Abstract
BACKGROUND
Biomphalaria glabrata is the major species used for the study of schistosomiasis-related parasite-host relationships, and understanding its gene regulation may aid in this endeavor. The ubiquitin-proteasome system (UPS) performs post-translational regulation in order to maintain cellular protein homeostasis and is related to several mechanisms, including immune responses.
OBJECTIVE
The aims of this work were to identify and characterise the putative genes and proteins involved in UPS using bioinformatic tools and also their expression on different tissues of B. glabrata.
METHODS
The putative genes and proteins of UPS in B. glabrata were predicted using BLASTp and as queries reference proteins from model organism. We characterised these putative proteins using PFAM and CDD software describing the conserved domains and active sites. The phylogenetic analysis was performed using ClustalX2 and MEGA5.2. Expression evaluation was performed from 12 snail tissues using RPKM.
FINDINGS
119 sequences involved in the UPS in B. glabrata were identified, which 86 have been related to the ubiquitination pathway and 33 to proteasome. In addition, the conserved domains found were associated with the ubiquitin family, UQ_con, HECT, U-box and proteasome. The main active sites were lysine and cysteine residues. Lysines are responsible and the starting point for the formation of polyubiquitin chains, while the cysteine residues of the enzymes are responsible for binding to ubiquitin. The phylogenetic analysis showed an organised distribution between the organisms and the clades of the sequences, corresponding to the tree of life of the animals, for all groups of sequences analysed. The ubiquitin sequence was the only one with a high expression profile found in all libraries, inferring its wide range of performance.
MAIN CONCLUSIONS
Our results show the presence, conservation and expression profile of the UPS in this mollusk, providing a basis and new knowledge for other studies involving this system. Due to the importance of the UPS and B. glabrata, this work may influence the search for new methodologies for the control of schistosomiasis.
Key words:
snail; UPS; signaling pathway; bioinformatics; schistosomiasis
Biomphalaria glabrata is a species of snail that has considerable epidemiological importance, since their presence represents a crucial condition for the dissemination of schistosomiasis, affecting more than 200 million people around the word. These mollusks are considered intermediate hosts of Schistosoma mansoni and present a high degree of susceptibility to helminth infection. In addition, they have a wide geographical distribution in the Americas.11. de Souza CP, Jannotti-Passos LK, de Freitas JR. Degree of host-parasite compatibility between Schistosoma mansoni and their intermediate molluscan hosts in Brazil. Mem Inst Oswaldo Cruz. 1995; 90(1): 5-10.,22. Carvalho OS, Coelho PMZ, Lenzi HL. Schistosoma mansoni e esquistossomose: uma visão multidisciplinar. Rio de Janeiro: Editora Fiocruz; 2008. p. 29-245. Due to the importance of this organism, many studies seek new knowledge about its biology. It is already known that the interaction between mollusk and trematode is complex, and the expression of genes involved in host susceptibility/resistance and parasite infectivity is fairly well-understood.33. Richards CS, Shade PC. The genetic variation of compatibility in Biomphalaria glabrata and Schistosoma mansoni. J Parasitol. 1987; 73(6): 1146-51. Thus, one of the ways to better understand this relationship is to deepen studies related to gene and protein regulation in B. glabrata, because the infection carried out by S. mansoni leads to changes in the expression profile of some proteins and, consequently, in the defense pattern of the mollusk.44. Negrão-Corrêa D, Mattos ACA, Pereira CAJ, Martins-Souza RL, Coelho PMZ. Interaction of Schistosoma mansoni sporocysts and hemocytes of Biomphalaria. J Parasitol Res. 2012; 2012(743920): 1-6.,55. Larson MK, Bender RC, Bayne CJ. Resistance of Biomphalaria glabrata 13-16-R1 snails to Schistosoma mansoni PR1 is a function of haemocyte abundance and constitutive levels of specific transcripts in haemocytes. Int J Parasitol. 2014; 44(6): 343-53. Thus, it is necessary to evaluate the regulatory systems of expression in these organisms, on the gene and protein level. One of the key components in protein regulation is the ubiquitin-proteasome system, as it performs specific post-translational regulation and assists in the maintenance of protein homeostasis in cells.
Given the epidemiological importance of B. glabrata, Adema and contributors,66. Adema CM, Hillier LW, Jones CS, Loker ES, Knight M, Minx P, et al. Whole genome analysis of a schistosomiasis-transmitting freshwater snail. Nat Commun. 2017; 8(15451): 1-11. including our working group, published a complete analysis of the snail genome. Genes involved in communication with the aquatic environment, stress, innate immunity and regulation of biological processes were identified and described, as well as several small RNAs related to gene regulation. In addition, transcripts involved in the ubiquitin-proteasome system were found, from genes encoding enzymes to accessory proteins and the subunits that form the proteolytic 26S proteasome.66. Adema CM, Hillier LW, Jones CS, Loker ES, Knight M, Minx P, et al. Whole genome analysis of a schistosomiasis-transmitting freshwater snail. Nat Commun. 2017; 8(15451): 1-11.
The ubiquitin-proteasome system (UPS) is composed of two main elements: the ubiquitination pathway and the proteolytic macromolecule 26S proteasome. First, the ubiquitination pathway labels proteins with one or more ubiquitin molecules that are then degraded by the 26S proteasome.77. Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002; 82(2): 373-428. This system is able to degrade mutated and defective proteins that are involved in important cellular processes, such as cell cycle regulation, stress response and extracellular modulators, DNA repair and the regulation of immune system and inflammatory responses.88. Ciechanover A. The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J. 1998; 17(24): 7151-60.,99. Ebner P, Versteeg GA, Ikeda F. Ubiquitin enzymes in the regulation of immune responses. Crit Rev Biochem Mol Biol. 2017; 52(4): 425-60.
The first class acting on the ubiquitination pathway are ubiquitin-activating enzymes (E1) that activate the ubiquitin molecule in an ATP-dependent manner and generate an ubiquitin E1-thioester. Subsequently, ubiquitin conjugating enzymes (E2) catalyse the covalent binding reaction of activated ubiquitin with target protein substrate. At the end of this cascade, the enzymes ubiquitin ligases (E3) recognise the specific protein to be degraded and assist in the transfer of the ubiquitin present in E2 to the substrate, being able to bind both E2-ubiquitin and target substrate concomitantly or at different times.77. Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002; 82(2): 373-428.,1010. Scheffner M, Nuber U, Huibregtse JM. Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature. 1995; 373(6509): 81-3. At the end of the first step, the labeled substrates are degraded by the proteolytic complex 26S proteasome. This protease is formed from an association between a 19S (PA700) regulatory particle divided between lid and base which are reversibly connected and ATP-dependent to the central component 20S.77. Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002; 82(2): 373-428.,1111. Tanaka K, Chiba T. The proteasome: a protein-destroying machine. Genes Cells. 1998; 3(8): 499-510. The regulatory particle recognises, unfolds and translocates the substrate to the central particle.1212. Budenholzer L, Cheng CL, Li Y, Hochstrasser M. Proteasome structure and assembly. J Mol Biol. 2017; 429(22): 3500-24. However, the central component 20S has proteolytic sites that play the role of degrading the target protein, since they have similar functions to caspase-like, chymotrypsin-like and trypsin-like.1313. Jung T, Catalgol B, Grune T. The proteasomal system. Mol Aspects Med. 2009; 30(4): 191-296. Soon after, the target proteins are degraded into smaller peptides and the ubiquitin molecules present in the tail are released.77. Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002; 82(2): 373-428.
The peptides generated from the digestion performed by the proteasome are related to immunity. In humans, these peptides are recognised as epitopes by MHC class I and, thus, the proteasome plays an essential role in this recognition.1414. Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, et al. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994; 78(5): 761-71. However, the immunoproteasome, a standard isoform of the proteasome, is directly involved with the immune system because it is more efficient for the generation of antigenic peptides. This isoform has three different subunits in the 20S nucleus as compared to the proteasome 26S, β1i (LMP2), β2i (MECL-1) and β5i (LMP7), respectively. They substitute constitutive subunits and are therefore assembled more rapidly, triggering a more agile immune response in hematopoietic cells, in addition to modifying peptidase activity and increasing epitope generation.1515. Murata S, Sasaki K, Kishimoto T, Niwa SI, Hayashi H, Takahama Y, et al. Regulation of CD8+T cell development by thymus-specific proteasomes. Science. 2007; 316(5829): 1349-53. In addition, the thymoproteasome is another isoform specifically expressed in the thymus; both are involved in cell-mediated immunity.1616. Murata S, Takahama Y, Kasahara M, Tanaka K. The immunoproteasome and thymoproteasome: functions, evolution and human disease. Nat Immunol. 2018; 19(1): 923-31. Thus, the relationship involving the catalytic proteasome and immunity has led us to believe that it is also related to the resistance/susceptibility that some organisms present when they are infected by pathogens.
Genes involved in the ubiquitination pathway and in proteasome formation have already been reported in studies involving the cells of the internal defense system, i.e. hemocytes. These genes demonstrated a different expression profile when analysed in infected and uninfected snails.1717. Lockyer AE, Spinks J, Kane RA, Hoffmann KF, Fitzpatrick JM, Rollinson D, et al. Biomphalaria glabrata transcriptome: cDNA microarray profiling identifies resistant- and susceptible-specific gene expression in haemocytes from snail strains exposed to Schistosoma mansoni. BMC Genomics. 2008; 643(9): 1-17.,1818. Zahoor Z, Lockyer AE, Davies AJ, Kirk RS, Emery AM, Rollinson D, et al. Differences in the gene expression profiles of haemocytes from schistosome-susceptible and -resistant Biomphalaria glabrata exposed to Schistosoma mansoni excretory-secretory products. PLoS One. 2014; 8(6): 1236-51.,1919. Dinguirard N, Cavalcanti MGS, Wu XJ, Bickham-Wright U, Sabat G, Yoshino TP. Proteomic analysis of Biomphalaria glabrata hemocytes during in vitro encapsulation of Schistosoma mansoni sporocysts. Front Immunol. 2018; 9(2773): 1-17. Thus, the hypothesis of this study is that the UPS has been conserved in the genome of B. glabrata and is expressed at the transcriptional level; that is, it is present in the transcriptome data in all the parts of the body of the uninfected adult snail. Thus, the aims of this work were to identify and characterise the UPS in the genome and transcriptome data of the snail using in silico analyses, as well as to characterise the classes involved in this system and to evaluate the expression profile of the sequences identified in different tissues of the adult snail.
MATERIALS AND METHODS
Identification of UPS genes in B. glabrata - The genome and transcriptome data used for analysis in silico were retrieved from the last version of VectorBase database, i.e. the BglaB1 genome of B. glabrata (https://www.vectorbase.org/organisms/biomphalaria-glabrata). Initially, the prediction of genes involved in the ubiquitin-proteasome pathway was performed through bibliographic searches and KEGG (https://www.genome.jp/kegg/pathway.html) in model organisms such as Drosophila melanogaster and Caenorhabditis elegans. The sequences belonging to the model organisms were obtained from the NCBI database (https://www.ncbi.nlm.nih.gov/) and later used as a queries against the genome and transcriptome of the snail, seeking to find UPS genes. Subsequently, the sequences identified in B. glabrata data were grouped according to the role they play and also analysed for their conserved domains, active sites and gene expression.
Sequence analysis - Sequences were fed into the protein family database, PFAM (https://pfam.xfam.org/), to identify the major conserved characteristic domains belonging to each previously organised sequence group. CDD (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) was used to search for amino acid residues involved in active site formations, structural motifs and catalytic clefts.
Phylogenetic analysis - To determine the evolutionary organisation and distribution of the sequences found, as well as to provide further evidence of the presence of probable proteins found in the snail, phylogenetic analysis was performed using MEGA5.2. For the analysis, amino acid sequences of deuterostomes and protostomes organisms such as Homo sapiens, Mus musculus, D. melanogaster, C. elegans and mollusks such as Aplysia californica and Lottia gigantea, obtained from NCBI were used. ClustalX 2.1 was used to perform multiple sequence alignment and their output fed in the MEGA 5.2 software, using the neighbor-joining method and JTT model for calculations of evolutionary distance. The consensus tree was inferred from a bootstrap of 1000 replicates to represent the evolutionary history of the study.2020. Queiroz FR, Silva LM, Jeremias WJ, Babá EH, Caldeira RL, Coelho PMZ, et al. Differential expression of small RNA pathway genes associated with the Biomphalaria glabrata/Schistosoma mansoni interaction. PLoS One. 2017; 12(7): 1-20.
Expression analysis - For the expression analysis, all transcripts identified in the B. glabrata genome data were individually submitted and analysed against an RNASeq data set for a library of 12 different snail tissues [Supplementary data (Table)].66. Adema CM, Hillier LW, Jones CS, Loker ES, Knight M, Minx P, et al. Whole genome analysis of a schistosomiasis-transmitting freshwater snail. Nat Commun. 2017; 8(15451): 1-11. Quality control and adapter removal were conducted using Trimmomatic (v 0.36). Single-end reads were aligned against pre-selected sequences using bowtie2 (v 2.3.0) with the “-very-sensitive-local parameter”. Alignment sam files were sorted and converted to bam files with samtools (v 1.6). Expression values were extracted from the alignment results using express (v 1.5.1) and RPKM (Reads Per Kilobase of transcript, per Million mapped reads) were calculated after library size normalisation with the Bioconductor package edgeR5 in the R statistical environment. The expression profile was plotted so that the more intense the red color, the more expressed the gene was presented to the corresponding library; a more intense green color indicates less expression found in the data used in the analysis.
RESULTS
Identification and characterisation of putative proteins of the UPS complex: alignment and phylogenetic analysis - In total, 119 sequences were found in the genome, transcriptome and predicted proteome of B. glabrata involved in the UPS. Among them, 86 were found to be involved with the ubiquitination pathway and 33 with 26S proteasome formation. Among the 86 genes of the ubiquitination pathway, one ubiquitin gene, six E1, 22 E2 and 39 E3 were identified, divided into the HECT, RING finger and U-box groups. In addition, 19 genes were found to be related to the forming of 26S proteasome and involved with the regulatory portion, while 14 involved in form the nucleus of the molecule (Table I). The genes were grouped according to their characteristics and functions based on the analysis of conserved domains, active sites and phylogenetic analysis.
Genes involved in ubiquitin-proteasome system (UPS) identified in Biomphalaria glabrata transcriptome and their orthologous organism
Ubiquitination pathway - The analysis performed by PFAM and CDD showed that all the proteins and enzymes found for the ubiquitination pathway in B. glabrata have conserved domains and active sites/catalytic clefts/structural motifs. These conserved domains identified for each set of sequences correspond to their previously known structures. In the ubiquitin (BGLB020284-PA) protein, the ubiquitin family domain was identified, as well as in the organisms D. melanogaster and C. elegans [Supplementary data (Fig. 1A)]. This domain is approximately 72 amino acids in size and shows some important residues for the molecule, such as those involved in the interaction of the molecule with E1 and E2. In addition, lysine residues at specific positions enable the formation of polyubiquitin chains [Supplementary data (Fig. 1B)].
In the genes identified as E1, some domains were found distributed along the sequence and were superimposed among them (Table II). These sequences showed the ThiF domain as the main representative showing a fairly characteristic size and location between the sequences of B. glabrata (Fig. 1). Additionally, it was possible to observe the presence of a cysteine residue as the active site of these enzymes and other amino acid residues relevant to the performance of the catalytic activity that it possess (Fig. 2). E2 are molecules capable of interacting with the other two classes of enzymes of the ubiquitination pathway because they are in the middle of this cascade. Genes found in the B. glabrata encode proteins that shared the UBQ_con domain and have a conserved cysteine residue as the active site, responsible for the role of conjugation of the ubiquitin molecule with the E3 [Supplementary data (Fig. 2A-B)].

scheme of the presence and distribution of the conserved domain ThiF (CL0063) in E1 sequences identified in the transcriptome of Biomphalaria glabrata.

amino acid residues involved in the formation of the active site of the ubiquitin activating enzymes (E1) identified in Biomphalaria glabrata. The cysteine residue (C) appears to be the active site (indicated by a red arrow), while the other amino acid residues are likely involved in mediating the catalytic activity (indicated by blue arrows).
The three ubiquitin ligases were divided into HECT, RING finger and U-box and grouped based on conserved domains HECT domain, U-box and domains homologous to RING finger, demonstrating the conservation of the catalytic activity of each group of enzymes [Supplementary data (Figs 3A, 4A, 5A)]. The analyses performed by the CDD show that the proteins of the HECT group had a catalytic cleft formed from the presence and distribution of some amino acid residues in specific and conserved positions [Supplementary data (Fig. 3B)]. The proteins classified in the RING finger and U-box groups evidenced the formation of a structural motif for each group. For the RING finger protein group, the cysteine residue was conserved in all B. glabrata genes [Supplementary data (Fig. 4B)]. On the other hand, the motif found in the U-box gene group was determined by the distribution of various residues located along the sequences, but there was a highly conserved aspartic acid residue [Supplementary data (Fig. 5B)].
Phylogenetic analysis was performed to evaluate the evolutionary relationship of the genes found in B. glabrata against the genes of orthologous organisms and to demonstrate the degree of conservation. For all classes of enzymes, this result showed that there was a very organised division between the clades of the enzymes and that they were distributed evolutionarily as presented by the evolutionary tree of life [Supplementary data (Figs 6, 7, 8, 9)]. This infers that the sequences found in the snail were likely enzymes involved in the ubiquitination pathway. In addition, the distribution found among the organisms used for this analysis was demonstrated in Fig. 3, showing the analysis done for E1.

phylogenetic distribution of the E1 sequences found in Biomphalaria glabrata data against their orthologous organisms.
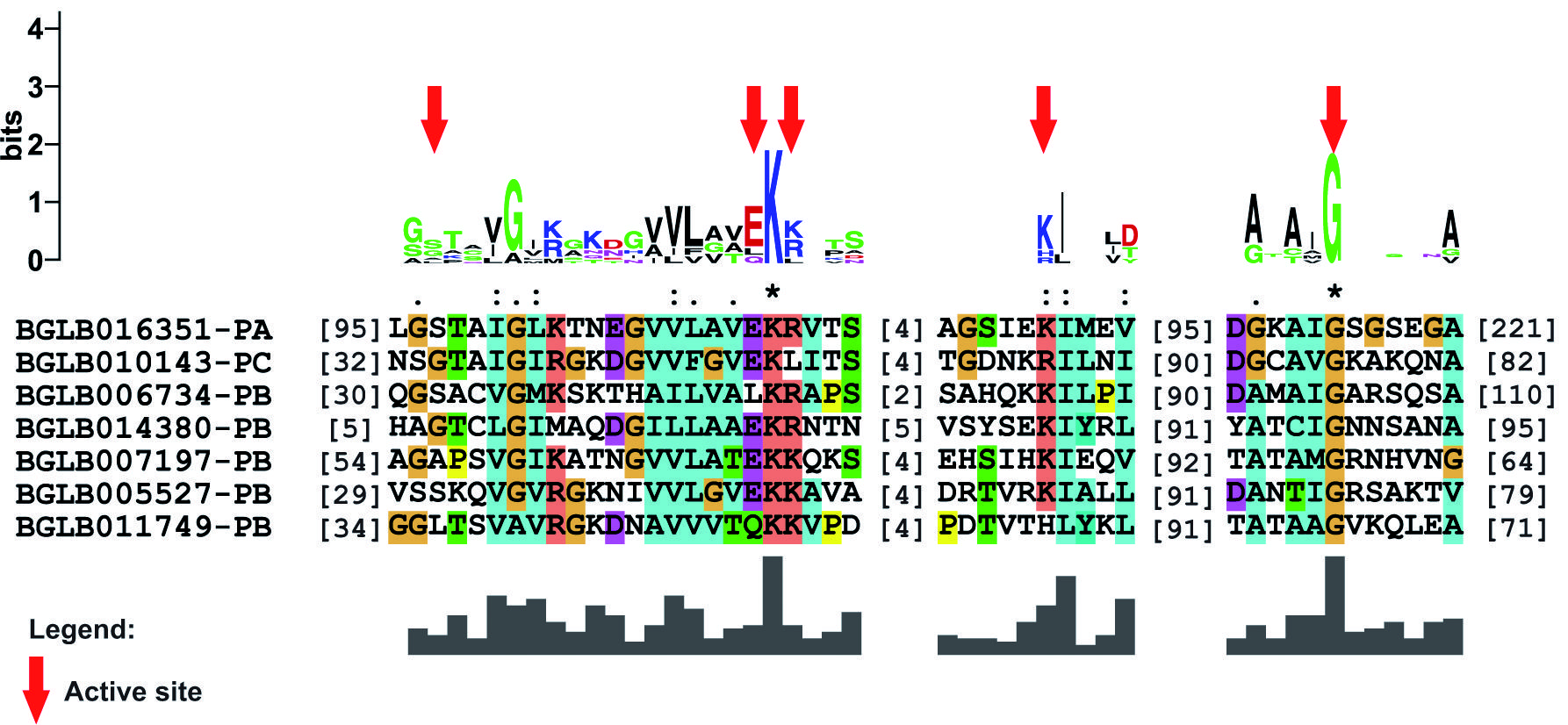
26S proteasome formation - The genes involved in proteasome formation identified in B. glabrata were divided between regulatory particles and core particles. Groups called PAS and PBS were related to the nucleus of the molecule, while the RPT and RPN groups form the recognition portions. All proteins encoded from the genes of the PAS showed the proteasome conserved domain (Fig. 4) and an active site consisting of amino acid residues distributed at five specific positions throughout the seven sequences (Fig. 5). In the same sense, the PBS group was also shown to share the proteasome conserved domain in their sequences and, in addition, showed the presence of an active site consisting of amino acid residues in seven distinct positions, but with low conservation [Supplementary data (Fig. 10A-B)]. This feature may indicate a direct site-position relationship and not between the site and amino acid conservation.

location and distribution of the proteasome domain identified in the sequences of the PAS group of Biomphalaria glabrata.

active site found in the sequences participating in the PAS group of the Biomphalaria glabrata data. The residues involved in the formation of the site are indicated by red arrows.
The other identified proteins were divided into the RPT and RPN groups that formed the regulatory and recognition portion of 26S proteasome. The protein group called RPT showed no amino acid residues relevant to active site formation; however, all six sequences presented the conserved domain AAA in the C-terminal portion [Supplementary data (Fig. 11)]. Unlike the RPT group, the sequences of the RPN group did not share the common domain [Supplementary data (Fig. 12)] and therefore did not show a conserved active site.
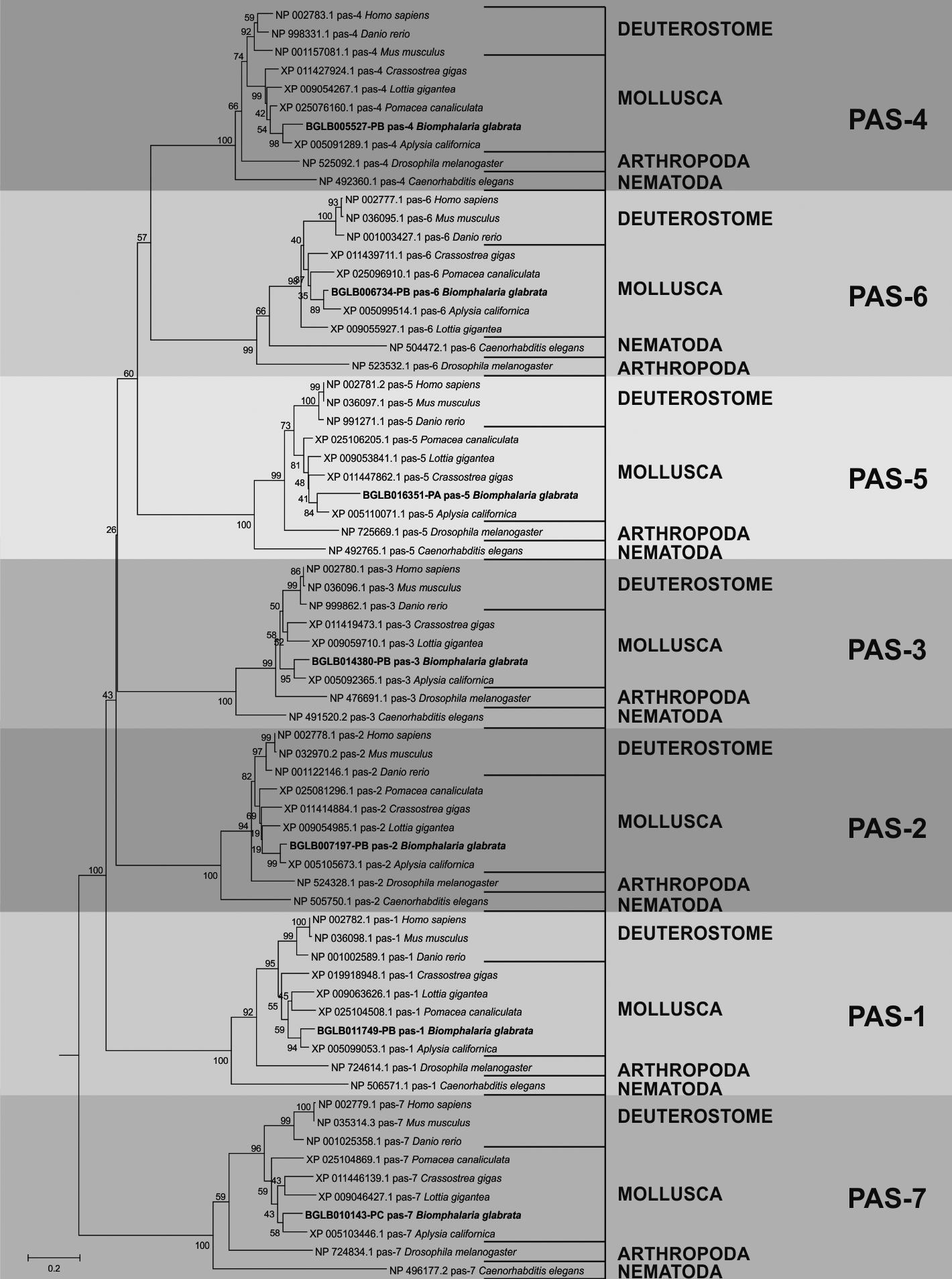
The phylogenetic analysis performed for the sequences involved with the formation of 26S proteasome showed an evolutionary distribution that corroborates with that known through the tree of life of the animals. In addition the genes identified in B. glabrata and their orthologous from other organisms were organised in deuterostomes and protostomes as seen for the results found in the phylogeny concerning the ubiquitination pathway [Supplementary data (Figs 13, 14, 15)]. Fig. 6 shows the phylogenetic distribution of the PAS group using those identified in the snail and the orthologous genes.

phylogenetic tree generated by the analyses using the sequences identified as PAS of mollusks versus model organisms and their orthologs.
Expression profile of the genes involved in the UPS identified in B. glabrata - The heatmap demonstrated the expression profile of all 119 sequences found in the B. glabrata in 12 libraries of tissues: albumen gland (AG), buccal mass (BUC), central nervous system (CNS), digestive gland/hepatopancreas (DG/HP), muscular part of the headfoot (FOOT), heart including amebocyte producing organ (HAPO), kidney (KID), mantle edge (MAN), ovotestis (OVO), salivary gland (SAL), stomach (STO) and terminal genitalia (TRG). The vast majority of the UPS related genes presented low to medium expression for all libraries used (Fig. 7).

The ubiquitin (ubq-1) gene, identified as BGLB020284-RA was the only one presented highly expressed in all libraries. Other genes such as rpt-2 (BGLB012308-RB), wwp-1-a (BGLB008139-RC) and wwp-1-b (BGLB008139-RB) also demonstrated sufficient, but not high, expression in all libraries. However, most other genes were not able to show a strong expression profile for any of the libraries. Only a few transcripts such as pbs-3 (BGLB010370-RB), uba1-a (BGLB013827-RB) and rpn-10 (BGLB003999-RB) were able to display good expression in specific libraries such as TRG, OVO and CNS, respectively (Fig. 7).
The genes with lower expression indicated in the results of the heatmap were related to the class of ubiquitin ligases (E3). All isoforms of the chn-1 genes (BGLB001813-PI, BGLB001813-PH, BGLB001813-PG, BGLB001813-PF, BGLB001813-PE, BGLB001813-PD, BGLB001813-PC and BGLB001813-PB) and traf-6 (BGLB026988-PC, BGLB026988-PB and BGLB026988-PA) demonstrated a very low RPKM-based expression profile. In the same sense, a very low expression was perceived for the isoforms of the fizzy genes (BGLB002564-PD, BGLB002564-PC, BGLB002564-PB, BGLB005726-PB and BGLB005726-PC) and skpt-1 (BGLB032982-PA) that were involved in the formation of E3 complexes as target recognising subunit (Fig. 7).
DISCUSSION
The ubiquitin-proteasome system (UPS) is one of the most conserved components among eukaryotic organisms and has been described in model organisms such as D. melanogaster, C. elegans and Homo sapiens. Most sequences identified in B. glabrata have an amount of amino acids close to or even equal to the number of amino acids found in the proteins of the model organism used, C. elegans and/or D. melanogaster. Ubiquitin is a polypeptide with 76 amino acids in its structure capable of labeling target substrates.2121. Müller WEG. Molecular phylogeny of metazoa (animals): monophyletic origin. Naturwissenschaften. 1995; 82(7): 321-9. However, the gene identified in B. glabrata encodes a gene product of 229 amino acids due to a polygene responsible for translating this molecule, a fusion of the ubiquitin peptides. In addition, the same situation in C. elegans and D. melanogaster occurs [Supplementary data (Fig. 1A)]. The difference found in the size of these sequences can be explained by the annotation performed for the mollusk sequence, suggesting that a new annotation process should be performed. This situation refers to the existence of polygenes that have tandem repeats of the coding region of ubiquitin in their sequences. Thus, most organisms present sequences of polygenes for ubiquitin and from that, they have become one of the best models for the study of the evolution of gene clusters.2121. Müller WEG. Molecular phylogeny of metazoa (animals): monophyletic origin. Naturwissenschaften. 1995; 82(7): 321-9. All three organisms used for this analysis have the conserved ubiquitin family domain (PF00240) in the same position within the polypeptide sequence, showing that the presence and location of the domain in the sequence may be of great importance for the role that this polypeptide plays.
The ubiquitin family domain in B. glabrata proteins shares very important and highly conserved amino acids against the orthologous organisms. Some of these residues have the ability to form a site of interaction with the ubiquitin conjugating enzymes (E2) and to catalyse the transfer [Supplementary data (Fig. 1B)]. Another three lysine (K) residues play a crucial role in labeling the target substrate. These residues at positions 29, 48 and 63 of the domain are considered binding sites for novel ubiquitin molecules capable of forming polyubiquitin chains. The chains formed at positions 29 and 48 induce degradation by the proteasome, while the chains originating at the lysine residue at position 63 are, for instance, related to DNA damage response signaling.2222. Ramos PC. A via proteolítica dependente de ubiquitina/proteassoma. Sociedade Portuguesa de Química. 2002; 1(96): 57-63.
It was not possible to trace the evolutionary history of ubiquitin through phylogenetic analysis because it is an extremely conserved sequence [Supplementary data (Fig. 1B)]. There is a high degree of conservation involving the ubiquitin encoding gene, suggesting that it is able to undergo few combined evolutionary events. The reason why this conservation is so high is still not fully understood, but the properties presented by the ubiquitin molecule in the cells were selected and fixed in a eukaryotic ancestor in the early stages of evolution.2323. Zuin A, Isasa M, Crosas B. Ubiquitin signaling: extreme conservation as a source of diversity. Cells. 2014; 3(3): 690-701.
E1 are the first enzymes recruited to the ubiquitination pathway and play the role of activating the ubiquitin molecule to be transferred to the substrate. Activation occurs by the ubiquitin adenylation from an ATP-dependent thioester bond between the E1 cysteine residue and the glycine of the C-terminal portion of ubiquitin. The activated ubiquitin molecule is transferred to an E2.77. Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002; 82(2): 373-428.,88. Ciechanover A. The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J. 1998; 17(24): 7151-60.,1010. Scheffner M, Nuber U, Huibregtse JM. Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature. 1995; 373(6509): 81-3. An existing hypothesis is that the E1 found in eukaryotes have evolved from the bacterial enzymes MoeB and ThiF.2424. Lois LM, Lima CD. Structures of the SUMO E1 provide mechanistic insights into SUMO activation and E2 recruitment to E1. EMBO J. 2005; 24(3): 439-51. All sequences identified as E1 in B. glabrata showed the ThiF domain with a similar amount of amino acids, showing a high degree of conservation, except in the sequence translated from the rfl-1 gene, which may have incomplete annotation. However, other conserved domains were also found in these sequences (Table II), capable of assisting in the process of ubiquitin activation. A highly conserved cysteine (C) residue was observed as an active site among all sequences identified as E1 in B. glabrata, the acceptor portion of ubiquitin in the enzyme. In addition, other amino acid residues in close positions are capable of aiding in the formation of the catalytic site and consequently in the activity that the enzyme plays (Fig. 2). Thus, these residues are probably involved with ATP binding and the formation of the thioester intermediate, activating the ubiquitin molecule for a subsequent transfer to E2.77. Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002; 82(2): 373-428.,88. Ciechanover A. The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J. 1998; 17(24): 7151-60.,1010. Scheffner M, Nuber U, Huibregtse JM. Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature. 1995; 373(6509): 81-3.,2323. Zuin A, Isasa M, Crosas B. Ubiquitin signaling: extreme conservation as a source of diversity. Cells. 2014; 3(3): 690-701.
E2 are enzymes from the centre of the ubiquitination pathway and are related to the other two enzymatic classes in the cascade. In addition, they play the role of catalysing the binding reaction of the ubiquitin molecule with the target substrate.2323. Zuin A, Isasa M, Crosas B. Ubiquitin signaling: extreme conservation as a source of diversity. Cells. 2014; 3(3): 690-701. The UQ_con domain was identified in all sequences identified as E2 present in the B. glabrata data, sharing close and highly conserved sizes. On the other hand, the UBA domain was found only in the sequence encoded from the ubc-20 gene (BGLB036058-PA) and has a role in limiting the formation of the polyubiquitins chain [Supplementary data (Fig. 2A)]. In addition, these enzymes have a cysteine residue as active site capable of binding the ubiquitin molecule to E2 and mediates the interaction with E3.2323. Zuin A, Isasa M, Crosas B. Ubiquitin signaling: extreme conservation as a source of diversity. Cells. 2014; 3(3): 690-701. Other amino acid residues were important in the sequences because they were related to the interaction E2 makes with E3; however, they did not show a high degree of conservation [Supplementary data (Fig. 2B)].
E3 performs the process of transferring the ubiquitin to the N-terminal portion of the lysine residue (K) belonging to the target substrate, and is more related to the target substrate it recognises than to the ubiquitin molecule. E3 is encoded in hundreds of proteins in eukaryotic cells, allowing the labeling of different proteins in specific ways; they are organised into different classes (Table I).2525. Rodrigo-Brenni MC, Morgan DO. Sequential E2s drive polyubiquitin chain assembly on APC targets. Cell. 2007; 130(1): 127-39. Thus, each of these classes shares specific domains that differ by recognition of the E2-ubiquitin complex. All E3-HECT sequences identified in B. glabrata showed a fairly conserved HECT domain in their C-terminal portion, maintaining a position pattern at the end of the protein chain. In the HECT domain, it was possible to find amino acid residues involved in the formation of a catalytic cleft and a cysteine as active site [Supplementary data (Fig. 3B)] where a thiol linkage between ubiquitin and ligase is performed. Although this residue was present in the C-terminal portion of the domain, its N-terminal portion is responsible for interacting with E2 and with the substrate that is bound in distinct regions along the enzyme sequence and outside the domain region.2626. Rotin D, Kumar S. Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol. 2009; 10(6): 398-409. However this binding does not happen directly, because a recruitment of the E2-ubiquitin complex is first made to the active site of the ligase. Subsequently, ubiquitin is transferred to the lysine residue belonging to the target substrate through a transesterification reaction.2222. Ramos PC. A via proteolítica dependente de ubiquitina/proteassoma. Sociedade Portuguesa de Química. 2002; 1(96): 57-63.,2323. Zuin A, Isasa M, Crosas B. Ubiquitin signaling: extreme conservation as a source of diversity. Cells. 2014; 3(3): 690-701.,2626. Rotin D, Kumar S. Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol. 2009; 10(6): 398-409.
The E3-RING finger class has a direct transfer of ubiquitin, since concomitant recruitment between the E2-ubiquitin complex and target substrate is performed. This mediates and facilitates the formation of the binding between ubiquitin and the target protein.2222. Ramos PC. A via proteolítica dependente de ubiquitina/proteassoma. Sociedade Portuguesa de Química. 2002; 1(96): 57-63.,2626. Rotin D, Kumar S. Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol. 2009; 10(6): 398-409.,2727. Pickart CM, Eddins MJ. Ubiquitin: structures, functions, mechanisms. Biochim Biophys Acta. 2004; 1695(1-3): 55-72. The conserved domains zf-C3HC4_3 (PF13920), zf-MIZ (PF02891), zf-RING_2 (PF13639), Sina (PF03145), Prok-RING_4 (PF13923) and zf-C3HC4_2 (PF14447) are homologous to the RING finger domain [Supplementary data (Fig. 4A)] and were found distributed among all the sequences identified in the mollusk. A highly conserved distribution of cysteine, cysteine, histidine and cysteine was found in the results of this work, providing evidence that there was a structural motif formation [Supplementary data (Fig. 4B)] and that it is related to the bond made with zinc.2828. Metzger MB, Hristova VA, Weissman AM. HECT and RING finger families of E3 ubiquitin ligases at a glance. J Cell Sci. 2012; 125(Pt 3): 531-7.
Like the E3-RING finger, the E3-U-box class also recruits the E2-ubiquitin complex and target substrate concomitantly. In addition, they share a common organisation and architecture, suggesting that U-box domains are modified RING domains because they do not bind to zinc and are formed from hydrogen bonds.2929. Aravind L, Koonin EV. The U box is a modified RING finger ? a common domain in ubiquitination. Curr Biol. 2000; 10(4); 132-4. Thirteen of the fifteen E3-U-box sequences identified in B. glabrata demonstrated the U-box domain (CL0229) and the other two inferred other correlated domains [Supplementary data (Fig. 5A)]. The ufd-2 (BGLB026024-PA) sequence showed only the Ufd2P_core domain (PF10408) which escapes ubiquitinated proteins to the proteasome and was directly linked to the U-box domain in the C-terminal portion of the sequence. The other sequence was designated cyn-4 (BGLB022189-PA), which was found the Rtf2 domain (PF04641), similar to the RING finger domain, but has a three-dimensional ring-like structure and only one site for binding with zinc. The absence of U-box domain may be related to the annotation process of these sequences. The results obtained showed that in the U-box domain there exists the formation of a structural motif in which not all amino acid residues were highly conserved, but there are some that were present in all sequences used in the analysis [Supplementary data (Fig. 5B)].
The proteasome is a proteolytic complex formed by an association between a 19S regulatory particle (PA700) and a central component 20S, composed of approximately 70 subunits. The 19S particle is divided into base and lid and are reversibly bound to 20S. The regulatory portion recognises the labeled substrate while the 20S nucleus is responsible for degrading the protein in minor peptides.77. Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002; 82(2): 373-428.,1414. Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, et al. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994; 78(5): 761-71.
Nineteen genes from the RPN and RPT groups were identified in our analysis (Table I), encompassing sequences of major importance such as RPN-11, RPN-15 and RPN-3, involved with target substrate recognition and removal of the ubiquitin chain formed.1515. Murata S, Sasaki K, Kishimoto T, Niwa SI, Hayashi H, Takahama Y, et al. Regulation of CD8+T cell development by thymus-specific proteasomes. Science. 2007; 316(5829): 1349-53.,3030. Kato K, Satoh T. Structural insights on the dynamics of proteasome formation. Biophys Rev. 2018; 10(2): 597-604. In the same sense, some RPT subunits, such as rpt-2 and rpt-5 were also found in the data of B. glabrata; however, they are related to activation of the 20S nucleus.3131. Papaevgeniou N, Chondrogianni N. The ubiquitin proteasome system in Caenorhabditis elegans and its regulation. Redox Biol. 2014; 2(1): 333-47. The results of this work show that RPT shares the conserved AAA domain, but not of an active site, structural motif or catalytic cleft, inferring that the function of these molecules was linked to the sequence that the conserved domain possesses [Supplementary data (Fig. 11)]. Conversely, sequences from the RPN group did not have a common domain and did not present any conserved active sites [Supplementary data (Fig. 12)]. A suggested explanation for this feature was that these subunits may be more involved with the target substrate than with the catalytic portion of the 26S proteasome and therefore do not share highly conserved sequences.
The nucleus of the proteasome is composed of α and β subunits. Subunits of the α-type have a structural and regulatory role, whereas β possesses proteolytic and degradation activity. Fourteen sequences involved in the formation of the 20S proteasome between α and β subunits were identified in C. elegans,3131. Papaevgeniou N, Chondrogianni N. The ubiquitin proteasome system in Caenorhabditis elegans and its regulation. Redox Biol. 2014; 2(1): 333-47. corroborating the number of sequences found in B. glabrata data (Table I). All sequences belonging to the PAS (α-subunit) and PBS (β-subunit) groups have the conserved proteasome domain in their N-terminal portions [Fig. 4 and Supplementary data (Fig. 10A)], demonstrating that the presence of this domain in this region is important for the activity they play. An active site composed of five relatively conserved amino acid residues was identified in the results obtained for the PAS group genes among all sequences (Fig. 5). Conversely, for the PBS group, a less conserved active site was found, which may be involved in the proteolytic function of this subunit [Supplementary data (Fig. 10B)].
Through the phylogenetic analysis, it was possible to describe the evolutionary history of the sequences under study and from these results to infer their identity by the location in the generated tree, as well as the proximity to orthologous organisms and model organisms. Our results show a clear and specific distribution among all data obtained from the analysis performed using MEGA5.2. The evolutionary organisation of each set of sequences, both the sequences involved in the ubiquitination pathway and those related to the formation of the 26S proteasome (Figs 3, 6), corresponds to what was already known in the animal tree of life. There was a well-defined division between the clade of deuterostomia and protostomia organisms, separated between phyla, and grouped according to the location of the species in the evolutionary process.3232. Donoghue MJ, Cracraft J, editors. Assembling the tree of life. Oxford University Press; 2004. p. 1-6. In the same sense, each sequence identified in B. glabrata presents well-defined ramifications involving its orthologous organisms, providing strong evidence that the putative sequences found in the mollusk transcriptome are real sequences involved in the ubiquitin-proteasome complex.
Our results demonstrate that the gene responsible for translating the ubiquitin (ubq-1) molecule was the only one to present a high expression profile in the RNAseq data of all 12 libraries used, that is, in all analysed tissues of the adult snail. Ubiquitin is a key molecule in UPS; however, it is also involved in other cellular signaling processes such as plasma membrane transport and DNA repair.2222. Ramos PC. A via proteolítica dependente de ubiquitina/proteassoma. Sociedade Portuguesa de Química. 2002; 1(96): 57-63. This only happens because of the role it plays in labeling target substrates with one or more ubiquitins, forming a polyubiquitin chain. In addition to these functions, ubiquitin is also involved in autophagy.1818. Zahoor Z, Lockyer AE, Davies AJ, Kirk RS, Emery AM, Rollinson D, et al. Differences in the gene expression profiles of haemocytes from schistosome-susceptible and -resistant Biomphalaria glabrata exposed to Schistosoma mansoni excretory-secretory products. PLoS One. 2014; 8(6): 1236-51.,2222. Ramos PC. A via proteolítica dependente de ubiquitina/proteassoma. Sociedade Portuguesa de Química. 2002; 1(96): 57-63.
The high expression that the ubq-1 transcript has demonstrated can be explained by the diversity of cellular processes in which ubiquitin is involved, being related to virtually all cellular processes, directly or indirectly. This also explains the high expression in all organs of B. glabrata used for analysis. In addition, the formation of polyubiquitin chains influences that a greater amount of this gene was transcribed and may be translated, since more than one molecule of ubiquitin is required to label only a target substrate.
Most of the genes involved in UPS and identified in the B. glabrata genome presented a median expression for all organs of uninfected adult snails evaluated. This expression profile infers that both the transcripts involved in the ubiquitination pathway and genes participating in the formation of the 26S proteasome do not show a high expression under normal conditions of mollusk survival. Thus, it can be inferred that these transcripts share behavior related to the expression very similar for all analysed organs. Our results indicate that this is a pathway capable of performing its role in the organism without large amounts of circulating molecules in its adult state. Our results were based on uninfected snails, although the hypothesis is supported that the results obtained for these transcripts may be different in infected snails, demonstrating degrees of expression different from those presented in this work, since the UPS plays a role related to immunity. To confirm this hypothesis, other transcriptomic analyses are required. Some transcripts appear to have a direct relationship with specific tissues because they have a very low expression profile for 11 libraries and relatively high for only one, suggesting that the function they play may be linked to their location in the organism. For example, rpn-10 (BGLB003999-RB) demonstrates better expression in the central nervous system (CNS) than in the other tissues (Fig. 7). The deletion of rpn-10 in D. melanogaster leads to lethality and demonstrates cause abnormalities in the mitotic cycle such as aneuploidies and absence of chromosomal segregation in CNS larval cells, besides accumulating multiubiquitin proteins in the tissue, since this is a gene participant of the 26S proteasome regulatory portion.3333. Szlanka T. Deletion of proteasomal subunit S5a/Rpn10/p54 causes lethality, multiple mitotic defects and overexpression of proteasomal genes in Drosophila melanogaster. J Cell Sci. 2003; 116(Pt 6): 1023-33. In view of this, it would be interesting to investigate in depth the relationship between the transcripts identified in B. glabrata and the snail tissues demonstrated in this work. This is due to the fact that they are tissue-specific transcripts in which their functions are performed more efficiently in exclusive tissues, demonstrating their importance in the development of these organisms.
Four families of genes showed a low expression for all libraries used. All were involved with ubiquitin ligases (E3), two of which refer to transcripts of E3 enzymes and two were genes encoding molecules that form part of complexes. The genes encoding the ubiquitin ligase enzymes are transcribed from the genome in the hundreds. This means that many different E3 are expressed, but not highly expressed, since they are directly related to recognition of the target substrate.77. Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002; 82(2): 373-428.,1010. Scheffner M, Nuber U, Huibregtse JM. Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature. 1995; 373(6509): 81-3. In the same sense, because of the specificity they present for the labeled substrate, they do not need to be transcribed in large quantities into the mollusk tissues in non-stress situations. traf-6 is in a family of genes that presented one of the lowest expression levels according to the results; however, others studies have related this transcript to cellular signaling in the immune system.3434. Li J, Chai QY, Liu CH. The ubiquitin system: a critical regulator of innate immunity and pathogen-host interactions. Cell Mol Immunol. 2016; 13(5): 560-76. Thus, the low expression can be explained by the fact that the data from the analysed libraries refer to uninfected adult snail transcripts. traf-6 becomes a possible gene to be investigated in future research using data from mollusks under infection conditions, suggesting that this expression may be increased in front of the parasite. In addition to traf-6, other genes involved in the ubiquitination pathway found in our results have also been described involved with immunity, such as diap1 (BGLB013412-RB) and ubc-13 (BGLB033527-RA and BGLB033527-RB). All presented a moderate expression profile, which suggests that genes may have a differential expression in case of infection.
Our results allow the beginning of the search for new knowledge and perspectives involving B. glabrata, increasing the amount of information on the regulation of the mollusk and the relation between parasite/host. This work provides evidence of the presence of the UPS in the genome and transcriptome of the snail, besides offering a delineation of the expression profile of the 119 sequences identified for mollusks in their normal development. Thus, it is concluded that the UPS is a system conserved in B. glabrata and that ubiquitin is indeed the key molecule of the system, demonstrating a high expression for all tissues analysed in adult snails. In the normal state of survival, the UPS is a moderately expressed system as a whole, but is believed to have a different expression in infected organs. To obtain these results, new transcriptomic analyses are required. Our work also provides a basis for new hypotheses to be developed, such as the evaluation of the expression profile of these sequences in different phases of the life of the organism, as well as new studies involving the analysis of the behavior of this pathway when the intermediate host is infected by S. mansoni. Therefore, these results may offer new ways of controlling infection and, consequently, schistosomiasis.
ACKNOWLEDGEMENTS
To the researchers responsible for moluscario Lobato Paraense, and the research group involved in the sequencing of the genome and transcriptome of Biomphalaria glabrata.
REFERENCES
-
1de Souza CP, Jannotti-Passos LK, de Freitas JR. Degree of host-parasite compatibility between Schistosoma mansoni and their intermediate molluscan hosts in Brazil. Mem Inst Oswaldo Cruz. 1995; 90(1): 5-10.
-
2Carvalho OS, Coelho PMZ, Lenzi HL. Schistosoma mansoni e esquistossomose: uma visão multidisciplinar. Rio de Janeiro: Editora Fiocruz; 2008. p. 29-245.
-
3Richards CS, Shade PC. The genetic variation of compatibility in Biomphalaria glabrata and Schistosoma mansoni. J Parasitol. 1987; 73(6): 1146-51.
-
4Negrão-Corrêa D, Mattos ACA, Pereira CAJ, Martins-Souza RL, Coelho PMZ. Interaction of Schistosoma mansoni sporocysts and hemocytes of Biomphalaria. J Parasitol Res. 2012; 2012(743920): 1-6.
-
5Larson MK, Bender RC, Bayne CJ. Resistance of Biomphalaria glabrata 13-16-R1 snails to Schistosoma mansoni PR1 is a function of haemocyte abundance and constitutive levels of specific transcripts in haemocytes. Int J Parasitol. 2014; 44(6): 343-53.
-
6Adema CM, Hillier LW, Jones CS, Loker ES, Knight M, Minx P, et al. Whole genome analysis of a schistosomiasis-transmitting freshwater snail. Nat Commun. 2017; 8(15451): 1-11.
-
7Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002; 82(2): 373-428.
-
8Ciechanover A. The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J. 1998; 17(24): 7151-60.
-
9Ebner P, Versteeg GA, Ikeda F. Ubiquitin enzymes in the regulation of immune responses. Crit Rev Biochem Mol Biol. 2017; 52(4): 425-60.
-
10Scheffner M, Nuber U, Huibregtse JM. Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature. 1995; 373(6509): 81-3.
-
11Tanaka K, Chiba T. The proteasome: a protein-destroying machine. Genes Cells. 1998; 3(8): 499-510.
-
12Budenholzer L, Cheng CL, Li Y, Hochstrasser M. Proteasome structure and assembly. J Mol Biol. 2017; 429(22): 3500-24.
-
13Jung T, Catalgol B, Grune T. The proteasomal system. Mol Aspects Med. 2009; 30(4): 191-296.
-
14Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, et al. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994; 78(5): 761-71.
-
15Murata S, Sasaki K, Kishimoto T, Niwa SI, Hayashi H, Takahama Y, et al. Regulation of CD8+T cell development by thymus-specific proteasomes. Science. 2007; 316(5829): 1349-53.
-
16Murata S, Takahama Y, Kasahara M, Tanaka K. The immunoproteasome and thymoproteasome: functions, evolution and human disease. Nat Immunol. 2018; 19(1): 923-31.
-
17Lockyer AE, Spinks J, Kane RA, Hoffmann KF, Fitzpatrick JM, Rollinson D, et al. Biomphalaria glabrata transcriptome: cDNA microarray profiling identifies resistant- and susceptible-specific gene expression in haemocytes from snail strains exposed to Schistosoma mansoni. BMC Genomics. 2008; 643(9): 1-17.
-
18Zahoor Z, Lockyer AE, Davies AJ, Kirk RS, Emery AM, Rollinson D, et al. Differences in the gene expression profiles of haemocytes from schistosome-susceptible and -resistant Biomphalaria glabrata exposed to Schistosoma mansoni excretory-secretory products. PLoS One. 2014; 8(6): 1236-51.
-
19Dinguirard N, Cavalcanti MGS, Wu XJ, Bickham-Wright U, Sabat G, Yoshino TP. Proteomic analysis of Biomphalaria glabrata hemocytes during in vitro encapsulation of Schistosoma mansoni sporocysts. Front Immunol. 2018; 9(2773): 1-17.
-
20Queiroz FR, Silva LM, Jeremias WJ, Babá EH, Caldeira RL, Coelho PMZ, et al. Differential expression of small RNA pathway genes associated with the Biomphalaria glabrata/Schistosoma mansoni interaction. PLoS One. 2017; 12(7): 1-20.
-
21Müller WEG. Molecular phylogeny of metazoa (animals): monophyletic origin. Naturwissenschaften. 1995; 82(7): 321-9.
-
22Ramos PC. A via proteolítica dependente de ubiquitina/proteassoma. Sociedade Portuguesa de Química. 2002; 1(96): 57-63.
-
23Zuin A, Isasa M, Crosas B. Ubiquitin signaling: extreme conservation as a source of diversity. Cells. 2014; 3(3): 690-701.
-
24Lois LM, Lima CD. Structures of the SUMO E1 provide mechanistic insights into SUMO activation and E2 recruitment to E1. EMBO J. 2005; 24(3): 439-51.
-
25Rodrigo-Brenni MC, Morgan DO. Sequential E2s drive polyubiquitin chain assembly on APC targets. Cell. 2007; 130(1): 127-39.
-
26Rotin D, Kumar S. Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol. 2009; 10(6): 398-409.
-
27Pickart CM, Eddins MJ. Ubiquitin: structures, functions, mechanisms. Biochim Biophys Acta. 2004; 1695(1-3): 55-72.
-
28Metzger MB, Hristova VA, Weissman AM. HECT and RING finger families of E3 ubiquitin ligases at a glance. J Cell Sci. 2012; 125(Pt 3): 531-7.
-
29Aravind L, Koonin EV. The U box is a modified RING finger ? a common domain in ubiquitination. Curr Biol. 2000; 10(4); 132-4.
-
30Kato K, Satoh T. Structural insights on the dynamics of proteasome formation. Biophys Rev. 2018; 10(2): 597-604.
-
31Papaevgeniou N, Chondrogianni N. The ubiquitin proteasome system in Caenorhabditis elegans and its regulation. Redox Biol. 2014; 2(1): 333-47.
-
32Donoghue MJ, Cracraft J, editors. Assembling the tree of life. Oxford University Press; 2004. p. 1-6.
-
33Szlanka T. Deletion of proteasomal subunit S5a/Rpn10/p54 causes lethality, multiple mitotic defects and overexpression of proteasomal genes in Drosophila melanogaster. J Cell Sci. 2003; 116(Pt 6): 1023-33.
-
34Li J, Chai QY, Liu CH. The ubiquitin system: a critical regulator of innate immunity and pathogen-host interactions. Cell Mol Immunol. 2016; 13(5): 560-76.
-
Financial support: CNPq (Project nº 479890/2013-7).
Publication Dates
-
Publication in this collection
03 June 2019 -
Date of issue
2019
History
-
Received
06 Feb 2019 -
Accepted
02 Mar 2019