engl J Med. 2018;379(1):22-31. Post-hoc analyses of subgroups of CA patients in these studies suggest positive effects on the progression of cardiomyopathy. Both classes of gene expression silencers are currently being tested in multicenter phase 3 trials in ATTR-CA patients (ClinicalTrials.gov Identifiers: NCT03997383 and NCT04136171).
8.2.4. Degradation and Resorption of Amyloid Fibrils
In vitro and experimental studies have shown that certain compounds based on hydrophobic molecules promote the degradation of amyloid tissue deposits, allowing their resorption through the macrophage system. 169169. Emdin M, Aimo A, Rapezzi C, Fontana M, Perfetto F, Seferovic PM, et al. Treatment of cardiac transthyretin amyloidosis: an update. Eur Heart J. 2019;40(45):3699-706. Experimental studies have found that doxycycline, an antibiotic of the tetracycline family, is one such compound, 170170. Cardoso I, Saraiva MJ. Doxycycline disrupts transthyretin amyloid: evidence from studies in a FAP transgenic mice model. FASEB J. 2006;20(2):234-9. having synergistic effects with tauroursodeoxycholic acid. 171171. Cardoso I, Martins D, Ribeiro T, Merlini G, Saraiva MJ. Synergy of combined doxycycline/TUDCA treatment in lowering Transthyretin deposition and associated biomarkers: studies in FAP mouse models. J Transl Med. 2010;8:74. Although promising in preclinical studies, clinical experience with this approach is limited and has not established its efficacy or provided recommendations for its use.
9. Heart Failure Syndrome Management
In addition to specific therapy for amyloidosis, supportive treatment for HF may be necessary. CA initially presents as HFpEF and a restrictive pattern of LV filling, which could lead to disease progression and reduced EF. This pathophysiological mechanism could explain the difficulties in clinical management of CA patients when using HFrEF medications. 2020. Lane T, Fontana M, Martinez-Naharro A, Quarta CC, Whelan CJ, Petrie A, et al. Natural History, Quality of Life, and Outcome in Cardiac Transthyretin Amyloidosis. Circulation. 2019;140(1):16-26. , 2121. Gonzalez-Lopez E, Gallego-Delgado M, Guzzo-Merello G, de Haro-Del Moral FJ, Cobo-Marcos M, Robles C, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36(38):2585-94.
Maintaining euvolemia through water restriction and medications is the focus. Loop diuretics, the most frequently used medication, are indicated to reduce pulmonary and systemic congestion, and they may be associated with aldosterone antagonists ( Table 12 ). Using diuretics to achieve euvolemia can be a challenge, since excessive dosage can impair renal function and/or result in low cardiac output due to reduced preload in hearts with already reduced stroke volume. 172172. Ritts AJ, Cornell RF, Swiger K, Singh J, Goodman S, Lenihan DJ. Current concepts of cardiac amyloidosis: diagnosis, clinical management, and the need for collaboration. Heart Failure Clinics. 2017;13(2):409-16. , 173173. Pollak A, Falk RH. Left ventricular systolic dysfunction precipitated by verapamil in cardiac amyloidosis. Chest. 1993;104(2):618-20. Furthermore, in patients with autonomic polyneuropathy, hypotension may impede diuretic use due to unstable preload conditions. 172172. Ritts AJ, Cornell RF, Swiger K, Singh J, Goodman S, Lenihan DJ. Current concepts of cardiac amyloidosis: diagnosis, clinical management, and the need for collaboration. Heart Failure Clinics. 2017;13(2):409-16.
Regarding HFrEF drugs, there is no scientific evidence that neurohormonal antagonists such as angiotensin II-converting enzyme inhibitors, angiotensin II receptor blockers, and beta blockers, or even substances recently described as neprilysin inhibitors, angiotensin II receptor antagonists, and SGLT2 inhibitors, have any effect on CA, and they also involve the risk of hypotension and autonomic dysfunction. In a retrospective, single-center study of 99 CA patients (33% AL-CA and 67% ATTR-CA), Aimo et al. 173173. Pollak A, Falk RH. Left ventricular systolic dysfunction precipitated by verapamil in cardiac amyloidosis. Chest. 1993;104(2):618-20. observed that an association of angiotensin II-converting enzyme inhibitors/angiotensin II receptor blockers and mineralocorticoid antagonists can be used safely and with gradual dose adjustments if there are no contraindications, as well as that tolerance for beta-blockers is lower in AL-CA patients with left or right ventricular dysfunction. Beta-blockers and non-dihydropyridine calcium channel blockers are usually not well tolerated, because, due to the low ejected systolic volume, cardiac output is maintained through increased heart rate. In addition, non-dihydropyridine calcium channel blockers should be avoided in in AL-CA patients, since they bind to amyloid fibrils, which can result in advanced blocks and cardiogenic shock. 172172. Ritts AJ, Cornell RF, Swiger K, Singh J, Goodman S, Lenihan DJ. Current concepts of cardiac amyloidosis: diagnosis, clinical management, and the need for collaboration. Heart Failure Clinics. 2017;13(2):409-16. , 173173. Pollak A, Falk RH. Left ventricular systolic dysfunction precipitated by verapamil in cardiac amyloidosis. Chest. 1993;104(2):618-20.
9.1. Arrhythmia Management
Arrhythmias are very common in CA patients and are usually symptomatic and poorly tolerated. Arrhythmia assessment in this population should involve three different situations: atrial arrhythmias, ventricular arrhythmias, and conduction system disease.
9.1.1. Atrial Arrhythmia and Anticoagulants
Amyloid deposition leads to atrial thickening, including changes in atrial relaxation and increased intracavitary pressure, which produces atrial dilatation that, in association with atrial fibrosis, predisposes patients to atrial fibrillation (AF) or other atrial arrhythmias. The prevalence of AF in CA patients can range from 11% to 71%, being even higher in ATTR-CA, possibly because it affects older men. 174174. Sanchis K, Cariou E, Colombat M, Ribes D, Huart A, Cintas P, et al. Atrial fibrillation and subtype of atrial fibrillation in cardiac amyloidosis: clinical and echocardiographic features, impact on mortality. Amyloid. 2019;26(3):128-38. , 175175. Longhi S, Quarta CC, Milandri A, Lorenzini M, Gagliardi C, Manuzzi L, et al. Atrial fibrillation in amyloidotic cardiomyopathy: prevalence, incidence, risk factors and prognostic role. Amyloid. 2015;22(3):147-55.
Managing AF in these patients is generally difficult, since they usually cannot tolerate drugs such as beta-blockers, non-dihydropyridine calcium channel blockers or digitalis due to the fact that they cause postural hypotension and HF decompensation. Given the need for these medications, it is advisable to use low doses with careful hemodynamic monitoring, in addition to serum level control, if digoxin is used. Regarding rhythm control, a retrospective analysis found no difference in survival between patients who received antiarrhythmic drugs and those treated with frequency control alone. 176176. Mints YY, Doros G, Berk JL, Connors LH, Ruberg FL. Features of atrial fibrillation in wild-type transthyretin cardiac amyloidosis: a systematic review and clinical experience. ESC heart failure. 2018;5(5):772-9. Recent data suggest that catheter ablation may be associated with lower mortality in CA patients, especially when performed early. However, these data are from small retrospective observational studies that also had a high rate of AF recurrence. 177177. Donnellan E, Wazni O, Kanj M, Elshazly MB, Hussein A, Baranowski B, et al. Atrial fibrillation ablation in patients with transthyretin cardiac amyloidosis. EP Europace. 2020;22(2):259-64.
Finally, the reduced contractility caused by amyloid infiltration in atrial tissue may also contribute to thrombus formation. Autopsy studies have found that up to 33% CA patients have intracavitary thrombi, 178178. Roberts WC, Waller BF. Cardiac amyloidosis causing cardiac dysfunction: analysis of 54 necropsy patients. The American journal of cardiology. 1983;52(1):137-46. while retrospective studies have found a prevalence of 15 to 33%. 179179. Feng D, Edwards WD, Oh JK, Chandrasekaran K, Grogan M, Martinez MW, et al. Intracardiac thrombosis and embolism in patients with cardiac amyloidosis. Circulation. 2007;116(21):2420-6. , 180180. Feng D, Syed IS, Martinez M, Oh JK, Jaffe AS, Grogan M, et al. Intracardiac thrombosis and anticoagulation therapy in cardiac amyloidosis. Circulation. 2009;119(18):2490. Therefore, anticoagulation is indicated in CA patients who develop AF, regardless of risk score calculations ( Table 12 ). In addition, left atrial thrombus has been described in up to 30% of patients who underwent transesophageal echocardiography prior to a planned electrical cardioversion, even with adequate anticoagulation. 181181. Donnellan E, Elshazly MB, Vakamudi S, Wazni OM, Cohen JA, Kanj M, et al. No association between CHADS-VASc score and left atrial appendage thrombus in patients with transthyretin amyloidosis. JACC: Clinical Electrophysiology. 2019;5(12):1473-4. , 182182. El-Am EA, Dispenzieri A, Melduni RM, Ammash NM, White RD, Hodge DO, et al. Direct current cardioversion of atrial arrhythmias in adults with cardiac amyloidosis. Journal of the American College of Cardiology. 2019;73(5):589-97. Thus, a transesophageal echocardiogram is recommended for every electrical cardioversion candidate. The role of anticoagulation in patients in sinus rhythm is still uncertain. However, even in sinus rhythm, changes in atrial contractility are common and are associated with atrial thrombus formation, especially in AL-CA patients. 180180. Feng D, Syed IS, Martinez M, Oh JK, Jaffe AS, Grogan M, et al. Intracardiac thrombosis and anticoagulation therapy in cardiac amyloidosis. Circulation. 2009;119(18):2490. , 182182. El-Am EA, Dispenzieri A, Melduni RM, Ammash NM, White RD, Hodge DO, et al. Direct current cardioversion of atrial arrhythmias in adults with cardiac amyloidosis. Journal of the American College of Cardiology. 2019;73(5):589-97.
9.1.2. Ventricular Arrhythmias
Ventricular arrhythmias are frequent in CA patients, especially AL-CA. Previous studies have detected complex ventricular arrhythmias in at least 50% of AL-CA patients, in whom non-sustained ventricular tachycardia was the most frequent arrhythmia and was associated with lower survival. 183183. Goldsmith YB, Liu J, Chou J, Hoffman J, Comenzo RL, Steingart RM. Frequencies and types of arrhythmias in patients with systemic light-chain amyloidosis with cardiac involvement undergoing stem cell transplantation on telemetry monitoring. The American journal of cardiology. 2009;104(7):990-4. , 184184. Palladini G, Malamani G, Co F, Pistorio A, Recusani F, Anesi E, et al. Holter monitoring in AL amyloidosis: prognostic implications. Pacing and Clinical Electrophysiology. 2001;24(8):1228-33.
In this context, implantable cardioverter-defibrillators (ICD) may have a role in preventing sudden death in CA patients. ICD may benefit patients with unstable ventricular tachycardia or who have survived cardiac arrest without a reversible cause and a life expectancy > 1 year with significant quality. 185185. Towbin JA, McKenna WJ, Abrams DJ, Ackerman MJ, Calkins H, Darrieux FC, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart rhythm. 2019;16(11):e301-e72. – 187187. Cappelli F, Perfetto F, Martone R, Di Mario C. Cardiac Amyloidosis in Patients Undergoing TAVR: Why We Need to Think About It. Cardiovascular Revascularization Medicine. 2021;22:109-14.
However, recommending ICD as primary prevention is difficult for a number of reasons. The first is that most causes of sudden death are related to electromechanical dissociation and not ventricular arrhythmias. 188188. Sayed RH, Rogers D, Khan F, Wechalekar AD, Lachmann HJ, Fontana M, et al. A study of implanted cardiac rhythm recorders in advanced cardiac AL amyloidosis. European heart journal. 2015;36(18):1098-105. The second reason is the historically low life expectancy of CA patients, especially those with AL-CA. Finally, traditional risk stratification tools, such as reduced EF, do not really seem applicable to CA patients, since severe systolic dysfunction is associated with the late stages of the disease, in which pump failure predominates as the cause of death. The challenge is, therefore, to identify at-risk patients in the early stage of the disease when arrhythmia predominates and can potentially be corrected with ICD. Prospective studies are necessary to determine when ICD can be beneficial in CA patients. 189189. Giancaterino S, Urey MA, Darden D, Hsu JC. Management of arrhythmias in cardiac amyloidosis. Clinical Electrophysiology. 2020;6(4):351-61.
9.2. Conduction Disorders
Conduction-system disease is highly prevalent among CA patients, with atrioventricular conduction being more commonly affected than the sinus node. The involved pathophysiology has not yet been fully defined, although small studies have suggested that the amyloid protein conduction system is involved. 190190. Ridolfi RL, Bulkley BH, Hutchins GM. The conduction system in cardiac amyloidosis: clinical and pathologic features of 23 patients. The American journal of medicine. 1977;62(5):677-86. There is evidence that these changes are the cause of death in a considerable number of CA patients, 191191. DePasquale EC, Nasir K, Jacoby DL. Outcomes of adults with restrictive cardiomyopathy after heart transplantation. The Journal of heart and lung transplantation. 2012;31(12):1269-75. for whom pacemakers are often indicated, especially ATTR-CA patients. 178178. Roberts WC, Waller BF. Cardiac amyloidosis causing cardiac dysfunction: analysis of 54 necropsy patients. The American journal of cardiology. 1983;52(1):137-46. In a Columbian cohort, a pacemaker was indicated in 43% of ATTRwt patients and 36% of ATTRv patients. 192192. Givens RC, Russo C, Green P, Maurer MS. Comparison of cardiac amyloidosis due to wild-type and V122I transthyretin in older adults referred to an academic medical center. Aging health. 2013;9(2):229-35. Pacemaker use in this population should follow traditional implantation recommendations. 193193. Topilsky Y, Pereira NL, Shah DK, Boilson B, Schirger JA, Kushwaha SS, et al. Left ventricular assist device therapy in patients with restrictive and hypertrophic cardiomyopathy. Circulation: Heart Failure. 2011;4(3):266-75.
9.3. Therapeutic Options in Advanced Heart Failure
In advanced HF associated with CA, advanced support strategies, such as mechanical circulatory assistance and transplantation, present challenges, especially since it is a multisystem disease. Moreover, due to the reduced size of the left ventricular cavity and the frequent involvement of the right ventricle, the use of long-term mechanical circulatory assistance devices may be limited. 193193. Topilsky Y, Pereira NL, Shah DK, Boilson B, Schirger JA, Kushwaha SS, et al. Left ventricular assist device therapy in patients with restrictive and hypertrophic cardiomyopathy. Circulation: Heart Failure. 2011;4(3):266-75. , 194194. Grupper A, Park SJ, Pereira NL, Schettle SD, Gerber Y, Topilsky Y, et al. Role of ventricular assist therapy for patients with heart failure and restrictive physiology: improving outcomes for a lethal disease. The Journal of Heart and Lung Transplantation. 2015;34(8):1042-9. Although heart transplantation has historically had a lower survival curve in CA, 191191. DePasquale EC, Nasir K, Jacoby DL. Outcomes of adults with restrictive cardiomyopathy after heart transplantation. The Journal of heart and lung transplantation. 2012;31(12):1269-75. the most recent results indicate that it is similar to other etiologies. 195195. Kristen AV, Kreusser MM, Blum P, Schönland SO, Frankenstein L, Dösch AO, et al. Improved outcomes after heart transplantation for cardiac amyloidosis in the modern era. The journal of heart and lung transplantation. 2018;37(5):611-8. This change is related to better patient selection and specific strategies, such as double transplantation for ATTRv patients and heart transplantation prior to bone marrow transplantation in AL-CA. 196196. Trachtenberg BH, Kamble RT, Rice L, Araujo-Gutierrez R, Bhimaraj A, Guha A, et al. Delayed autologous stem cell transplantation following cardiac transplantation experience in patients with cardiac amyloidosis. American Journal of Transplantation. 2019;19(10):2900-9.
Table 12 summarizes the recommendations for CA treatment.
10. Centers of Excellence/Reference and New Treatment Remuneration Models
Access to care and a sustained commitment to improving care quality are fundamental for achieving excellent results, which are critical for complex diseases such as CA. Developing a course of care that provides the right care at the right time for CA requires the involvement of public and private health care managers, health professionals, and everyone involved in the care of these patients.
CA, particularly AL-CA, must be approached with a view to rapid diagnosis, and multidisciplinary teams with proven experience and clinical protocols based on the best scientific evidence must be available. In addition, comprehensive care and the monitoring and publishing of care results should be promoted. An integrated and collaborative practice model should be observed at these reference centers, with a telemedicine structure to support diagnosis at more distant centers, patient monitoring, and clinical research, which is the basis for emerging therapies. Such initiatives have been undertaken at a number of centers worldwide, including some in Brazil. 187187. Cappelli F, Perfetto F, Martone R, Di Mario C. Cardiac Amyloidosis in Patients Undergoing TAVR: Why We Need to Think About It. Cardiovascular Revascularization Medicine. 2021;22:109-14. – 199199. Brasil. Ministério da Saúde. Política Nacional de Atenção Integral às Pessoas com Doenças Raras. Portaria GM nº 199, de 30 de janeiro de 2014. Disponível em: http://bvsmssaudegovbr/bvs/saudelegis/gm/2014/prt0199_30_01_2014html.
http://bvsmssaudegovbr/bvs/saudelegis/gm...
Additionally, such reference centers should contribute to a National Registry of Cardiac Amyloidosis, allowing better insight into regional epidemiology and care quality, in addition to measuring patient-centered clinical outcomes and contributing to new public policies.
New financing models for high-cost rare disease treatments have been discussed, tested in pharmacoeconomic studies, and implemented around the world. 200200. INTERFARMA. Doenças Raras: A urgência do acesso à saúde. Fevereiro de 2018. Disponível em: https://www.interfarma.org.br/public/files/biblioteca/doencas-raras--a-urgencia-do-acesso-a-saude-interfarma.pdf. Acesso em: 14 abr. 2019.
https://www.interfarma.org.br/public/fil...
A risk-sharing strategy for pharmaceutical companies, funders, and healthcare providers must involve clinical research, being based on outcome assessment and the impact of clinical protocols. This new paradigm is under development in Brazil for rare diseases, and it can be used for CA. 201201. Novaes HMD, Soárez PC. Doenças raras, drogas órfãs e as políticas para avaliação e incorporação de tecnologias nos sistemas de saúde. Sociologias. 2019;21(5):332-64. , 202202. Nicod E, Annemans L, Bucsics A, Lee A, Upadhyaya S, Facey K. HTA programme response to the challenges of dealing with orphan medicinal products: Process evaluation in selected European countries. Health Policy. 2019;123(2):140-51. Ensuring the sustainability of the health system and access to excellent treatment for CA patients will increasingly involve the Brazilian Society of Cardiology and its scientific departments/study groups. The Society is promoting debate on good care practices in all of these critical dimensions in order to build a health system that focuses on patient needs and fights waste.
An assessment of new treatments for rare diseases by the National Commission for Technological Incorporation in the Unified Health System (CONITEC) indicated that approximately 52% of the evaluated medicines have already been incorporated. In our vision were are building a new, technologically-based scenario developed through robust data with the support of clinical protocols and reference centers that can replace the current model, where access is sought through judicialization, which has a heavy impact on the federal budget. 197197. Biglia LV, Mendes SJ, Lima TM, Aguiar PM. Incorporações de medicamentos para doenças raras no Brasil: é possível acesso integral a estes pacientes? Cien Saúde Colet. 2020(julho/2020 - Está disponível em: http://www.cienciaesaudecoletiva.com.br/artigos/incorporacoes-de-medicamentos-para-doencas-raras-no-brasil-e-possivel-acesso-integral-a-estes-pacientes/17706?id=17706&id=17706).
http://www.cienciaesaudecoletiva.com.br/...
11. Knowledge Gaps and Future Perspectives
Despite recent advances in CA research, there are still numerous knowledge gaps to be filled regarding this complex and multifaceted disease. Both the diagnosis and treatment of amyloidosis are evolving, which is exemplified by the 638 studies registered in the clinicaltrials.gov site that have recently been completed or are in progress.
Current cardiovascular imaging for CA diagnosis involves cardiac scintigraphy with bone-seeking radiotracers, such as99mTc-pyrophosphate, which allows molecular images of amyloid fibril deposition in the myocardium. Although not yet available in Brazil, another promising technique is PET imaging with 1818. Kyle RA, Linos A, Beard CM, Linke RP, Gertz MA, O’Fallon WM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood. 1992;79(7):1817-22. F-Florbetapir in peripheral nerves and/or other extracardiac sites. 203203. Kircher M, Ihne S, Brumberg J, Morbach C, Knop S, Kortum KM, et al. Detection of cardiac amyloidosis with (18)F-Florbetaben-PET/CT in comparison to echocardiography, cardiac MRI and DPD-scintigraphy. Eur J Nucl Med Mol Imaging. 2019;46(7):1407-16. , 204204. Morgenstern R, Yeh R, Castano A, Maurer MS, Bokhari S. (18)Fluorine sodium fluoride positron emission tomography, a potential biomarker of transthyretin cardiac amyloidosis. J Nucl Cardiol. 2018;25(5):1559-67. Elastography, ultrasound, and CMR are also being used to assess the degree of myocardial fibrosis and may be additional and promising tools to aid in diagnosis and prognosis.
Recent studies have focused on transcriptome analysis, seeking differences in expression between healthy and sick individuals by means of molecular analysis and integrative genomics. The transcriptome of AL-CA patients is similar to that of patients with monoclonal gammopathy of undetermined significance. Furthermore, the level of circulating microRNA, which is known to correlate with cardiac damage, is increased in AL patients. Through principal component analysis, highly overlapping phenotypic profiles have been found between AL, monoclonal gammopathy of undetermined significance, and multiple myeloma. 205205. Kufova Z, Sevcikova T, Growkova K, Vojta P, Filipova J, Adam Z, et al. Biomarkers in Immunoglobulin Light Chain Amyloidosis. Klin Onkol.30(Suppl 2):60-7. , 206206. Paiva B, Martinez-Lopez J, Corchete LA, Sanchez-Vega B, Rapado I, Puig N, et al. Phenotypic, transcriptomic, and genomic features of clonal plasma cells in light-chain amyloidosis. Blood. 2016;127(24):3035-9.
Additionally, using artificial intelligence to analyze data in medical databases is a promising strategy for identifying individuals with warning signs. This strategy could lead to earlier CA diagnosis and reduce treatment delay.
New therapies specifically for ATTR-CA are under intense investigation. TTR gene silencing therapies that have been found effective for hereditary amyloid polyneuropathy are currently being tested in ATTR-CA patients in large multicenter studies. The APOLLO-B study is testing the RNA interference agent patisiran (ClinicalTrials.gov Identifier: NCT03997383). The CARDIO-TTRansform clinical trial is testing antisense oligonucleotide technology in a new second-generation drug, AKCEA-TTR-LRx (ClinicalTrials.gov Identifier: NCT04136171).
Since amyloid deposition has arrhythmogenic potential and can damage the conduction system, implantable devices such as pacemakers or defibrillators are frequently used to reduce mortality and increase survival in this population. On the other hand, individuals with CA and atrial fibrillation or flutter are at high risk for cardioembolic events, and treatment with anticoagulants has been recommended. Although there is a pathophysiological rationale for using these interventions in CA patients, they must still be tested in appropriate clinical trials, and this is an important area for future clinical investigation. In addition, heart transplantation has proven to be a safe strategy in these patients, although artificial ventricles and combined therapies must still be assessed in clinical trials.
To define the best treatment options and combinations for this disease, we must wait for the results of studies that are testing different interventions and specific therapies, bearing in mind that: 1) we are not fully aware of the details of the disease’s pathophysiology; 2) we lack a broad understanding of how medications work in this disease; 3) we still do not have an accurate, detailed and long-term assessment of the risks and benefits of different treatments; 4) and we also do not know the dose-response relationship of different medications for this clinical scenario.
Thus, we believe that the following basic steps are relevant and should be implemented:
-
Creating new centers of reference/excellence in CA.
-
Training professionals in early CA recognition and referral to specialized centers.
-
Promoting the development of a National Registry of Cardiac Amyloidosis.
-
Discussing the safety and care quality challenges involved in the journey of CA patients.
-
Discussing new remuneration and care models for CA.
-
Encouraging clinical research on CA in Brazil.
-
1Sipe JD, Cohen AS. Review: history of the amyloid fibril. J Struct Biol. 2000;130(2-3):88-98.
-
2Benson MD, Buxbaum JN, Eisenberg DS, Merlini G, Saraiva MJM, Sekijima Y, et al. Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid. 2018;25(4):215-9.
-
3Falk RH, Alexander KM, Liao R, Dorbala S. AL (Light-Chain) Cardiac Amyloidosis: A Review of Diagnosis and Therapy. J Am Coll Cardiol. 2016;68(12):1323-41.
-
4Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126(10):1286-300.
-
5Zhao L, Buxbaum JN, Reixach N. Age-related oxidative modifications of transthyretin modulate its amyloidogenicity. Biochemistry. 2013;52(11):1913-26.
-
6Buxbaum JN, Tagoe C, Gallo G, Walker JR, Kurian S, Salomon DR. Why are some amyloidoses systemic? Does hepatic “chaperoning at a distance” prevent cardiac deposition in a transgenic model of human senile systemic (transthyretin) amyloidosis? FASEB J. 2012;26(6):2283-93.
-
7Gertz MA, Dispenzieri A, Sher T. Pathophysiology and treatment of cardiac amyloidosis. Nat Rev Cardiol. 2015;12(2):91-102.
-
8Koike H, Katsuno M. Ultrastructure in Transthyretin Amyloidosis: From Pathophysiology to Therapeutic Insights. Biomedicines. 2019;7(1):11.
-
9Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019;73(22):2872-91.
-
10Mankad AK, Sesay I, Shah KB. Light-chain cardiac amyloidosis. Curr Probl Cancer. 2017;41(2):144-56.
-
11Wechalekar AD, Gillmore JD, Hawkins PN. Systemic amyloidosis. Lancet. 2016;387(10038):2641-54.
-
12Gertz MA, Dispenzieri A. Systemic Amyloidosis Recognition, Prognosis, and Therapy: A Systematic Review. JAMA. 2020;324(1):79-89.
-
13Muchtar E, Dispenzieri A, Magen H, Grogan M, Mauermann M, McPhail ED, et al. Systemic amyloidosis from A (AA) to T (ATTR): a review. J Intern Med. 2021;289(3):268-92.
-
14Milandri A, Farioli A, Gagliardi C, Longhi S, Salvi F, Curti S, et al. Carpal tunnel syndrome in cardiac amyloidosis: implications for early diagnosis and prognostic role across the spectrum of aetiologies. Eur J Heart Fail. 2020;22(3):507-15.
-
15Witteles RM, Bokhari S, Damy T, Elliott PM, Falk RH, Fine NM, et al. Screening for Transthyretin Amyloid Cardiomyopathy in Everyday Practice. JACC Heart Fail. 2019;7(8):709-16.
-
16Jacobson D, Tagoe C, Schwartzbard A, Shah A, Koziol J, Buxbaum J. Relation of clinical, echocardiographic and electrocardiographic features of cardiac amyloidosis to the presence of the transthyretin V122I allele in older African-American men. Am J Cardiol. 2011;108(3):440-4.
-
17Parman Y, Adams D, Obici L, Galan L, Guergueltcheva V, Suhr OB, et al. Sixty years of transthyretin familial amyloid polyneuropathy (TTR-FAP) in Europe: where are we now? A European network approach to defining the epidemiology and management patterns for TTR-FAP. Curr Opin Neurol. 2016;29 Suppl 1:S3-S13.
-
18Kyle RA, Linos A, Beard CM, Linke RP, Gertz MA, O’Fallon WM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood. 1992;79(7):1817-22.
-
19Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, et al. Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation. 2016;133(24):2404-12.
-
20Lane T, Fontana M, Martinez-Naharro A, Quarta CC, Whelan CJ, Petrie A, et al. Natural History, Quality of Life, and Outcome in Cardiac Transthyretin Amyloidosis. Circulation. 2019;140(1):16-26.
-
21Gonzalez-Lopez E, Gallego-Delgado M, Guzzo-Merello G, de Haro-Del Moral FJ, Cobo-Marcos M, Robles C, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36(38):2585-94.
-
22Cornwell GG, 3rd, Murdoch WL, Kyle RA, Westermark P, Pitkanen P. Frequency and distribution of senile cardiovascular amyloid. A clinicopathologic correlation. Am J Med. 1983;75(4):618-23.
-
23Tanskanen M, Peuralinna T, Polvikoski T, Notkola IL, Sulkava R, Hardy J, et al. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: a population-based autopsy study. Ann Med. 2008;40(3):232-9.
-
24Cruz MW, Pinto MV, Pinto LF, Gervais R, Dias M, Perez C, et al. Baseline disease characteristics in Brazilian patients enrolled in Transthyretin Amyloidosis Outcome Survey (THAOS). Arq Neuropsiquiatr. 2019;77(2):96-100.
-
25Gilstrap LG, Dominici F, Wang Y, El-Sady MS, Singh A, Di Carli MF, et al. Epidemiology of Cardiac Amyloidosis-Associated Heart Failure Hospitalizations Among Fee-for-Service Medicare Beneficiaries in the United States. Circ Heart Fail. 2019;12(6):e005407.
-
26Lousada I, Comenzo RL, Landau H, Guthrie S, Merlini G. Light Chain Amyloidosis: Patient Experience Survey from the Amyloidosis Research Consortium. Adv Ther. 2015;32(10):920-8.
-
27Adams D, Polydefkis M, Gonzalez-Duarte A, Wixner J, Kristen AV, Schmidt HH, et al. Long-term safety and efficacy of patisiran for hereditary transthyretin-mediated amyloidosis with polyneuropathy: 12-month results of an open-label extension study. Lancet Neurol. 2021;20(1):49-59.
-
28Plante-Bordeneuve V, Said G. Familial amyloid polyneuropathy. Lancet Neurol. 2011;10(12):1086-97.
-
29Sekijima Y. Transthyretin (ATTR) amyloidosis: clinical spectrum, molecular pathogenesis and disease-modifying treatments. J Neurol Neurosurg Psychiatry. 2015;86(9):1036-43.
-
30Pinto MV, Pinto LF, Dias M, Rosa RS, Mundayat R, Pedrosa RC, et al. Late-onset hereditary ATTR V30M amyloidosis with polyneuropathy: Characterization of Brazilian subjects from the THAOS registry. J Neurol Sci. 2019;403:1-6.
-
31Rapezzi C, Quarta CC, Riva L, Longhi S, Gallelli I, Lorenzini M, et al. Transthyretin-related amyloidoses and the heart: a clinical overview. Nat Rev Cardiol. 2010;7(7):398-408.
-
32Kapoor M, Rossor AM, Laura M, Reilly MM. Clinical Presentation, Diagnosis and Treatment of TTR Amyloidosis. J Neuromuscul Dis. 2019;6(2):189-99.
-
33Lahuerta-Pueyo CL, Tubar Arregui MA, Gracia- Gutierrez AG, Buena Juana E, Guillén SM. Estimating the prevalence of allelic variants in the transthyretin gene by analysing large-scale sequencing data. Eur J Hum Genet. 2019;27(5):783-91.
-
34Park GY, Jamerlan A, Shim KH, An SSA. Diagnostic and Treatment Approaches Involving Transthyretin in Amyloidogenic Diseases. Int J Mol Sci. 2019;20(12):2982.
-
35Maurer MS, Bokhari S, Damy T, Dorbala S, Drachman BM, Fontana M, et al. Expert Consensus Recommendations for the Suspicion and Diagnosis of Transthyretin Cardiac Amyloidosis. Circ Heart Fail. 2019;12(9):e006075.
-
36Obici L, Kuks JB, Buades J, Adams D, Suhr OB, Coelho T, et al. Recommendations for presymptomatic genetic testing and management of individuals at risk for hereditary transthyretin amyloidosis. Curr Opin Neurol. 2016;29 Suppl 1:S27-35.
-
37Maurer MS, Elliott P, Comenzo R, Semigran M, Rapezzi C. Addressing Common Questions Encountered in the Diagnosis and Management of Cardiac Amyloidosis. Circulation. 2017;135(14):1357-77.
-
38Castano A, Drachman BM, Judge D, Maurer MS. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20(2):163-78.
-
39Dungu JN, Anderson LJ, Whelan CJ, Hawkins PN. Cardiac transthyretin amyloidosis. Heart. 2012;98(21):1546-54.
-
40Mohty D, Damy T, Cosnay P, Echahidi N, Casset-Senon D, Virot P, et al. Cardiac amyloidosis: updates in diagnosis and management. Arch Cardiovasc Dis. 2013;106(10):528-40.
-
41Mohammed SF, Mirzoyev SA, Edwards WD, Dogan A, Grogan DR, Dunlay SM, et al. Left ventricular amyloid deposition in patients with heart failure and preserved ejection fraction. JACC Heart Fail. 2014;2(2):113-22.
-
42Rapezzi C, Lorenzini M, Longhi S, Milandri A, Gagliardi C, Bartolomei I, et al. Cardiac amyloidosis: the great pretender. Heart Fail Rev. 2015;20(2):117-24.
-
43Yilmaz A, Bauersachs J, Bengel F, Buchel R, Kindermann I, Klingel K, et al. Diagnosis and treatment of cardiac amyloidosis: position statement of the German Cardiac Society (DGK). Clin Res Cardiol. 2021;110(4):479-506.
-
44Nakagawa M, Sekijima Y, Yazaki M, Tojo K, Yoshinaga T, Doden T, et al. Carpal tunnel syndrome: a common initial symptom of systemic wild-type ATTR (ATTRwt) amyloidosis. Amyloid. 2016;23(1):58-63.
-
45Geller HI, Singh A, Alexander KM, Mirto TM, Falk RH. Association Between Ruptured Distal Biceps Tendon and Wild-Type Transthyretin Cardiac Amyloidosis. JAMA. 2017;318(10):962-3.
-
46Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349(6):583-96.
-
47Conceicao I, Gonzalez-Duarte A, Obici L, Schmidt HH, Simoneau D, Ong ML, et al. “Red-flag” symptom clusters in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst. 2016;21(1):5-9.
-
48Sekijima Y, Ueda M, Koike H, Misawa S, Ishii T, Ando Y. Diagnosis and management of transthyretin familial amyloid polyneuropathy in Japan: red-flag symptom clusters and treatment algorithm. Orphanet J Rare Dis. 2018;13(1):6.
-
49Martinez-Naharro A, Treibel TA, Abdel-Gadir A, Bulluck H, Zumbo G, Knight DS, et al. Magnetic Resonance in Transthyretin Cardiac Amyloidosis. J Am Coll Cardiol. 2017;70(4):466-77.
-
50Pibarot P, Lancellotti P, Narula J. Concomitant Cardiac Amyloidosis in Severe Aortic Stenosis: The Trojan Horse? J Am Coll Cardiol. 2021;77(2):140-3.
-
51Cyrille NB, Goldsmith J, Alvarez J, Maurer MS. Prevalence and prognostic significance of low QRS voltage among the three main types of cardiac amyloidosis. Am J Cardiol. 2014;114(7):1089-93.
-
52Quarta CC, Solomon SD, Uraizee I, Kruger J, Longhi S, Ferlito M, et al. Left ventricular structure and function in transthyretin-related versus light-chain cardiac amyloidosis. Circulation. 2014;129(18):1840-9.
-
53Carroll JD, Gaasch WH, McAdam KP. Amyloid cardiomyopathy: characterization by a distinctive voltage/mass relation. Am J Cardiol. 1982;49(1):9-13.
-
54Kittleson MM, Maurer MS, Ambardekar AV, Bullock-Palmer RP, Chang PP, Eisen HJ, et al. Cardiac Amyloidosis: Evolving Diagnosis and Management: A Scientific Statement From the American Heart Association. Circulation. 2020;142(1):e7-e22.
-
55Mitchell C, Rahko PS, Blauwet LA, Canaday B, Finstuen JA, Foster MC, et al. Guidelines for Performing a Comprehensive Transthoracic Echocardiographic Examination in Adults: Recommendations from the American Society of Echocardiography. J Am Soc Echocardiogr. 2019;32(1):1-64.
-
56Kolias TJ, Aaronson KD, Armstrong WF. Doppler-derived dP/dt and -dP/dt predict survival in congestive heart failure. J Am Coll Cardiol. 2000;36(5):1594-9.
-
57Voigt JU, Pedrizzetti G, Lysyansky P, Marwick TH, Houle H, Baumann R, et al. Definitions for a common standard for 2D speckle tracking echocardiography: consensus document of the EACVI/ASE/Industry Task Force to standardize deformation imaging. J Am Soc Echocardiogr. 2015;28(2):183-93.
-
58Collier P, Phelan D, Klein A. A Test in Context: Myocardial Strain Measured by Speckle-Tracking Echocardiography. J Am Coll Cardiol. 2017;69(8):1043-56.
-
59Pagourelias ED, Vassilikos VP, Voigt JU. Left Ventricular Pressure Strain-Derived Myocardial Work at Rest and during Exercise in Patients with Cardiac Amyloidosis. J Am Soc Echocardiogr. 2020;33(10):1295-6.
-
60Di Bella G, Pizzino F. Myocardial Deformation Analysis and Late-Gadolinium Enhancement: Important Markers of Cardiac Amyloidosis Involvement That Can Masquerade as a False-Negative Diagnosis. Circ J. 2018;82(10):2687.
-
61Bravo PE, Fujikura K, Kijewski MF, Jerosch-Herold M, Jacob S, El-Sady MS, et al. Relative Apical Sparing of Myocardial Longitudinal Strain Is Explained by Regional Differences in Total Amyloid Mass Rather Than the Proportion of Amyloid Deposits. JACC Cardiovasc Imaging. 2019;12(7 Pt 1):1165-73.
-
62Rapezzi C, Fontana M. Relative Left Ventricular Apical Sparing of Longitudinal Strain in Cardiac Amyloidosis: Is it Just Amyloid Infiltration? JACC Cardiovasc Imaging. 2019;12(7 Pt 1):1174-6.
-
63Monivas Palomero V, Durante-Lopez A, Sanabria MT, Cubero JS, Gonzalez-Mirelis J, Lopez-Ibor JV, et al. Role of Right Ventricular Strain Measured by Two-Dimensional Echocardiography in the Diagnosis of Cardiac Amyloidosis. J Am Soc Echocardiogr. 2019;32(7):845-53 e1.
-
64Brand A, Frumkin D, Hubscher A, Dreger H, Stangl K, Baldenhofer G, et al. Phasic left atrial strain analysis to discriminate cardiac amyloidosis in patients with unclear thick heart pathology. Eur Heart J Cardiovasc Imaging. 2020 Apr 3;jeaa043. Doi 10.1093/ehjci/jeaa043
» https://doi.org/10.1093/ehjci/jeaa043 -
65Syed IS, Glockner JF, Feng D, Araoz PA, Martinez MW, Edwards WD, et al. Role of cardiac magnetic resonance imaging in the detection of cardiac amyloidosis. JACC Cardiovascular imaging. 2010;3(2):155-64.
-
66Kwong RY, Falk RH. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2005;111(2):122-4.
-
67Maceira AM, Joshi J, Prasad SK, Moon JC, Perugini E, Harding I, et al. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2005;111(2):186-93.
-
68Kotecha T, Martinez-Naharro A, Treibel TA, Francis R, Nordin S, Abdel-Gadir A, et al. Myocardial Edema and Prognosis in Amyloidosis. Journal of the American College of Cardiology. 2018;71(25):2919-31.
-
69Fontana M, Corovic A, Scully P, Moon JC. Myocardial Amyloidosis: The Exemplar Interstitial Disease. JACC Cardiovascular imaging. 2019;12(11 Pt 2):2345-56.
-
70Mongeon FP, Jerosch-Herold M, Coelho-Filho OR, Blankstein R, Falk RH, Kwong RY. Quantification of extracellular matrix expansion by CMR in infiltrative heart disease. JACC Cardiovascular imaging. 2012;5(9):897-907.
-
71Banypersad SM, Sado DM, Flett AS, Gibbs SD, Pinney JH, Maestrini V, et al. Quantification of myocardial extracellular volume fraction in systemic AL amyloidosis: an equilibrium contrast cardiovascular magnetic resonance study. Circulation Cardiovascular imaging. 2013;6(1):34-9.
-
72Martinez-Naharro A, Kotecha T, Norrington K, Boldrini M, Rezk T, Quarta C, et al. Native T1 and Extracellular Volume in Transthyretin Amyloidosis. JACC Cardiovascular imaging. 2019;12(5):810-9.
-
73Fontana M, Banypersad SM, Treibel TA, Maestrini V, Sado DM, White SK, et al. Native T1 mapping in transthyretin amyloidosis. JACC Cardiovascular imaging. 2014;7(2):157-65.
-
74Fontana M, Chung R, Hawkins PN, Moon JC. Cardiovascular magnetic resonance for amyloidosis. Heart Fail Rev. 2015;20(2):133-44.
-
75Baggiano A, Boldrini M, Martinez-Naharro A, Kotecha T, Petrie A, Rezk T, et al. Noncontrast Magnetic Resonance for the Diagnosis of Cardiac Amyloidosis. JACC Cardiovasc Imaging. 2020;13(1 Pt 1):69-80.
-
76Pozo E, Kanwar A, Deochand R, Castellano JM, Naib T, Pazos-Lopez P, et al. Cardiac magnetic resonance evaluation of left ventricular remodelling distribution in cardiac amyloidosis. Heart. 2014;100(21):1688-95.
-
77Jurcut R, Onciul S, Adam R, Stan C, Coriu D, Rapezzi C, et al. Multimodality imaging in cardiac amyloidosis: a primer for cardiologists. European heart journal cardiovascular Imaging. 2020;21(8):833-44.
-
78Dorbala S, Ando Y, Bokhari S, Dispenzieri A, Falk RH, Ferrari VA, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI Expert Consensus Recommendations for Multimodality Imaging in Cardiac Amyloidosis: Part 1 of 2-Evidence Base and Standardized Methods of Imaging. J Card Fail.2019;25(11):e 1-e39.
-
79Zhao L, Tian Z, Fang Q. Diagnostic accuracy of cardiovascular magnetic resonance for patients with suspected cardiac amyloidosis: a systematic review and meta-analysis. BMC Cardiovasc Dis. 2016;16:129.
-
80Fontana M, Pica S, Reant P, Abdel-Gadir A, Treibel TA, Banypersad SM, et al. Prognostic Value of Late Gadolinium Enhancement Cardiovascular Magnetic Resonance in Cardiac Amyloidosis. Circulation. 2015;132(16):1570-9.
-
81Boynton SJ, Geske JB, Dispenzieri A, Syed IS, Hanson TJ, Grogan M, et al. LGE Provides Incremental Prognostic Information Over Serum Biomarkers in AL Cardiac Amyloidosis. JACC Cardiovascular imaging. 2016;9(6):680-6.
-
82Lin L, Li X, Feng J, Shen KN, Tian Z, Sun J, et al. The prognostic value of T1 mapping and late gadolinium enhancement cardiovascular magnetic resonance imaging in patients with light chain amyloidosis. Journal of cardiovascular magnetic resonance . J Cardiovasc Magn Reson. 2018;20(1):2.
-
83Fontana M, White SK, Banypersad SM, Sado DM, Maestrini V, Flett AS, et al. Comparison of T1 mapping techniques for ECV quantification. Histological validation and reproducibility of ShMOLLI versus multibreath-hold T1 quantification equilibrium contrast CMR. Journal of cardiovascular magnetic resonance : J Cardiovasc Magn Reson. 2012;14:88.
-
84White JA, Kim HW, Shah D, Fine N, Kim KY, Wendell DC, et al. CMR imaging with rapid visual T1 assessment predicts mortality in patients suspected of cardiac amyloidosis. JACC Cardiovasc Imaging. 2014;7(2):143-56.
-
85Dorbala S, Cuddy S, Falk RH. How to Image Cardiac Amyloidosis: A Practical Approach. JACC Cardiovascular imaging. 2020;13(6):1299-310.
-
86Pan JA, Kerwin MJ, Salerno M. Native T1 Mapping, Extracellular Volume Mapping, and Late Gadolinium Enhancement in Cardiac Amyloidosis: A Meta-Analysis. JACC Cardiovascular imaging. 2020;13(6):1299-310.
-
87Singh V, Falk R, Di Carli MF, Kijewski M, Rapezzi C, Dorbala S. State-of-the-art radionuclide imaging in cardiac transthyretin amyloidosis. J Nucl Cardiol. 2019;26(1):158-73.
-
88Stats MA, Stone JR. Varying levels of small microcalcifications and macrophages in ATTR and AL cardiac amyloidosis: implications for utilizing nuclear medicine studies to subtype amyloidosis. Cardiovasc Pathol. 2016;25(5):413-7.
-
89Perugini E, Guidalotti PL, Salvi F, Cooke RM, Pettinato C, Riva L, et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol. 2005;46(6):1076-84.
-
90Dorbala S, Ando Y, Bokhari S, Dispenzieri A, Falk RH, Ferrari VA, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: Part 1 of 2-evidence base and standardized methods of imaging. J Nucl Cardiol. 2019;26(6):2065-123.
-
91Hanna M, Ruberg FL, Maurer MS, Dispenzieri A, Dorbala S, Falk RH, et al. Cardiac Scintigraphy With Technetium-99m-Labeled Bone-Seeking Tracers for Suspected Amyloidosis: JACC Review Topic of the Week. J Am Coll Cardiol. 2020;75(22):2851-62.
-
92Hutt DF, Quigley AM, Page J, Hall ML, Burniston M, Gopaul D, et al. Utility and limitations of 3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy in systemic amyloidosis. Eur Heart J Cardiovasc Imaging. 2014;15(11):1289-98.
-
93Pepys MB, Dyck RF, de Beer FC, Skinner M, Cohen AS. Binding of serum amyloid P-component (SAP) by amyloid fibrils. Clin Exp Immunol. 1979;38(2):284-93.
-
94Castano A, Haq M, Narotsky DL, Goldsmith J, Weinberg RL, Morgenstern R, et al. Multicenter Study of Planar Technetium 99m Pyrophosphate Cardiac Imaging: Predicting Survival for Patients With ATTR Cardiac Amyloidosis. JAMA Cardiol. 2016;1(8):880-9.
-
95Vranian MN, Sperry BW, Hanna M, Hachamovitch R, Ikram A, Brunken RC, et al. Technetium pyrophosphate uptake in transthyretin cardiac amyloidosis: Associations with echocardiographic disease severity and outcomes. J Nucl Cardiol. 2018;25(4):1247-56.
-
96Kyriakou P, Mouselimis D, Tsarouchas A, Rigopoulos A, Bakogiannis C, Noutsias M, et al. Diagnosis of cardiac amyloidosis: a systematic review on the role of imaging and biomarkers. BMC Cardiovasc Disord. 2018;18(1):221.
-
97Kristen AV, Maurer MS, Rapezzi C, Mundayat R, Suhr OB, Damy T, et al. Impact of genotype and phenotype on cardiac biomarkers in patients with transthyretin amyloidosis - Report from the Transthyretin Amyloidosis Outcome Survey (THAOS). PLoS One. 2017;12(4):e0173086.
-
98Perfetto F, Bergesio F, Grifoni E, Fabbri A, Ciuti G, Frusconi S, et al. Different NT-proBNP circulating levels for different types of cardiac amyloidosis. J Cardiovasc Med (Hagerstown). 2016;17(11):810-7.
-
99Hahn VS, Yanek LR, Vaishnav J, Ying W, Vaidya D, Lee YZJ, et al. Endomyocardial Biopsy Characterization of Heart Failure With Preserved Ejection Fraction and Prevalence of Cardiac Amyloidosis. JACC Heart Fail. 2020;8(9):712-24.
-
100Kociol RD, Pang PS, Gheorghiade M, Fonarow GC, O’Connor CM, Felker GM. Troponin elevation in heart failure prevalence, mechanisms, and clinical implications. J Am Coll Cardiol. 2010;56(14):1071-8.
-
101Takashio S, Yamamuro M, Izumiya Y, Hirakawa K, Marume K, Yamamoto M, et al. Diagnostic utility of cardiac troponin T level in patients with cardiac amyloidosis. ESC Heart Fail. 2018;5(1):27-35.
-
102Hafeez AS, Bavry AA. Diagnosis of Transthyretin Amyloid Cardiomyopathy. Cardiol Ther. 2020;9(1):85-95.
-
103Rajkumar SV. Multiple myeloma: 2011 update on diagnosis, risk-stratification, and management. Am J Hematol. 2011;86(1):57-65.
-
104Comenzo RL, Reece D, Palladini G, Seldin D, Sanchorawala V, Landau H, et al. Consensus guidelines for the conduct and reporting of clinical trials in systemic light-chain amyloidosis. Leukemia. 2012;26(11):2317-25.
-
105Duston MA, Skinner M, Meenan RF, Cohen AS. Sensitivity, specificity, and predictive value of abdominal fat aspiration for the diagnosis of amyloidosis. Arthritis Rheum. 1989;32(1):82-5.
-
106van G, II, Hazenberg BP, Bijzet J, van Rijswijk MH. Diagnostic accuracy of subcutaneous abdominal fat tissue aspiration for detecting systemic amyloidosis and its utility in clinical practice. Arthritis Rheum. 2006;54(6):2015-21.
-
107Andrews TR, Colon-Otero G, Calamia KT, Menke DM, Boylan KB, Kyle RA. Utility of subcutaneous fat aspiration for diagnosing amyloidosis in patients with isolated peripheral neuropathy. Mayo Clin Proc. 2002;77(12):1287-90.
-
108Fine NM, Arruda-Olson AM, Dispenzieri A, Zeldenrust SR, Gertz MA, Kyle RA, et al. Yield of noncardiac biopsy for the diagnosis of transthyretin cardiac amyloidosis. Am J Cardiol. 2014;113(10):1723-7.
-
109Phull P, Sanchorawala V, Connors LH, Doros G, Ruberg FL, Berk JL, et al. Monoclonal gammopathy of undetermined significance in systemic transthyretin amyloidosis (ATTR). Amyloid. 2018;25(1):62-7.
-
110Vrana JA, Gamez JD, Madden BJ, Theis JD, Bergen HR, 3rd, Dogan A. Classification of amyloidosis by laser microdissection and mass spectrometry-based proteomic analysis in clinical biopsy specimens. Blood. 2009;114(24):4957-9.
-
111Musumeci MB, Cappelli F, Russo D, Tini G, Canepa M, Milandri A, et al. Low Sensitivity of Bone Scintigraphy in Detecting Phe64Leu Mutation-Related Transthyretin Cardiac Amyloidosis. JACC Cardiovasc Imaging. 2020;13(6):1314-21.
-
112Binder C, Duca F, Stelzer PD, Nitsche C, Rettl R, Aschauer S, et al. Mechanisms of heart failure in transthyretin vs. light chain amyloidosis. Eur Heart J Cardiovasc Imaging. 2019;20(5):512-24.
-
113Podduturi V, Armstrong DR, Hitchcock MA, Roberts WC, Guileyardo JM. Isolated atrial amyloidosis and the importance of molecular classification. Proc (Bayl Univ Med Cent). 2013;26(4):387-9.
-
114Gertz MA. Immunoglobulin light chain amyloidosis: 2018 Update on diagnosis, prognosis, and treatment. Am J Hematol. 2018;93(9):1169-80.
-
115Kumar S, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Colby C, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. 2012;30(9):989-95.
-
116Koyama J, Falk RH. Prognostic significance of strain Doppler imaging in light-chain amyloidosis. JACC Cardiovasc Imaging. 2010;3(4):333-42.
-
117Pislaru C, Ionescu F, Alashry M, Petrescu I, Pellikka PA, Grogan M, et al. Myocardial Stiffness by Intrinsic Cardiac Elastography in Patients with Amyloidosis: Comparison with Chamber Stiffness and Global Longitudinal Strain. J Am Soc Echocardiogr. 2019;32(8):958-68 e4.
-
118Kuruvilla S, Adenaw N, Katwal AB, Lipinski MJ, Kramer CM, Salerno M. Late gadolinium enhancement on cardiac magnetic resonance predicts adverse cardiovascular outcomes in nonischemic cardiomyopathy: a systematic review and meta-analysis. Circ Cardiovasc Imaging. 2014;7(2):250-8.
-
119Raina S, Lensing SY, Nairooz RS, Pothineni NV, Hakeem A, Bhatti S, et al. Prognostic Value of Late Gadolinium Enhancement CMR in Systemic Amyloidosis. JACC Cardiovasc Imaging. 2016;9(11):1267-77.
-
120Banypersad SM, Fontana M, Maestrini V, Sado DM, Captur G, Petrie A, et al. T1 mapping and survival in systemic light-chain amyloidosis. Eur Heart J. 2015;36(4):244-51.
-
121Rapezzi C, Merlini G, Quarta CC, Riva L, Longhi S, Leone O, et al. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation. 2009;120(13):1203-12.
-
122Chacko L, Martone R, Bandera F, Lane T, Martinez-Naharro A, Boldrini M, et al. Echocardiographic phenotype and prognosis in transthyretin cardiac amyloidosis. Eur Heart J. 2020;41(14):1439-47.
-
123Siddiqi OK, Ruberg FL. Cardiac amyloidosis: An update on pathophysiology, diagnosis, and treatment. Trends Cardiovasc Med. 2018;28(1):10-21.
-
124Sugiura A, Kitahara H, Iwahana T, Suzuki N, Okada S, Miyauchi H, et al. Association of heart failure duration with clinical prognosis in advanced heart failure. Clin Res Cardiol. 2020;109(3):350-7.
-
125Kristen AV, Perz JB, Schonland SO, Hegenbart U, Schnabel PA, Kristen JH, et al. Non-invasive predictors of survival in cardiac amyloidosis. Eur J Heart Fail. 2007;9(6-7):617-24.
-
126Pinney JH, Whelan CJ, Petrie A, Dungu J, Banypersad SM, Sattianayagam P, et al. Senile systemic amyloidosis: clinical features at presentation and outcome. J Am Heart Assoc. 2013;2(2):e000098.
-
127Maurer MS, Hanna M, Grogan M, Dispenzieri A, Witteles R, Drachman B, et al. Genotype and Phenotype of Transthyretin Cardiac Amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J Am Coll Cardiol. 2016;68(2):161-72.
-
128Grogan M, Scott CG, Kyle RA, Zeldenrust SR, Gertz MA, Lin G, et al. Natural History of Wild-Type Transthyretin Cardiac Amyloidosis and Risk Stratification Using a Novel Staging System. J Am Coll Cardiol. 2016;68(10):1014-20.
-
129Hanson JLS, Arvanitis M, Koch CM, Berk JL, Ruberg FL, Prokaeva T, et al. Use of Serum Transthyretin as a Prognostic Indicator and Predictor of Outcome in Cardiac Amyloid Disease Associated With Wild-Type Transthyretin. Circ Heart Fail. 2018;11(2):e004000.
-
130Gillmore JD, Damy T, Fontana M, Hutchinson M, Lachmann HJ, Martinez-Naharro A, et al. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J. 2018;39(30):2799-806.
-
131Cappelli F, Martone R, Gabriele M, Taborchi G, Morini S, Vignini E, et al. Biomarkers and Prediction of Prognosis in Transthyretin-Related Cardiac Amyloidosis: Direct Comparison of Two Staging Systems. Can J Cardiol. 2020;36(3):424-31.
-
132Rubin J, Steidley DE, Carlsson M, Ong ML, Maurer MS. Myocardial Contraction Fraction by M-Mode Echocardiography Is Superior to Ejection Fraction in Predicting Mortality in Transthyretin Amyloidosis. J Card Fail. 2018;24(8):504-11.
-
133Senapati A, Sperry BW, Grodin JL, Kusunose K, Thavendiranathan P, Jaber W, et al. Prognostic implication of relative regional strain ratio in cardiac amyloidosis. Heart. 2016;102(10):748-54.
-
134Knight DS, Zumbo G, Barcella W, Steeden JA, Muthurangu V, Martinez-Naharro A, et al. Cardiac Structural and Functional Consequences of Amyloid Deposition by Cardiac Magnetic Resonance and Echocardiography and Their Prognostic Roles. JACC Cardiovasc Imaging. 2019;12(5):823-33.
-
135Palladini G, Milani P, Merlini G. Management of AL amyloidosis in 2020. Blood. 2020;136(23):2620-7.
-
136Palladini G, Dispenzieri A, Gertz MA, Kumar S, Wechalekar A, Hawkins PN, et al. New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. J Clin Oncol. 2012;30(36):4541-9.
-
137Mahmood S, Palladini G, Sanchorawala V, Wechalekar A. Update on treatment of light chain amyloidosis. Haematologica. 2014;99(2):209-21.
-
138Palladini G, Hegenbart U, Milani P, Kimmich C, Foli A, Ho AD, et al. A staging system for renal outcome and early markers of renal response to chemotherapy in AL amyloidosis. Blood. 2014;124(15):2325-32.
-
139Palladini G, Kastritis E, Maurer MS, Zonder J, Minnema MC, Wechalekar AD, et al. Daratumumab plus CyBorD for patients with newly diagnosed AL amyloidosis: safety run-in results of ANDROMEDA. Blood. 2020;136(1):71-80.
-
140Cornell RF, Zhong X, Arce-Lara C, Atallah E, Blust L, Drobyski WR, et al. Bortezomib-based induction for transplant ineligible AL amyloidosis and feasibility of later transplantation. Bone Marrow Transplant. 2015;50(7):914-7.
-
141Jaccard A, Moreau P, Leblond V, Leleu X, Benboubker L, Hermine O, et al. High-dose melphalan versus melphalan plus dexamethasone for AL amyloidosis. N Engl J Med. 2007;357(11):1083-93.
-
142Moreau P, Leblond V, Bourquelot P, Facon T, Huynh A, Caillot D, et al. Prognostic factors for survival and response after high-dose therapy and autologous stem cell transplantation in systemic AL amyloidosis: a report on 21 patients. Br J Haematol. 1998;101(4):766-9.
-
143Comenzo RL, Vosburgh E, Falk RH, Sanchorawala V, Reisinger J, Dubrey S, et al. Dose-intensive melphalan with blood stem-cell support for the treatment of AL (amyloid light-chain) amyloidosis: survival and responses in 25 patients. Blood. 1998;91(10):3662-70.
-
144Cordes S, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Dingli D, et al. Ten-year survival after autologous stem cell transplantation for immunoglobulin light chain amyloidosis. Cancer. 2012;118(24):6105-9.
-
145Landau H, Smith M, Landry C, Chou JF, Devlin SM, Hassoun H, et al. Long-term event-free and overall survival after risk-adapted melphalan and SCT for systemic light chain amyloidosis. Leukemia. 2017;31(1):136-42.
-
146Sanchorawala V, Sun F, Quillen K, Sloan JM, Berk JL, Seldin DC. Long-term outcome of patients with AL amyloidosis treated with high-dose melphalan and stem cell transplantation: 20-year experience. Blood. 2015;126(20):2345-7.
-
147Sidiqi MH, Aljama MA, Buadi FK, Warsame RM, Lacy MQ, Dispenzieri A, et al. Stem Cell Transplantation for Light Chain Amyloidosis: Decreased Early Mortality Over Time. J Clin Oncol. 2018;36(13):1323-9.
-
148Gertz MA, Lacy MQ, Dispenzieri A, Kumar SK, Dingli D, Leung N, et al. Refinement in patient selection to reduce treatment-related mortality from autologous stem cell transplantation in amyloidosis. Bone Marrow Transplant. 2013;48(4):557-61.
-
149Gertz MA, Lacy MQ, Dispenzieri A, Hayman SR, Kumar SK, Leung N, et al. Effect of hematologic response on outcome of patients undergoing transplantation for primary amyloidosis: importance of achieving a complete response. Haematologica. 2007;92(10):1415-8.
-
150Kourelis TV, Kumar SK, Gertz MA, Lacy MQ, Buadi FK, Hayman SR, et al. Coexistent multiple myeloma or increased bone marrow plasma cells define equally high-risk populations in patients with immunoglobulin light chain amyloidosis. J Clin Oncol. 2013;31(34):4319-24.
-
151Venner CP, Lane T, Foard D, Rannigan L, Gibbs SD, Pinney JH, et al. Cyclophosphamide, bortezomib, and dexamethasone therapy in AL amyloidosis is associated with high clonal response rates and prolonged progression-free survival. Blood. 2012;119(19):4387-90.
-
152Mikhael JR, Schuster SR, Jimenez-Zepeda VH, Bello N, Spong J, Reeder CB, et al. Cyclophosphamide-bortezomib-dexamethasone (CyBorD) produces rapid and complete hematologic response in patients with AL amyloidosis. Blood. 2012;119(19):4391-4.
-
153Palladini G, Sachchithanantham S, Milani P, Gillmore J, Foli A, Lachmann H, et al. A European collaborative study of cyclophosphamide, bortezomib, and dexamethasone in upfront treatment of systemic AL amyloidosis. Blood. 2015;126(5):612-5.
-
154Jaccard A, Comenzo RL, Hari P, Hawkins PN, Roussel M, Morel P, et al. Efficacy of bortezomib, cyclophosphamide and dexamethasone in treatment-naive patients with high-risk cardiac AL amyloidosis (Mayo Clinic stage III). Haematologica. 2014;99(9):1479-85.
-
155Kastritis E, Roussou M, Gavriatopoulou M, Migkou M, Kalapanida D, Pamboucas C, et al. Long-term outcomes of primary systemic light chain (AL) amyloidosis in patients treated upfront with bortezomib or lenalidomide and the importance of risk adapted strategies. Am J Hematol. 2015;90(4):E60-5.
-
156Kastritis E, Leleu X, Arnulf B, Zamagni E, Cibeira MT, Kwok F, et al. Bortezomib, Melphalan, and Dexamethasone for Light-Chain Amyloidosis. J Clin Oncol. 2020;38(28):3252-60.
-
157Holmgren G, Steen L, Ekstedt J, Groth CG, Ericzon BG, Eriksson S, et al. Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP-met30). Clin Genet. 1991;40(3):242-6.
-
158Liepnieks JJ, Zhang LQ, Benson MD. Progression of transthyretin amyloid neuropathy after liver transplantation. Neurology. 2010;75(4):324-7.
-
159Okamoto S, Zhao Y, Lindqvist P, Backman C, Ericzon BG, Wijayatunga P, et al. Development of cardiomyopathy after liver transplantation in Swedish hereditary transthyretin amyloidosis (ATTR) patients. Amyloid. 2011;18(4):200-5.
-
160Sack FU, Kristen A, Goldschmidt H, Schnabel PA, Dengler T, Koch A, et al. Treatment options for severe cardiac amyloidosis: heart transplantation combined with chemotherapy and stem cell transplantation for patients with AL-amyloidosis and heart and liver transplantation for patients with ATTR-amyloidosis. Eur J Cardiothorac Surg. 2008;33(2):257-62.
-
161Bulawa CE, Connelly S, Devit M, Wang L, Weigel C, Fleming JA, et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci U S A. 2012;109(24):9629-34.
-
162Coelho T, Maia LF, Martins da Silva A, Waddington Cruz M, Plante-Bordeneuve V, Lozeron P, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79(8):785-92.
-
163Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington-Cruz M, et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N Engl J Med. 2018;379(11):1007-16.
-
164Damy T, Garcia-Pavia P, Hanna M, Judge DP, Merlini G, Gundapaneni B, et al. Efficacy and safety of tafamidis doses in the Tafamidis in Transthyretin Cardiomyopathy Clinical Trial (ATTR-ACT) and long-term extension study. Eur J Heart Fail. 2020.
-
165Hammarstrom P, Jiang X, Hurshman AR, Powers ET, Kelly JW. Sequence-dependent denaturation energetics: A major determinant in amyloid disease diversity. Proc Natl Acad Sci U S A. 2002;99 Suppl 4:16427-32.
-
166Judge DP, Heitner SB, Falk RH, Maurer MS, Shah SJ, Witteles RM, et al. Transthyretin Stabilization by AG10 in Symptomatic Transthyretin Amyloid Cardiomyopathy. J Am Coll Cardiol. 2019;74(3):285-95.
-
167Adams D, Gonzalez-Duarte A, O’Riordan WD, Yang CC, Ueda M, Kristen AV, et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med. 2018;379(1):11-21.
-
168Benson MD, Waddington-Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N Engl J Med. 2018;379(1):22-31.
-
169Emdin M, Aimo A, Rapezzi C, Fontana M, Perfetto F, Seferovic PM, et al. Treatment of cardiac transthyretin amyloidosis: an update. Eur Heart J. 2019;40(45):3699-706.
-
170Cardoso I, Saraiva MJ. Doxycycline disrupts transthyretin amyloid: evidence from studies in a FAP transgenic mice model. FASEB J. 2006;20(2):234-9.
-
171Cardoso I, Martins D, Ribeiro T, Merlini G, Saraiva MJ. Synergy of combined doxycycline/TUDCA treatment in lowering Transthyretin deposition and associated biomarkers: studies in FAP mouse models. J Transl Med. 2010;8:74.
-
172Ritts AJ, Cornell RF, Swiger K, Singh J, Goodman S, Lenihan DJ. Current concepts of cardiac amyloidosis: diagnosis, clinical management, and the need for collaboration. Heart Failure Clinics. 2017;13(2):409-16.
-
173Pollak A, Falk RH. Left ventricular systolic dysfunction precipitated by verapamil in cardiac amyloidosis. Chest. 1993;104(2):618-20.
-
174Sanchis K, Cariou E, Colombat M, Ribes D, Huart A, Cintas P, et al. Atrial fibrillation and subtype of atrial fibrillation in cardiac amyloidosis: clinical and echocardiographic features, impact on mortality. Amyloid. 2019;26(3):128-38.
-
175Longhi S, Quarta CC, Milandri A, Lorenzini M, Gagliardi C, Manuzzi L, et al. Atrial fibrillation in amyloidotic cardiomyopathy: prevalence, incidence, risk factors and prognostic role. Amyloid. 2015;22(3):147-55.
-
176Mints YY, Doros G, Berk JL, Connors LH, Ruberg FL. Features of atrial fibrillation in wild-type transthyretin cardiac amyloidosis: a systematic review and clinical experience. ESC heart failure. 2018;5(5):772-9.
-
177Donnellan E, Wazni O, Kanj M, Elshazly MB, Hussein A, Baranowski B, et al. Atrial fibrillation ablation in patients with transthyretin cardiac amyloidosis. EP Europace. 2020;22(2):259-64.
-
178Roberts WC, Waller BF. Cardiac amyloidosis causing cardiac dysfunction: analysis of 54 necropsy patients. The American journal of cardiology. 1983;52(1):137-46.
-
179Feng D, Edwards WD, Oh JK, Chandrasekaran K, Grogan M, Martinez MW, et al. Intracardiac thrombosis and embolism in patients with cardiac amyloidosis. Circulation. 2007;116(21):2420-6.
-
180Feng D, Syed IS, Martinez M, Oh JK, Jaffe AS, Grogan M, et al. Intracardiac thrombosis and anticoagulation therapy in cardiac amyloidosis. Circulation. 2009;119(18):2490.
-
181Donnellan E, Elshazly MB, Vakamudi S, Wazni OM, Cohen JA, Kanj M, et al. No association between CHADS-VASc score and left atrial appendage thrombus in patients with transthyretin amyloidosis. JACC: Clinical Electrophysiology. 2019;5(12):1473-4.
-
182El-Am EA, Dispenzieri A, Melduni RM, Ammash NM, White RD, Hodge DO, et al. Direct current cardioversion of atrial arrhythmias in adults with cardiac amyloidosis. Journal of the American College of Cardiology. 2019;73(5):589-97.
-
183Goldsmith YB, Liu J, Chou J, Hoffman J, Comenzo RL, Steingart RM. Frequencies and types of arrhythmias in patients with systemic light-chain amyloidosis with cardiac involvement undergoing stem cell transplantation on telemetry monitoring. The American journal of cardiology. 2009;104(7):990-4.
-
184Palladini G, Malamani G, Co F, Pistorio A, Recusani F, Anesi E, et al. Holter monitoring in AL amyloidosis: prognostic implications. Pacing and Clinical Electrophysiology. 2001;24(8):1228-33.
-
185Towbin JA, McKenna WJ, Abrams DJ, Ackerman MJ, Calkins H, Darrieux FC, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart rhythm. 2019;16(11):e301-e72.
-
186Members ATF, Priori SG, Blomström-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC) Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Ep Europace. 2015;17(11):1601-87.
-
187Cappelli F, Perfetto F, Martone R, Di Mario C. Cardiac Amyloidosis in Patients Undergoing TAVR: Why We Need to Think About It. Cardiovascular Revascularization Medicine. 2021;22:109-14.
-
188Sayed RH, Rogers D, Khan F, Wechalekar AD, Lachmann HJ, Fontana M, et al. A study of implanted cardiac rhythm recorders in advanced cardiac AL amyloidosis. European heart journal. 2015;36(18):1098-105.
-
189Giancaterino S, Urey MA, Darden D, Hsu JC. Management of arrhythmias in cardiac amyloidosis. Clinical Electrophysiology. 2020;6(4):351-61.
-
190Ridolfi RL, Bulkley BH, Hutchins GM. The conduction system in cardiac amyloidosis: clinical and pathologic features of 23 patients. The American journal of medicine. 1977;62(5):677-86.
-
191DePasquale EC, Nasir K, Jacoby DL. Outcomes of adults with restrictive cardiomyopathy after heart transplantation. The Journal of heart and lung transplantation. 2012;31(12):1269-75.
-
192Givens RC, Russo C, Green P, Maurer MS. Comparison of cardiac amyloidosis due to wild-type and V122I transthyretin in older adults referred to an academic medical center. Aging health. 2013;9(2):229-35.
-
193Topilsky Y, Pereira NL, Shah DK, Boilson B, Schirger JA, Kushwaha SS, et al. Left ventricular assist device therapy in patients with restrictive and hypertrophic cardiomyopathy. Circulation: Heart Failure. 2011;4(3):266-75.
-
194Grupper A, Park SJ, Pereira NL, Schettle SD, Gerber Y, Topilsky Y, et al. Role of ventricular assist therapy for patients with heart failure and restrictive physiology: improving outcomes for a lethal disease. The Journal of Heart and Lung Transplantation. 2015;34(8):1042-9.
-
195Kristen AV, Kreusser MM, Blum P, Schönland SO, Frankenstein L, Dösch AO, et al. Improved outcomes after heart transplantation for cardiac amyloidosis in the modern era. The journal of heart and lung transplantation. 2018;37(5):611-8.
-
196Trachtenberg BH, Kamble RT, Rice L, Araujo-Gutierrez R, Bhimaraj A, Guha A, et al. Delayed autologous stem cell transplantation following cardiac transplantation experience in patients with cardiac amyloidosis. American Journal of Transplantation. 2019;19(10):2900-9.
-
197Biglia LV, Mendes SJ, Lima TM, Aguiar PM. Incorporações de medicamentos para doenças raras no Brasil: é possível acesso integral a estes pacientes? Cien Saúde Colet. 2020(julho/2020 - Está disponível em: http://www.cienciaesaudecoletiva.com.br/artigos/incorporacoes-de-medicamentos-para-doencas-raras-no-brasil-e-possivel-acesso-integral-a-estes-pacientes/17706?id=17706&id=17706).
» http://www.cienciaesaudecoletiva.com.br/artigos/incorporacoes-de-medicamentos-para-doencas-raras-no-brasil-e-possivel-acesso-integral-a-estes-pacientes/17706?id=17706&id=17706) -
198Silva EN, Sousa TR. Economic evaluation in the context of rare diseases: is it possible? Cad Saude Publica. 2015;31(3):496-506.
-
199Brasil. Ministério da Saúde. Política Nacional de Atenção Integral às Pessoas com Doenças Raras. Portaria GM nº 199, de 30 de janeiro de 2014. Disponível em: http://bvsmssaudegovbr/bvs/saudelegis/gm/2014/prt0199_30_01_2014html
» http://bvsmssaudegovbr/bvs/saudelegis/gm/2014/prt0199_30_01_2014html -
200INTERFARMA. Doenças Raras: A urgência do acesso à saúde. Fevereiro de 2018. Disponível em: https://www.interfarma.org.br/public/files/biblioteca/doencas-raras--a-urgencia-do-acesso-a-saude-interfarma.pdf Acesso em: 14 abr. 2019.
» https://www.interfarma.org.br/public/files/biblioteca/doencas-raras--a-urgencia-do-acesso-a-saude-interfarma.pdf -
201Novaes HMD, Soárez PC. Doenças raras, drogas órfãs e as políticas para avaliação e incorporação de tecnologias nos sistemas de saúde. Sociologias. 2019;21(5):332-64.
-
202Nicod E, Annemans L, Bucsics A, Lee A, Upadhyaya S, Facey K. HTA programme response to the challenges of dealing with orphan medicinal products: Process evaluation in selected European countries. Health Policy. 2019;123(2):140-51.
-
203Kircher M, Ihne S, Brumberg J, Morbach C, Knop S, Kortum KM, et al. Detection of cardiac amyloidosis with (18)F-Florbetaben-PET/CT in comparison to echocardiography, cardiac MRI and DPD-scintigraphy. Eur J Nucl Med Mol Imaging. 2019;46(7):1407-16.
-
204Morgenstern R, Yeh R, Castano A, Maurer MS, Bokhari S. (18)Fluorine sodium fluoride positron emission tomography, a potential biomarker of transthyretin cardiac amyloidosis. J Nucl Cardiol. 2018;25(5):1559-67.
-
205Kufova Z, Sevcikova T, Growkova K, Vojta P, Filipova J, Adam Z, et al. Biomarkers in Immunoglobulin Light Chain Amyloidosis. Klin Onkol.30(Suppl 2):60-7.
-
206Paiva B, Martinez-Lopez J, Corchete LA, Sanchez-Vega B, Rapado I, Puig N, et al. Phenotypic, transcriptomic, and genomic features of clonal plasma cells in light-chain amyloidosis. Blood. 2016;127(24):3035-9.
-
Development:The Brazilian Society of Cardiology's Department of Heart Failure (Departamento de Insuficiência Cardíaca – DEIC) and the Study Group on Cardiomyopathies (Grupo de Estudos em Miocardiopatias – GEMIC) of the Brazilian Society of Cardiology (Sociedade Brasileira de Cardiologia – SBC)
-
Norms and Guidelines Council (2020-2021):Antonio Carlos Sobral Sousa, Aurora Felice de Castro Issa, Bruno Ramos Nascimento, Harry Corrêa Filho, Marcelo Luiz Campos Vieira
-
Norms and Guidelines Coordinator (2020-2021):Brivaldo Markman Filho
-
How to cite this statement:Simões MV, et al. Posicionamento sobre Diagnóstico e Tratamento da Amiloidose Cardíaca – 2021. Arq Bras Cardiol. 2021; 117(3):561-598
-
Note:These statements are intended to support, not replace, the clinical judgment of physicians who, ultimately, must determine the appropriate treatment for their patients.
-
ErratumSeptember 2021 Issue, vol. 117 (3), pages 561-598In “Position Statement on Diagnosis and Treatment of Cardiac Amyloidosis – 2021”, com número de DOI: https://doi.org/10.36660/abc.20210718, published in the Journal Arquivos Brasileiros de Cardiologia, 117(3):561-598, on page 561, author Flávio Henrique Valicelli, institution number 1, after the author Carlos Eduardo Rochitte was included. On page 564, the conflict of interest was included: “Nothing to be declared”, under the name Fernando Bacal.
Publication Dates
-
Publication in this collection
20 Sept 2021 -
Date of issue
Sept 2021