Abstract
This report describes the development of a SYBR Green I based real time polymerase chain reaction (PCR) protocol for detection on the ABI Prism 7000 instrument. Primers targeting the gene encoding the SSU rRNA were designed to amplify with high specificity DNA from Schistosoma mansoni, in a real time quantitative PCR system. The limit of detection of parasite DNA for the system was 10 fg of purified genomic DNA, that means less than the equivalent to one parasite cell (genome ~580 fg DNA). The efficiency was 0.99 and the correlation coefficient (R²) was 0.97. When different copy numbers of the target amplicon were used as standards, the assay could detect at least 10 copies of the specific target. The primers used were designed to amplify a 106 bp DNA fragment (Tm 83ºC). The assay was highly specific for S. mansoni, and did not recognize DNA from closely related non-schistosome trematodes. The real time PCR allowed for accurate quantification of S. mansoni DNA and no time-consuming post-PCR detection of amplification products by gel electrophoresis was required. The assay is potentially able to quantify S. mansoni DNA (and indirectly parasite burden) in a number of samples, such as snail tissue, serum and feces from patients, and cercaria infested water. Thus, these PCR protocols have potential to be used as tools for monitoring of schistosome transmission and quantitative diagnosis of human infection.
polymerase chain reaction (PCR); molecular diagnosis; schistosomiasis; real-time PCR
Development of a real time polymerase chain reaction for quantitation of Schistosoma mansoni DNA
Ana Lisa do Vale Gomes; Fábio L Melo; Roberto P Werkhauser; Frederico GC Abath1 1 Corresponding author and recipient of a research scholarship from CNPq. E-mail: fabath@cpqam.fiocruz.br
Departamento de Imunologia, Centro de Pesquisas Aggeu Magalhães-Fiocruz, Av. Prof. Moraes Rego s/nº, Cidade Universitária, 50670-420 Recife, PE, Brasil
ABSTRACT
This report describes the development of a SYBR Green I based real time polymerase chain reaction (PCR) protocol for detection on the ABI Prism 7000 instrument. Primers targeting the gene encoding the SSU rRNA were designed to amplify with high specificity DNA from Schistosoma mansoni, in a real time quantitative PCR system. The limit of detection of parasite DNA for the system was 10 fg of purified genomic DNA, that means less than the equivalent to one parasite cell (genome ~580 fg DNA). The efficiency was 0.99 and the correlation coefficient (R2) was 0.97. When different copy numbers of the target amplicon were used as standards, the assay could detect at least 10 copies of the specific target. The primers used were designed to amplify a 106 bp DNA fragment (Tm 83ºC). The assay was highly specific for S. mansoni, and did not recognize DNA from closely related non-schistosome trematodes. The real time PCR allowed for accurate quantification of S. mansoni DNA and no time-consuming post-PCR detection of amplification products by gel electrophoresis was required. The assay is potentially able to quantify S. mansoni DNA (and indirectly parasite burden) in a number of samples, such as snail tissue, serum and feces from patients, and cercaria infested water. Thus, these PCR protocols have potential to be used as tools for monitoring of schistosome transmission and quantitative diagnosis of human infection.
Key words: polymerase chain reaction (PCR) - molecular diagnosis - schistosomiasis - real-time PCR
In Brazil, Schistosoma mansoni is the only causative species of schistosomiasis, and there are three species of intermediary hosts: Biomphalaria glabrata, B. straminea, and B. tenagophila. During the course of its life cycle, S. mansoni differentiates into several stages. Adult schistosomes release eggs with host's feces that in contact with water gives rise to miracidia, which is the infective form for the snail hosts. Miracidia differentiate into sporocysts immediately after penetration, migrating through snail tissues, and giving rise to cercariae. Humans become infected by contact with water infested with cercariae, which are capable of actively penetrating the skin. Molecular detection of different life cycle stages of the parasite is possible, although these proposals have been scarce. Indeed, few studies were published for the detection of the parasite in snails (Hanelt et al. 1997, Jannotti-Passos et al. 1997, Hamburger et al. 1998a, Melo et al. 2006), monitoring of cercariae in water bodies (Hamburger et al. 1998b), and diagnosis of human infection (Pontes et al. 2002).
Recently, real-time PCR (polymerase chain reaction) assays have been developed for the detection of a number of infectious organisms (Bankowski & Anderson 2004). Real-time PCR is a fluorescent-based technology, which is performed in a closed system. The World Health Organization recommends that research should concentrate on developing and evaluation of new strategies and tools for control of the disease (WHO 2006). To our knowledge this is the first report on the development of a sensitive and specific quantitative real-time PCR for the detection of S. mansoni DNA. This system is potentially useful for quantitating parasite burden in human infection. In addition, it can be used for sensitive and accurate monitoring of transmission sites of schistosomiasis by detecting and quantitating infection in the snail vector.
Sequences of SSU rRNA (small subunit ribosomal RNA) of Schistosoma sp., Mus musculus, Homo sapiens, B. glabrata, and other molluscs were aligned using tools available at the European ribosomal RNA database (http://www.psb.ugent.be/rRNA/index.html), maintained by the University of Gent, Belgium, and also using Clustal W (http://www.ebi.ac.uk/clustalw). This allowed the selection of possible targets for PCR amplification. Target regions were further refined by additional alignments of sequences from trematodes closely related to S. mansoni. Primers Schfo111 (5'- cgatcaggaccagtgttcagc -3') and Schre111 (5'- gacaggtcaacaagacgaactcg -3') were judiciously designed using the software Lasergene (DNASTAR, Inc., Madison,WI, US), so that S. mansoni DNA could be discriminated from other closely related parasites, and particular attention was given to the 3' end of the primers. DNA from S. mansoni adult worms, mice and B. glabrata were purified using the GenomicPrep Cells and Tissue DNA Isolation Kit TM (Amersham Biosciences, Uppsala, Sweden), following the instructions of the supplier.
DNA samples from Echinostoma paraensis, S. haematobium, S. bovis, S. japonicum, S. rhodaini, Cercaria minensis, C. macrogranulosa, and C. caratinguensis were kindly provided by A Maldonado (Instituto Oswaldo Cruz- Fiocruz, Rio de Janeiro), G Oliveira, and O Carvalho (Centro de Pesquisas René-Rachou-Fiocruz, Belo Horizonte).
Real-time PCR was carried out on the ABI PRISM 7000 system (Applied Biosystems, CA, US) using the sequence unspecific SYBR Green I dye, that binds to any double stranded DNA (and ROX as passive reference). PCR products were detected with the melting curve analysis, which was subsequently performed after the PCR run, by increasing the temperature slowly from 60 to 95ºC (0.1ºC/s) and by measuring the fluorescence continuously. The ABI PRISM software (version 1.1) was used for the analysis and interpretation of the results. The reactions included 2 µl of the extracted DNA, primers (2.5 pmols each) and the Sybr Green Master Mix (Applied Biosystems, CA, US), as well as water added to a final volume of 50 µl. The PCR was performed under the following cycling conditions: an initial denaturation step at 95ºC for 10 min, followed by 40 cycles of amplification (95ºC for 15 s, 60ºC for 30 s, and 72ºC for 30 s). Several non template controls (NTC) and quantitative standards were included each time PCR was undertaken to detect false positive results due to contamination and to construct a standard curve. All assays were performed in duplicates.
As PCR is an exponential process, it can be described by the equation Nn=N0+ (1+e)n, where Nn is the number of target molecules at cycle n, N0 is the initial number of target molecules, e is the efficiency of amplification, and n is the number of cycles. The efficiency of amplification (e) of a target molecule can be calculated from the slope of the standard curve (plot of Ct versus the negative log10 concentration of the target). High efficiencies of amplifications have slopes approaching the value of 3.32 for every 10-fold dilution of the target. To compare the specificities of any assays, it is critical to compare the differences in the Ct values of the defined target and the templates (D Ct) as well as the efficiencies of the amplifications of the target and templates within each assay. Hence, specificity (s) can be defined by the equation: s = (1+ e D Ct), where (1+ e) is 10 1/slope (Too 2003). Hence, the larger the value s, the more specific the assays are in discriminating the target over the test templates. The sensitivity of the detection limit of an assay is defined by the amplification of the highest dilution of the target when compared to the formation of primer-dimer in samples without template. A log 10 dilution series of purified DNA from S. mansoni was amplified to assess the limits of detection of the PCR assays. In addition, the detection limit in copy numbers was determined analyzing amplicons corresponding to the target region, that were previously purified and quantified. To determine the specificity, M. musculus, H. sapiens, B. glabrata, and several parasites closely related to S. mansoni, and eventually infective to snails were analyzed. As we had access to a limited range of non-schistosome trematodes for experimentation, the study was complemented by a theoretical analysis of SSU rRNA sequences from a number of parasites.
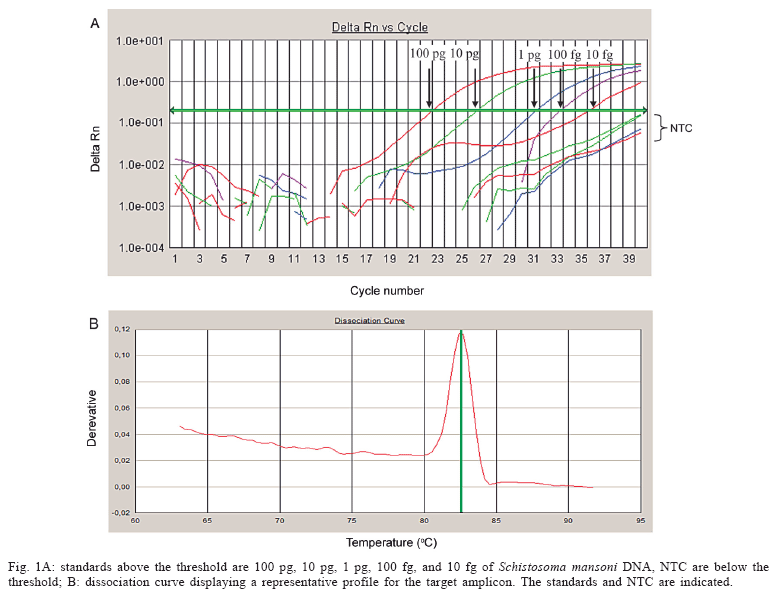
A standard curve was constructed, resulting in a detection limit of 10 fg of S. mansoni genomic DNA (Fig. 1). The efficiency was 0.99 and the correlation coefficient (R2) was 0.97. When different copy numbers of the target amplicon were used as standards, the assay could detect at least 10 copies of the specific target (data not shown).
The novel PCR system developed was highly specific for the detection of Schistosoma DNA. As can be observed in Fig. 2, the PCR system composed by primers Schfo111 and Schre111 amplified only S. mansoni DNA, but not DNA from their vertebrate or invertebrate hosts. None of the other trematodes were amplified by these systems, except S. bovis. When universal primers were used, amplicons from all species analyzed were obtained, indicating that the samples were appropriate for PCR (data not shown).
Real-time PCR offers several advantages compared to conventional nested PCR. Real-time PCR is a closed system, in which DNA amplification and detection is carried out in a single tube or well that remain sealed during the whole PCR run. Since no post amplification processing as gel electrophoresis has to be done, the risk of carry over contamination is minimized (Bankowski & Anderson 2004). The melting curve analysis is directly and automatically performed after the PCR run within a couple of minutes, to discriminate between the specific and nonspecific PCR products like primer dimers. A total PCR run, including the identification of the PCR products by melting curve analysis can be carried out within approximately 45 min. Another advantage and interesting feature of real-time PCR is the possibility for quantitative analysis, because of the negative correlation between Ct values and number of target copies.
The S. mansoni real-time PCR detection system protocol developed is sensitive and highly specific. Since the genome of S. mansoni contains ~ 580 fg, theoretically our PCR system can detect DNA corresponding to less than a single cell of the multicelular parasite S. mansoni. The approach can be potentially used to detect S. mansoni DNA in several biological samples: human feces, snails, water. As this technique is able to accurately quantify target DNA, the approach can be useful to quantify parasite burden in human and snail infection. Indirectly, it can be used to monitor the potential of infection of transmission sites by estimating the intensity of infestation of water bodies and intensity of infection in snail.
ACKNOWLEDGEMENTS
To Rosineide Lira, Wlademir Melo, and Barnabe Tabosa for technical assistance.
Received 25 May 2006
Accepted 26 June 2006
Financial support: Fiocruz/PDTIS, Facepe, CNPq
- Bankowski MJ, Anderson SM 2004. Real-time nucleic acid amplification in clinical microbiology. Clin Microbiol Newsletter 26: 9-15.
- Hamburger J, He N, Xin XY, Ramzy RM, Jourdane J, Ruppel A 1998a. A polymerase chain reaction assay for detecting snails infected with bilharzia parasites (Schistosoma mansoni) from very early prepatency. Am J Trop Med Hyg 59: 872-876.
- Hamburger J, Yu-Xin X, Ramzy RM, Jourdane J, Ruppel A 1998b. Development and laboratory evaluation of a polymerase chain reaction for monitoring Schistosoma mansoni infestation of water. Am J Trop Med Hyg 59: 468-473.
- Hanelt B, Adema CM, Mansour MH, Loker ES 1997. Detection of Schistosoma mansoni in Biomphalaria using nestd PCR. J Parasitol 83: 387-394.
- Jannotti-Passos LK, Vidigal THDA, Dias-Neto E, Pena SDJ, Simpson AJG, Dutra WO, Souza CP, Carvalho-Parra JF 1997. PCR amplification of the mitocondrial DNA minisatellite region to detect Schistosoma mansoni infection in Biomphalaria glabrata snails. Parasitology 83: 395-399.
- Melo FL, Gomes ALG, Barbosa CS, Werkhauser RP, Abath FGC 2006. Development of molecular approaches for the identification of transmission sites of schistosomiasis. Trans R Soc Trop Med Hyg (in press).
- Pontes LA, Dias-Neto E, Rabello ALT 2002. Detection by polymerase chain reaction of Schistosoma mansoni DNA in human serum and feces. Am J Trop Med Hyg 66: 157-162.
- Too HP 2003. Real time PCR quantification of GFR-2 alternatively spliced isoforms in murine brain and peripheral tissues. Mol Brain Res 114: 146-154.
- WHO 2006. World Health Organization Special Programme for Research and Training in Tropical Disease. TDR home page at http://www.who.int/tdr/
Publication Dates
-
Publication in this collection
12 Feb 2007 -
Date of issue
Oct 2006
History
-
Accepted
26 June 2006 -
Received
25 May 2006