Abstract
Trypanosoma cruzi infection of the adipose tissue of mice triggers the local expression of inflammatory mediators and a reduction in the expression of the adipokine adiponectin. T. cruzi can be detected in adipose tissue by PCR 300 days post-infection. Infection of cultured adipocytes results in increased expression of cytokines and chemokines and a reduction in the expression of adiponectin and the peroxisome proliferator-activated receptor ³, both of which are negative regulators of inflammation. Infection also results in the upregulation of cyclin D1, the Notch pathway, and extracellular signal-regulated kinase and a reduction in the expression of caveolin-1. Thus, T. cruzi infection of cultured adipocytes leads to an upregulation of the inflammatory process. Since adiponectin null mice have a cardiomyopathic phenotype, it is possible that the reduction in adiponectin contributes to the pathogenesis of chagasic cardiomyopathy. Adipose tissue may serve as a reservoir for T. cruzi from which parasites can become reactivated during periods of immunosuppression. T. cruzi infection of mice often results in hypoglycemia. In contrast, hyperglycemia as observed in diabetes results in increased parasitemia and mortality. Adipose tissue is an important target tissue of T. cruzi and the infection of this tissue is associated with a profound impact on systemic metabolism, increasing the risk of metabolic syndrome.
adipose tissue; adipocyte; adiponectin; Trypanosoma cruzi; Chagas disease
Chagas disease, adipose tissue and the metabolic syndrome
Fnu NagajyothiI; Mahalia S DesruisseauxI, II, III; Louis M WeissI, II; Streamson ChuaI, III; Chris AlbaneseIV; Fabiana S MachadoV; Lisia EsperV; Michael P LisantiVI; Mauro M TeixeiraV; Philipp E SchererVII; Herbert B TanowitzI, II,+ + Corresponding author: tanowitz@aecom.yu.edu
IDepartment of Pathology
IIDepartment of Medicine
IIIDepartment of Neuroscience, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY 10461, USA
IVDepartment of Oncology, Lombardi Comprehensive Cancer Center, Georgetown University Medical Center, Washington DC, USA
VDepartamento de Bioquímica e Imunologia, Instituto de Ciência Biológicas, Universidade Federal de Minas Gerais, Belo Horizonte, MG, Brasil
VIDepartment of Cancer Biology and the Kimmel Cancer Center, Thomas Jefferson University, Philadelphia, PA, USA
VIITouchstone Diabetes Center, Departments of Internal Medicine and Cell Biology, University of Texas Southwestern, Dallas, TX, USA
ABSTRACT
Trypanosoma cruzi infection of the adipose tissue of mice triggers the local expression of inflammatory mediators and a reduction in the expression of the adipokine adiponectin. T. cruzi can be detected in adipose tissue by PCR 300 days post-infection. Infection of cultured adipocytes results in increased expression of cytokines and chemokines and a reduction in the expression of adiponectin and the peroxisome proliferator-activated receptor γ, both of which are negative regulators of inflammation. Infection also results in the upregulation of cyclin D1, the Notch pathway, and extracellular signal-regulated kinase and a reduction in the expression of caveolin-1. Thus, T. cruzi infection of cultured adipocytes leads to an upregulation of the inflammatory process. Since adiponectin null mice have a cardiomyopathic phenotype, it is possible that the reduction in adiponectin contributes to the pathogenesis of chagasic cardiomyopathy. Adipose tissue may serve as a reservoir for T. cruzi from which parasites can become reactivated during periods of immunosuppression. T. cruzi infection of mice often results in hypoglycemia. In contrast, hyperglycemia as observed in diabetes results in increased parasitemia and mortality. Adipose tissue is an important target tissue of T. cruzi and the infection of this tissue is associated with a profound impact on systemic metabolism, increasing the risk of metabolic syndrome.
Key words: adipose tissue - adipocyte - adiponectin - Trypanosoma cruzi - Chagas disease
Chagas disease caused by Trypanosoma cruzi remains an important cause of morbidity and mortality in endemic areas of Mexico, Central and South America. Ten to thirty percent of individuals with this infection eventually succumb to chronic manifestations such as cardiomyopathy and/or mega syndromes. This infection is also regarded as an opportunistic infection during cases of immunosuppression, for instance affecting individuals with HIV/AIDS. The pathogenesis of Chagas disease has been explored in detail by many laboratories over the past decades, but the role of the adipocyte and of adipose tissue, long ignored, is only now being investigated.
The adipocyte and adipose tissue: general considerations
The adipocyte, or fat cell, contributes to the pathogenesis of diabetes, obesity and the metabolic syndrome (Rajala et al. 2003, Wernstedt-Asterholm et al. 2007) and its secretory products have been implicated in many physiological processes (Nawrocki & Scherer 2005, Attie & Scherer 2009). Until recently, adipose tissue was considered to be a mere storage compartment for triglycerides. However, adipocytes are active endocrine cells that play a central role in overall energy homeostasis and are important contributors to some aspects of the immune system (Halberg et al. 2008). They do so by influencing systemic lipid homeostasis and also through the production and release of a host of adipocyte-specific and adipocyte-enriched hormonal factors, inflammatory mediators such as cytokines and chemokines and extracellular matrix components also known as adipokines. There has been little attention given to the role of adipose tissue and adipocytes in infectious disease (Desruisseaux et al. 2007), but the strong pro-inflammatory potential of adipose tissue suggests that it may have an important role in the systemic innate immune response.
Adipocytes are the most prominent cell type in adipose tissue. However, adipose tissue is also composed of other cell types including fibroblasts, endothelial cells, smooth muscle cells and, especially in the setting of infection, inflammatory cells. Since different adipose tissue depots show distinct gene expression patterns and vary widely in their size and proximity to neighboring organs, individual depots have been viewed as "mini-organs." Despite differences between the different fat pads, the depots share similarity with respect to their ability to store lipids and secrete adipose tissue-derived hormones. Adipose tissue stores lipid in the form of triglycerides. It also stores mostly non-esterified cholesterol on the surface of lipid droplets that act as specialized organelles inside the adipocyte. Since the lipid droplet is such a large component of the adipocyte, changes in the amount of lipid stored within it affect fat cell size (which can range from 25-250 microns).
The potential endocrine function of adipose tissue was first recognized in the 1980s with the report that the serine protease adipsin was secreted by cultured 3T3-L1 adipocytes (Kook et al. 1987). Subsequently, several additional adipokines have been discovered, including adiponectin (Scherer et al. 1995), leptin, resistin, SAA3, omentin, visfatin and RBP4 (Desruisseaux et al. 2007). These adipokines contribute to the regulation of energy homeostasis through effects on both central and peripheral tissues. Several of these adipokines also contribute to non-metabolic processes in the body, highlighting the fact that adipokines participate in the coordination of multiple physiological functions in a variety of tissues. The most adipocyte-specific adipokine is adiponectin. Other adipokines can also be synthesized by tissues other than adipose tissue and/or by cells other than adipocytes.
Systemic energy homeostasis is maintained by the competing effects of a number of different hormonal factors, some of which originate in adipose tissue. These adipocyte-derived factors (adipokines), influence processes such as food intake, energy expenditure and insulin sensitivity in a variety of tissues. Two adipokines, resistin and adiponectin have opposing effects on whole-body glucose homeostasis (Combs et al. 2001, Rajala et al. 2003). Pharmacological doses of recombinant resistin hyper-activate gluconeogenesis through decreased hepatic insulin sensitivity. Adiponectin, a hormone exclusively produced by the adipocytes, is a 30-kDa molecule with three defined domains. The N-terminus contains a hypervariable region, which is commonly used as the antigenic site for species-specific antibody generation. The collagenous stalk containing 22 GXY repeats is followed by a globular domain at the C-terminus. Both intracellularly and extracellularly, adiponectin exists in three different higher-order complexes: a high molecular weight form (HMW; 12-36 mer), a low molecular weight form (hexamer), and a trimeric form. The different complexes have distinct functions, and the ratio of HMW to the other forms serves as an independent predicting factor of metabolic disorders. The total level and HMW ratio are decreased in obese patients and obese mouse models. This suggests that adiponectin, especially the HMW form, may be involved in obesity-related disorders. It has been demonstrated that adiponectin increases insulin sensitivity by inhibiting hepatic glucose output. Lower levels of circulating adiponectin are associated with increased susceptibility to a variety of diseases of metabolic dysfunction, including diabetes, hypertension and obesity.
Many studies have demonstrated an association between circulating adiponectin levels and various metabolic parameters that regulate insulin sensitivity in different patient populations. For example, Arita et al. (1999) demonstrated decreased plasma adiponectin concentration in obese humans; follow-up studies confirmed that this finding could be extended to obese rodents and other animal models. The pattern of decreased adiponectin secretion with increasing adiposity, though contrary to what is observed for the majority of adipose-specific secretory proteins (i.e., leptin), has been well recognized. There is a reduction in the levels of adiponectin in diabetics with coronary artery disease compared to diabetics without coronary artery disease and adiponectin levels in serum are negatively correlated with basal metabolic rate, plasma glucose, insulin and serum triglycerides (Hottoa et al. 2000). Furthermore, a relatively moderate weight loss led to a significant increase in circulating adiponectin levels in both diabetics and non-diabetics. In morbidly obese individuals (Yang et al. 2001) undergoing gastric partition surgery, it was demonstrated that post-surgical decreases in basic metabolic rate and fasting glucose and insulin levels were associated with a similar increase in circulating levels of adiponectin, together with an increase in insulin sensitivity. The paradox of why adiponectin levels tend to increase with decreasing adiposity has yet to be explained. After weight loss, either through conventional or surgical means, the remaining adipocytes may be correspondingly more insulin sensitive and therefore secrete increased amounts of adiponectin. Alternatively, adiponectin expression and/or secretion may be directly or indirectly regulated by plasma insulin levels. Supporting this view, previous studies have demonstrated that insulin treatment of 3T3-L1 adipocytes results in significantly decreased adiponectin expression (Fasshauer et al. 2002) and serum adiponectin levels are inversely proportional to fasting insulin levels. A corollary is that an inhibitory feedback pathway must exist to down-regulate the expression and secretion of adiponectin in the obese.
Central adipose pads (intra-abdominal/mesenteric) are the predominant sources of systemic adiponectin in the lean state. The production of adiponectin by this tissue in the obese state is reduced. Individuals with the highest levels of adiponectin had a reduced risk of myocardial infarction compared with those with the lowest adiponectin levels. This relationship persisted even when controlling for several variables. Animal models have corroborated these observations, demonstrating the importance of adiponectin for preventing diet-induced progression of atherosclerosis.
Adiponectin, inflammation and heart disease
The mechanism of the anti-atherosclerotic activity of adiponectin has not been completely elucidated. It has been hypothesized that adiponectin has inflammation-modulating activities and clinical studies have demonstrated inverse associations between adiponectin levels and serum markers of inflammation (Ouchi et al. 2003, Goldstein & Scalia 2004). Several studies have reported that the physiologically relevant, full-length form of adiponectin has anti-inflammatory effects on both endothelium and macrophages. These studies used a recombinant mammalian cell-produced full length form of the protein that has potential as a pharmacologic agent but differs in its bioactivity from the endogenous post-translationally modified and multimerized hormone (Nawrocki & Scherer 2005, Desruisseaux et al. 2007). Although it is unclear how or whether adiponectin itself has anti-inflammatory properties, it is clear that adiponectin production by adipose tissue can be inhibited by systemic inflammation in at least some circumstances.
Adiponectin production by cultured adipocytes is inhibited by inflammatory cytokines such as TNF-α (Desruisseaux et al. 2007, Ruan & Lodish 2003). This inhibition may be mediated in part by NFκB signaling. In cultured adipocytes as well as in obese diabetic mice, the inhibition of adipocyte inflammatory NFκB signaling by an IκB Kinase inhibitor resulted in a reduction of cytokine levels and increased plasma levels of adiponectin. Thus, IκB Kinase inhibition leads to increased plasma adiponectin levels and an improvement in systemic insulin sensitivity (Keller et al. 2003). The anti-inflammatory activity of adiponectin may be mediated by its principal signaling target, the AMP-activated protein kinase (AMPK) as the truncated bacterial form of an adiponectin has anti-inflammatory effects on endothelium via AMPK-mediated modulation of NFκB and Akt/PKB (Ouchi et al. 2000).
Chemokines positively control the secretion of leptin, suggesting a role for these molecules in the regulation of adipose tissue. Targeting chemokines may provide a novel therapeutic basis for the treatment of obesity, diabetes and cachexia (Gerhardt et al. 2001). A high-fat diet increases the expression of inflammatory genes, including the early induction of MCP-1 and MCP-3 (Chen et al. 2005). Some of the proven anti-atheromatous effects of adiponectin may be mediated by anti-inflammatory actions directly on the vasculature. Okamoto et al. (2008) recently reported that adiponectin inhibits the production of CXCR-3 chemokine ligands in macrophages and causes a reduction in T-lymphocyte recruitment.
Adiponectin has a role in protecting against cardiac hypertrophy in cardiac overload states such as hypertension, hypertrophic cardiomyopathy and ischemic heart disease. In mice, adiponectin protects against myocardial ischemia-reperfusion injury and overload- and adrenergically-induced cardiac myocyte hypertrophy by inhibiting hypertrophic signals via AMPK (Shibata et al. 2004, 2005, Ouchi et al. 2006). Interestingly, adiponectin null mice have a cardiomyopathic phenotype (Ouchi et al. 2006, Shibata et al. 2009). The present information is consistent with the idea that adiponectin is anti-inflammatory and reduced levels of adiponectin are pro-inflammatory.
Adipose tissue adipocytes and infection
The relationship between adipocytes and infection has only recently received attention. For example, there have been several investigations into the infectious etiologies of obesity (Pasarica & Dhurandhar 2007). A role for adipose tissue in infection was underscored by the work of the Scherer laboratory, which demonstrated that the injection of LPS into mice that were rendered fatless by manipulation of the apoptotic pathway did not cause the immediate death of the mice, as it did in control mice with a normal amount of adipose tissue (Pajvani et al. 2005). These data suggested that the inflammatory mediators associated with adipose tissue play an important role in the inflammatory response to infection. One of the most intensively investigated areas in the interface between infection and adipose tissue has been in HIV/AIDS.
Adipocytes are now regarded as a direct target for HIV infection. In that regard, Hazan et al. (2002) demonstrated the receptors for HIV entry, such as CD4, CXCR4 and CCR5, were expressed in vitro on pre-adipocytes and adipocytes. In vivo expression of these receptors was also shown in sections of human white adipose tissue. Maurin et al. (2005) demonstrated HIV receptor expression in freshly isolated pre-adipocytes and in mature adipocytes from subcutaneous fat depots. They also verified HIV infection of adipocytes by observing HIV-1 transcriptional activity in these cells. Additionally, they showed that Gag p24 antigen expression was significantly increased upon stimulation of these cells with pro-inflammatory cytokines, indicating that HIV-1 infects human adipocytes in vitro. These observations further suggest that the initial infection may be overcome by treatment with pro-inflammatory cytokine stimulation. Alterations in the differentiation and morphology of fat (Jan et al. 2004), as well as increased levels of collagen fibers and vessel density, have been described in HIV patients. Leptin levels are significantly higher in HIV infected patients with lipoatrophy and these levels are negatively correlated with insulin resistance. Our laboratory (Scherer) demonstrated that adiponectin levels were lower in HIV-infected individuals with lipoatrophy and that this decrease was positively correlated with insulin resistance (Mynarcik et al. 2002). Patients with highly active antiretrovial therapy-associated lipodystrophy have leptin levels that are correlated more closely with nutritional status. Increased levels of TNF-α and IL-6 during HIV infection may directly affect the insulin signaling pathways and induce insulin resistance. Moreover, the lipolytic effects of these cytokines may increase free fatty acids and cause insulin resistance through lipotoxic effects in the muscle and liver. Recent evidence suggests that the development of HIV-related lipoatrophy is related to mitochondrial dysfunction in adipose tissue (Mallon et al. 2008).
T. cruzi infection, diabetes and adipose tissue
Diseases caused by nematodes and protozoa have been reported to be associated with nutritional deficiencies, wasting and diabetes. An association between human T. cruzi infection and obesity and diabetes has been suspected and there has been general belief, although not proven, that the incidence of diabetes may be increased in the chagasic population.
There may be a relationship between Chagas disease and both obesity and underweightedness; the common factor appears to be poverty (Barreto et al. 2003). Previous studies from our laboratory demonstrated that when mice with chemical-induced diabetes were infected with T. cruzi, they had a higher parasitemia and mortality (Tanowitz et al. 1988). The same observation was noted in infected diabetic db/db mice (Tanowitz et al. 1988). The underlying pathophysiological mechanisms of these observations remain unknown. A personal communication from a physician in Argentina (MM Aranda) stated that there are groups of "aboriginal people" where Chagas disease afflicts over 50% of the population and that there is a strong co-existence with the features of the metabolic syndrome in the same population. These observations need to be examined more systematically.
We determined the metabolic consequences of T. cruzi infection on basal glucose levels and insulin sensitivity. Acute infection of CD-1 mice with the Brazil-strain of T. cruzi is usually associated with severe hypoglycemia and generally correlated with mortality (Combs et al. 2005). Interestingly, the metabolic response to bacterial sepsis is often associated with hyperglycemia, insulin resistance, profound negative nitrogen balance and the diversion of protein from skeletal muscle to splanchnic tissues. Thus, the response to this infection differs from that generally observed in bacterial sepsis. It is possible that there is an effect on glucose metabolism due to invasion of the liver by the parasite. During acute infection, glucose levels in all of the T. cruzi-infected mice were below those measured in the control mice. Even though the baseline glucose levels in the infected animals were lower, the oral glucose tolerance test indicated a relatively normal ability to clear ingested glucose despite the high degree of inflammation associated with this infection.
The level of adiponectin was decreased during T. cruzi infection of CD-1 mice (Combs et al. 2005) and FVB mice (unpublished observations). Reduced levels of adiponectin are often associated with insulin resistance, hyperglycemia and obesity, i.e., the metabolic syndrome. Decreased levels of adiponectin are observed in some conditions of inflammation and cardiovascular disease, as noted above. Importantly, acute inflammation induced by endotoxemia does not affect adiponectin levels (Keller et al. 2003). The infection-induced hypoglycemia cannot be readily explained by changes in adiponectin. This is an example of a physiologically relevant condition that combines hypoglycemia and normal glucose tolerance with significantly reduced adiponectin levels. The decreased insulin levels observed 30 days post-infection in the mouse model of T. cruzi infection are consistent with a physiological response to very low glucose levels during that time. In addition, leptin levels were significantly reduced in infected mice compared to controls. Resistin levels, another fat cell-specific secretory factor with insulin-desensitizing properties, were not affected by infection. Levels of plasminogen activator inhibitor-1, which is also prominently expressed in adipocytes, were also completely unaffected by infection. However, proinflammatory markers such as cytokines (TNF-α, IL-1β, IFN-γ) and chemokines were markedly elevated in the adipose tissue of acutely infected mice (unpublished observations). This elevation often persisted into the chronic phase.
The significant decrease in leptin levels was initially surprising since the infected mice gained more weight than the control mice. Magnetic resonance imaging studies, as well as body composition studies using an ECHO magnetic resonance spectrometry body composition instrument, revealed a decrease in abdominal adipose tissue. Mice that had marked right ventricular dilation had a greater loss of fat deposits. The weight gain in infected mice appeared to be related to edema, which may have been a consequence of right-sided heart failure (Combs et al. 2005).
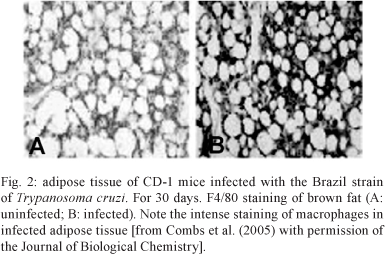
CD-1 and FVB mice infected with the Brazil strain of T. cruzi displayed a reduction in plasma levels of adiponectin, suggesting that the infection of adipocytes may also have consequences on other proteins synthesized in adipose tissue. Consistent with the reduction of plasma adiponectin, the level of adiponectin in adipose tissue was reduced during acute infection in a number of fat pads known to be important sources of adiponectin. At 30 days post-infection, the acute-phase reactants α-1 acid glycoprotein and SAA3, which are expressed in adipocytes, were upregulated. Consistent with the infection-induced increase in macrophages and inflammatory mediators (cytokines and chemokines) (Figs 1, 2), there was a concomitant reduction in adiponectin and peroxisome proliferator-activated receptor-γ (PPAR-γ). Both of these proteins are negative regulators of the inflammatory pathway (unpublished observations).
We have previously demonstrated by qPCR that T. cruzi DNA in adipose tissue 300 days post-infection was present at the same levels as seen in the heart (Combs et al. 2005) (Fig. 3). This observation suggests that the adipocyte proper may serve as an important target for T. cruzi and in chronic Chagas disease adipocytes may represent an important long-term reservoir for parasites from which relapse of infection can occur.
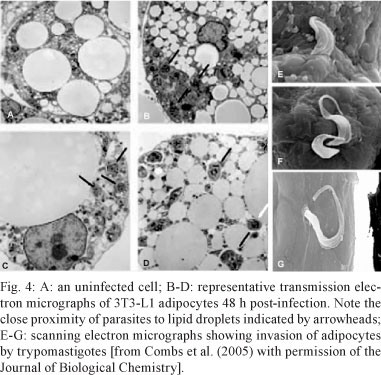
In vitro studies were subsequently performed to evaluate the role of adipocytes in T. cruzi infection in a model system devoid of many of the other cell types ordinarily found in adipose tissue. Although we were not the first group to observe T. cruzi in fat cells (Andrade & Silva 1995), we published the first systematic analysis of the detailed events occurring during acute and chronic stages of T. cruzi infection in fat (Combs et al. 2005, Nagajyothi et al. 2008). Intracellular amastigotes were monitored by electron microscopy (Fig. 4), revealing that adipocytes infected for 96 h maintained their integrity. T. cruzi infection of cultured adipocytes induced an inflammatory phenotype. For example, there was increased expression of chemokines, such as CCL2, CCL3, CCL5 and CXCL10, as well as the cytokines TNF-α, IL-10 and interferon-γ. The expression of STAT3, an important downstream mediator of cytokine signaling, was also increased. Toll-like receptors, which have been reported to be activated during T. cruzi infection of other cell types, were upregulated in cultured adipocytes (TLR-2 and -9) and there was also activation of components of the Mitogen-Activated Protein Kinase (MAPK pathway). Specifically extracellular signal-regulated kinase (ERK) and p38 MAPK were activated, whereas Jun N-terminal Kinase was not (Nagajyothi et al. 2008).
T. cruzi infection of cultured adipocytes also resulted in the increased expression of cyclin D1. Cyclin D1 is generally associated with cell proliferation; however, cultured adipocytes are usually terminally differentiated. The increased expression of cyclin D1 is of interest because it is upregulated by ERK and inversely regulated by caveolin-1 (Hulit et al. 2000). Indeed, we have demonstrated that infection resulted in a reduction in the expression of caveolin-1 and the activation of ERK. Both of these events increase the expression of cyclin D1. A reduction in caveolin-1 expression has also been demonstrated to be associated with an increased proinflammatory cytokine response (Cohen et al. 2003, 2004). Interestingly, infection activates the Notch pathway, which regulates, in part, the expression of cyclin D1 (Stahl et al. 2006).
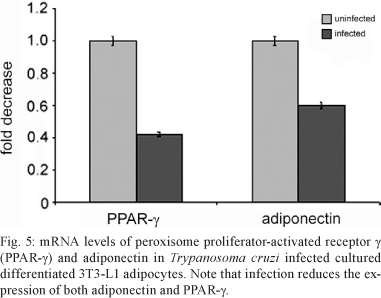
PPAR-γ is expressed in adipose tissue and reduces the inflammatory process, similar to adiponectin (Nawrocki et al. 2006, Kim et al. 2007). As noted, a reduction in the level of adiponectin is associated with an increase in inflammation (Desruisseaux et al. 2007). In addition, there is an inverse relationship between PPAR-γ and inflammation, as well as between PPAR-γ and cyclin D1 (Wang et al. 2003). It has been demonstrated that increased expression of cyclin D1 is associated with a reduction in PPAR-γ. Recent evidence suggests a similar relationship between adiponectin and PPAR-γ (Nawrocki et al. 2006, Kim et al. 2007). Our observations clearly demonstrated that T. cruzi infection resulted in a reduction in the expression of adiponectin and PPAR-γ (Fig. 5) and an increase in the expression of cyclin D1 and inflammatory mediators.
Interestingly, T. cruzi infection also results in the increased expression of PI3kinase and the activation of AKT/PKB (thymoma viral protoco-oncogene), suggesting that this infection may induce components of the insulin/IGF-1 receptor cascade. This is surprising since the upregulation of proinflammatory pathways is usually associated with a downregulation of the insulin signal transduction pathway. It is not clear what is responsible for this phenomenon. T. cruzi is likely to have an impact on lipid pathways in vivo, yet these issues have not been examined to date. These observations are significant because there is usually a correlation between inflammation and insulin resistance. However, the infection of adipocytes with a parasite that resides intracellularly can be viewed as different from exposing adipocytes to an endotoxin. The continued intracellular presence of the parasites clearly has a differential effect on insulin sensitivity, perhaps by lowering the levels of one of the critical lipid mediators of insulin resistance. Although IRS-1 levels are not necessarily indicative of activity, it is generally accepted that pAkt levels downstream are an excellent reflection of the cellular insulin signaling activity in 3T3-L1 adipocytes.
Infection also results in increased expression of PI3kinase and the activation of AKT, suggesting that this infection may induce components of the insulin/IGF-1 receptor cascade. This would be surprising as the upregulation of proinflammatory pathways is generally associated with a downregulation of the insulin signal transduction pathway (Hotamisligil 2006, Ferrante 2007). It is not clear what is responsible for this phenomenon, but it can be observed with a high degree of reproducibility in these cells. Despite the upregulation of some of the components of the pathway, there were no differences with respect to a dose-response to insulin in infected cells (unpublished observations). It remains to be determined whether other pathways influenced by insulin, such as events leading to differences in the rate of lipid accumulation or lipolysis, may be affected. T. cruzi is likely to have an impact on lipid pathways in vivo, yet these issues have not been examined to date.
In summary, fat and glucose metabolism are interrelated and dysregulated in T. cruzi infection. We have clearly demonstrated that adipocytes and adipose tissue represent an important target of and reservoir for infection. This is a reservoir from which parasites can be reactivated during periods of immunosuppression. In addition, the infection of adipocytes and adipose tissue create an inflammatory phenotype that affects a variety of metabolic processes. The reduction in the expression of adiponectin and PPAR-γ perpetuate this inflammatory phenotype. Since adiponectin null mice have a cardiomyopathic phenotype, it is tempting to suggest that the reduction in adiponectin and PPAR-γ contributes to the cardiomyopathy of Chagas disease. Recently Coura (2007) has commented on the need for new approaches to the study of Chagas disease. Although the study of adipose tissue was not specifically mentioned, we believe that this is a new and fruitful area of research.
Received 15 March 2009
Accepted 22 May 2009
Financial support: NIH AI-06538 (HBT), AI-052739 (HBT), R01-DK55758 (SC), R01-CA112023 (PES)
- Andrade ZA, Silva HRA 1995. Parasitism of adipocytes by Trypanosoma cruzi Mem Inst Oswaldo Cruz 90: 521-522.
- Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, Hotta K, Shimomura I, Nakamura T, Miyaoka K, Kuriyama H, Nishida M, Yamashita S, Okubo K, Matsubara K, Muraguchi M, Ohmoto Y, Funahashi T, Matsuzawa Y 1999. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. BBRC 257: 79-83.
- Attie AD, Scherer PE 2009. Adipocyte metabolism and obesity. J Lipid Res 50: 5395-5399.
- Barreto SM, Passos VM, Lima-Costa MF 2003. Obesity and underweight among Brazilian elderly: the Bambui Health and aging study. Cad Saude Publica 19: 605-612.
- Chen A, Mumick S, Zhang C, Lamb J, Dai H, Weingarth D, Mudgett J, Chen H, MacNeil DJ, Reitman ML, Qian S 2005. Diet induction of monocyte chemoattractant protein-1 and its impact on obesity. Obes Res 13: 1311-1320.
- Cohen AW, Hnasko R, Schubert W, Lisanti MP 2004. Role of caveolae and caveolins in health and disease. Physiol Rev 84: 1341-1379.
- Cohen AW, Park DS, Woodman SE, Williams TM, Chandra M, Shirani J, Pereira de Souza A, Kitsis RN, Russell RG, Weiss LM, Tang B, Jelicks LA, Factor SM, Shtutin V, Tanowitz HB, Lisanti MP 2003. Caveolin-1 null mice develop cardiac hypertrophy with hyperactivation of p42/44 MAP kinase in cardiac fibroblasts. Am J Physiol Cell Physiol 284: C457-C474.
- Combs TP, Berg AH, Obici S, Scherer PE, Rossetti L 2001. Endogenous glucose production is inhibited by the adipose-derived protein Acrp30. J Clin Inv 108: 1875-1881.
- Combs TP, Nagajyothi, Mukherjee S, de Almeida CJ, Jelicks LA, Schubert W, Lin Y, Jayabalan DS, Zhao D, Braunstein VL, Landskroner-Eiger S, Cordero A, Factor SM, Weiss LM, Lisanti MP, Tanowitz HB, Scherer PE 2005. The adipocyte as an important target cell for Trypanosoma cruzi infection. J Biol Chem 280: 24085-24094.
- Coura JR 2007. Chagas disease: what is known and what is needed - a background article. Mem Inst Oswaldo Cruz 102: (Suppl. I): 113-122.
- Desruisseaux MS, Nagajyothi, Trujillo ME, Tanowitz HB, Scherer PE 2007. Adipocyte and adipose tissue and infectious disease. Inf Immun 75: 1066-1078.
- Fasshauer M, Klein J, Neumann S, Eszlinger M, Paschke R 2002. Hormonal regulation of adiponectin gene expression in 3T3-L1 adipocytes. BBRC 290: 1084-1089.
- Ferrante AW Jr 2007. Obesity-induced inflammation: a metabolic dialogue in the language of inflammation. J Intern Med 262: 408-414.
- Gerhardt CC, Romero IA, Cancello R, Camoin L, Strosberg AD 2001. Chemokines control fat accumulation and leptin secretion by cultured human adipocytes. Mol Cell Endocrinol 175: 81-92.
- Goldstein BJ, Scalia R 2004. Adiponectin: a novel adipokine linking adipocytes and vascular function. J Clin Endocrinol Metab 89: 2563-2568.
- Halberg N, Wernstedt-Asterholm I, Scherer PE 2008. The adipocyte as an endocrine cell. Endocrinol Metab Clin North Am 37: 753-768.
- Hazan U, Romero IA, Cancello R, Valente S, Perrin V, Mariot V, Dumonceaux J, Gerhardt CC, Strosberg AD, Couraud PO, Pietri-Rouxel F 2002. Human adipose cells express CD4, CXCR4 and CCR5 [corrected] receptors: a new target cell type for the immunodeficiency virus-1? FASEB J 16: 1254-1256.
- Hotamisligil GS 2006. Inflammation and metabolic disorders. Nature 444: 860-867.
- Hotta K, Funahashi T, Arita Y, Takahashi M, Matsuda M, Okamoto Y, Iwahashi H, Kuriyama H, Ouchi N, Maeda K 2000. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc Biol 20: 1595-1599.
- Hulit J, Bash T, Fu M, Galbiati F, Albanese C, Sage DR, Schlegel A, Zhurinsky J, Shtutman M, Ben-Ze'ev A, Lisanti MP, Pestell RG 2000. The Cyclin D1 gene is transcriptionally repressed by caveolin-1. J Biol Chem 275: 21203-1209.
- Jan V, Cervera P, Maachi M, Baudrimont M, Kim M, Vidal H, Girard PM, Levan P, Rozenbaum W, Lombes A, Capeau J, Bastard JP 2004. Altered fat differentiation and adipocytokine expression are inter-related and linked to morphological changes and insulin resistance in HIV-1-infected lipodystrophic patients. Antivir Ther 9: 555-564.
- Keller P, Moller K, Krabbe KS, Pedersen BK 2003. Circulating adiponectin levels during human endotoxaemia. Clin Exp Immunol 134: 107-110.
- Kim JY, van de Wall E, Laplante M, Azzara A, Trujillo ME, Hofmann SM, Schraw T, Durand JL, Li H, Li G, Jelicks LA, Mehler MF, Hui DY, Deshaies Y, Shulman GI, Schwartz GJ, Scherer PE 2007. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest 117: 2621-2637.
- Kook KS, Min HY, Johnson D, Chaplinsky RJ, Flier JS, Hunt CR, Spiegelman BM 1987. Adipsin: a circulating serine protease homolog secreted by adipose tissue and sciatic nerve. Science 237: 402-405.
- Mallon PW, Sedwell R, Rogers G, Nolan D, Unemori P, Hoy J, Samaras K, Kelleher A, Emery S, Cooper DA, Carr A 2008. Effect of rosiglitazone on peroxisome proliferator-activated receptor γ gene expression in human adipose tissue is limited by antiretroviral drug-induced mitochondrial dysfunction. J Infect Dis 198: 1794-1803.
- Maurin T, Saillan-Barreau C, Cousin B, Casteilla L, Doglio A, Penicaud L 2005. Tumor necrosis factor-α stimulates HIV-1 production in primary culture of human adipocytes. Exp Cell Res 304: 544-551.
- Mynarcik DC, Combs T, McNurlan MA, Scherer PE, Komaroff E, Gelato MC 2002. Adiponectin and leptin levels in HIV-infected subjects with insulin resistance and body fat redistribution. J Acquir Immune Defic Syndr 31: 514-520.
- Nagajyothi F, Desruisseaux MS, Thiruvur N, Weiss LM, Braunstein VL, Albanese C, Teixeira MM, de Almeida CJ, Lisanti MP, Scherer PE, Tanowitz HB 2008. Trypanosoma cruzi infection of cultured adipocytes results in an inflammatory phenotype. Obesity 169: 1992-1997.
- Nawrocki AR, Rajala MW, Tomas E, Pajvani UB, Saha AK, Trumbauer ME, Pang Z, Chen AS, Ruderman NB, Chen H, Rossetti L, Scherer PE 2006. Mice lacking adiponectin show decreased hepatic insulin sensitivity and reduced responsiveness to peroxisome proliferator-activated receptor γ agonists. J Biol Chem 281: 2654-2660.
- Nawrocki AR, Scherer PE 2005. The adipocyte as a drug discovery target. Drug Discov Today 10: 1219-1230.
- Okamoto Y, Folco EJ, Minami M, Wara AK, Feinberg MW, Sukhova GK, Colvin RA, Kihara S, Funahashi T, Luster AD, Libby P 2008. Adiponectin inhibits the production of CXC receptor 3 chemokine ligands in macrophages and reduces T-lymphocyte recruitment in atherogenesis. Circ Res 102: 218-225.
- Ouchi N, Kihara S, Arita Y, Okamoto Y, Maeda K, Kuriyama H, Hotta K, Nishida M, Takahashi M, Muraguchi M, Ohmoto Y, Nakamura T, Yamashita S, Funahashi T, Matsuzawa Y 2000. Adiponectin, an adipocyte-derived plasma protein, inhibits endothelial NF-κB signaling through a cAMP-dependent pathway. Circulation 102: 1296-1301.
- Ouchi N, Kihara S, Funahashi T, Matsuzawa Y, Walsh K 2003. Obesity, adiponectin and vascular inflammatory disease. Curr Opin Lipidol 14: 561-566.
- Ouchi N, Shibata R, Walsh K 2006. Cardioprotection by adiponectin. Trends Cardiovasc Med 16: 141-146.
- Pajvani UB, Trujillo ME, Combs TP, Iyengar P, Jelicks L, Roth KA, Kitsis RN, Scherer PE 2005. Fat apoptosis through targeted activation of caspase 8: a new mouse model of inducible and reversible lipoatrophy. Nat Med 11: 797-803.
- Pasarica M, Dhurandhar NV 2007. Infectobesity: obesity of infectious origin. Adv Food Nutr Res 52: 61-102.
- Rajala MW, Obici S, Scherer PE, Rossetti L 2003. Adipose-derived resistin and gut-derived resistin-like molecule-β selectively impair insulin action on glucose production. J Clin Invest 111: 225-230.
- Rajala MW, Scherer PE 2003. Minireview: the adipocyte at the crossroads of energy homeostasis, inflammation and atherosclerosis. Endocrinology 144: 3765-3773.
- Ruan H, Lodish HF 2003. Insulin resistance in adipose tissue: direct and indirect effects of tumor necrosis factor-α. Cytokine Growth Factor Rev 14: 447-455.
- Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF 1995. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem 270: 26746-26749.
- Shibata R, Ouchi N, Ito M, Kihara S, Shiojima I, Pimentel DR, Kumada M, Sato K, Schiekofer S, Ohashi K, Funahashi T, Colucci WS, Walsh K 2004. Adiponectin-mediated modulation of hypertrophic signals in the heart. Nat Med 10: 1384-1399.
- Shibata R, Ouchi N, Murohara T 2009. Adiponectin and cardiovascular disease circulation, in press Circulation J 73: 608-614.
- Shibata R, Sato K, Pimentel DR, Takemura Y, Kihara S, Ohashi K, Funahashi T, Ouchi N, Walsh K 2005. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat Med 11: 1096-1103.
- Stahl M, Ge C, Shi S, Pestell RG, Stanley P 2006. Notch1-induced transformation of RKE-1 cells requires up-regulation of cyclin D1. Cancer Res 66: 7562-7570.
- Tanowitz HB, Amole B, Hewlett D, Wittner M 1988. Trypanosoma cruzi infection in diabetic. Trans R Soc Trop Med Hyg 82: 90-93.
- Wang C, Pattabiraman N, Zhou J-F, Fu M, Sakamaki T, Albanese C, Li Z, Wu K, Hulit J, Neumeister P, Novikoff PM, Brownlee M, Scherer PE, Jones JG, Whitney KD, Donehower LA, Harris EL, Rohan T, Johns DC, Pestell RG 2003. Cyclin D1 repression of peroxisome proliferator-activated receptor-γ expression and transactivation. Mol Cell Biol 23: 6159-6173.
- Wernstedt-Asterholm I, Halberg N, Scherer PE 2007. Mouse models of lipodystrophy key reagents for the understanding of the metabolic syndrome. Drug Discov Today Dis Models 4: 17-24.
- Yang WS, Lee WJ, Funahashi T, Tanaka S, Matsuzawa Y, Chao CL, Chen CL, Tai TY, Chuang LM 2001. Weight reduction increases plasma levels of an adipose-derived anti- inflammatory protein, adiponectin. J Clin Endocrinol Metab 86: 3815-3819.
Publication Dates
-
Publication in this collection
21 July 2009 -
Date of issue
July 2009
History
-
Received
15 Mar 2009 -
Accepted
22 May 2009