Abstract
BACKGROUND
Healthcare-associated infections caused by bacteria such as Pseudomonas aeruginosa are a major public health problem worldwide. Gene regulatory networks (GRN) computationally represent interactions among regulatory genes and their targets. They are an important approach to help understand bacterial behaviour and to provide novel ways of overcoming scientific challenges, including the identification of potential therapeutic targets and the development of new drugs.
OBJECTIVES
The goal of this study was to reconstruct the multidrug-resistant (MDR) P. aeruginosa GRN and to analyse its topological properties.
METHODS
The methodology used in this study was based on gene orthology inference using the reciprocal best hit method. We used the genome of P. aeruginosa CCBH4851 as the basis of the reconstruction process. This MDR strain is representative of the sequence type 277, which was involved in an endemic outbreak in Brazil.
FINDINGS
We obtained a network with a larger number of regulatory genes, target genes and interactions as compared to the previously reported network. Topological analysis results are in accordance with the complex network representation of biological processes.
MAIN CONCLUSIONS
The properties of the network were consistent with the biological features of P. aeruginosa. To the best of our knowledge, the P. aeruginosa GRN presented here is the most complete version available to date.
Key words:
Pseudomonas aeruginosa; gene regulatory network; multidrug resistance
Healthcare-associated infections (HAI) are a major global public health problem as they increase the morbidity and mortality rates of hospitalised individuals. HAI are often caused by multidrug-resistant (MDR) bacteria, such as Pseudomonas aeruginosa, especially in immunocompromised patients. In Brazil, P. aeruginosa was ranked as the fifth most common causative agent of HAI in patients hospitalised in adult and paediatric intensive care units, and nearly 35% of the reported strains are resistant to carbapenems, a class of antibiotics widely used to treat P. aeruginosa infections.11. Agência Nacional de Vigilância Sanitária. Boletim Segurança do Paciente e Qualidade em Serviços de Saúde no 16. Avaliação dos indicadores nacionais das Infecções Relacionadas à Assistência à Saúde (IRAS) e Resistência microbiana do ano de 2016 [Internet]. Brasília, DF: Anvisa; 2017 [cited 2019 Jan 17]. Available from: http://portal.anvisa.gov.br/documents/33852/271855/Boletim+Seguran%C3%A7a+do+Paciente+e+Qualidade+em+Servi%C3%A7os+de+Sa%C3%BAde+n%C2%BA+16+-+Avalia%C3%A7%C3%A3o+dos+indicadores+nacionais+das+Infec%C3%A7%C3%B5es+Relacionadas+%C3%A0+Assist%C3%AAncia+%C3%A0+Sa%C3%BAde+%28IRAS%29+e+Resist%C3%AAncia+microbiana+do+ano+de+2016+%28REVISADO%29/e8ec4ea2-1832-489d-8354-0dbc7e3c2f7b.
http://portal.anvisa.gov.br/documents/33...
In fact, individuals infected with MDR P. aeruginosa clones have a higher mortality rate (44.6%) compared to those infected with non-MDR clones (24.8%).22. Matos ECO de, Andriolo RB, Rodrigues YC, Lima PDL de, Carneiro IC do RS, Lima KVB. Mortality in patients with multidrug-resistant Pseudomonas aeruginosa infections: a meta-analysis. Rev Soc Bras Med Trop. 2018; 51(4): 415-20.
P. aeruginosa is a versatile pathogen that can cause several types of infections affecting the lower respiratory tract, skin, urinary tract and eyes, leading to bacteraemia, endocarditis and other complications. P. aeruginosa infections are difficult to treat as the therapeutic options are becoming increasingly limited. Biofilm formation and the presence of intrinsic resistance-associated genes are examples of the mechanisms that P. aeruginosa employs to resist chemotherapy. In addition, this bacterium can become resistant to a broad range of antibiotics through the acquisition of new resistance mechanisms via horizontal gene transfer.33. Lister PD, Wolter DJ, Hanson ND. Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin Microbiol Rev. 2009; 22(4): 582-610.
4. Mulcahy LR, Isabella VM, Lewis K. Pseudomonas aeruginosa biofilms in disease. Microb Ecol. 2014; 68(1): 1-12.-55. Neves PR, Mamizuka EM, Levy CE, Lincopan N. Pseudomonas aeruginosa multirresistente: um problema endêmico no Brasil. J Bras Patol e Med Lab. 2011; 47: 409-20.
In 2000, the genome sequence of P. aeruginosa PAO1 strain was published, providing data regarding its genetic complexity and ecological versatility.66. Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature. 2000; 406(6799): 959-64. The PAO1 strain is sensitive to most clinically used antimicrobial agents and has been extensively studied since the publication of its genomic sequence.
In 2003, the first clinical isolate of an MDR P. aeruginosa strain carrying the carbapenemase gene, bla
SPM-1, was identified in Brazil. The SPM-1 protein is a metallo-β-lactamase that confers resistance to almost all classes of beta-lactams.77. Toleman MA, Simm AM, Murphy TA, Gales AC, Biedenbach DJ, Jones RN, et al. Molecular characterization of SPM-1, a novel metallo-beta-lactamase isolated in Latin America: report from the SENTRY antimicrobial surveillance programme. J Antimicrob Chemother. 2002; 50(5): 673-9. Most of the SPM-producing isolates belong to sequence type (ST) 277, as indicated by the multilocus sequence typing (MLST). The ST277 has been associated with hospital outbreaks in several Brazilian states and has been isolated from hospital sewage systems and rivers.88. Carvalho APD, Albano RM, de Oliveira DN, Cidade DA de P, Teixeira LM, Marques E de A. Characterization of an epidemic carbapenem-resistant Pseudomonas aeruginosa producing SPM-1 metallo-beta-lactamase in a hospital located in Rio de Janeiro, Brazil. Microb Drug Resist Larchmt N. 2006; 12(2): 103-8.
9. Fontes LC, Neves PR, Oliveira S, Silva KC, Hachich EM, Sato MIZ, et al. Isolation of Pseudomonas aeruginosa coproducing metallo-beta-lactamase SPM-1 and 16S rRNA methylase RmtD1 in an urban river. Antimicrob Agents Chemother. 2011; 55(6): 3063-4.-1010. Nascimento APB, Ortiz MF, Martins WMBS, Morais GL, Fehlberg LCC, Almeida LGP, et al. Intraclonal genome stability of the metallo-ß-lactamase SPM-1-producing Pseudomonas aeruginosa ST277, an endemic clone disseminated in Brazilian hospitals. Front Microbiol [Internet]. 2016. [cited 2019 Apr 10]. Available from: https://www.frontiersin.org/articles/10.3389/fmicb.2016.01946/full.
https://www.frontiersin.org/articles/10....
In recent years, researchers have applied mathematical methods to generate computational models to study the behaviour of certain organisms. This approach has contributed to the development of new products, the improvement and acceleration of existing health policies and research into novel ways of overcoming scientific challenges. It is often based on the construction of biological networks and the analysis of gene regulatory, metabolic and signal transduction pathways and/or protein-protein interactions.1111. Tatarinova TV, Nikolsky Y, editors. Biological networks and pathway analysis [Internet]. Humana Press; 2017 [cited 2019 Mar 6]. (Methods in Molecular Biology). Available from: https://www.springer.com/gb/book/9781493970254.
https://www.springer.com/gb/book/9781493...
A gene regulatory network (GRN) is a collection of transcription factors (TFs) that interact with each other and with other molecules in the cell to regulate mRNA and protein expression levels. In 2011, Galán-Vásquez et al.1212. Galán-Vásquez E, Luna B, Martínez-Antonio A. The regulatory network of Pseudomonas aeruginosa. Microb Inform Exp. 2011; 14(1): 3. published the first P. aeruginosa GRN and ana-lysed its main topological properties and interactions between its regulatory components.
In the present study, we describe the reconstruction of the GRN of an MDR P. aeruginosa strain, including all currently available curated biological data. This reconstruction was based on P. aeruginosa CCBH4851, a strain representative of the ST277, which was involved in an endemic outbreak in Brazil. This strain shows resistance to all antimicrobials of clinical importance with the exception of polymyxin B, has several mechanisms of resistance and mobile genetic elements.1313. Silveira M, Albano R, Asensi M, Assef APC. The draft genome sequence of multidrug-resistant Pseudomonas aeruginosa strain CCBH4851, a nosocomial isolate belonging to clone SP (ST277) that is prevalent in Brazil. Mem Inst Oswaldo Cruz. 2014; 109(8): 1086-7. The implications of the choice of an MDR strain as the basis of the GRN reconstruction are discussed in this study. In addition, the topological properties of GRN were analysed, and regulators, target genes (TGs), TFs, auto-activation mechanisms, influential genes and network motifs were characterised.
MATERIALS AND METHODS
Bacterial strains - In this study, we reconstructed a GRN for P. aeruginosa CCBH4851. This strain is deposited at the Culture Collection of Hospital-Acquired Bacteria (CCBH) located at the Laboratório de Pesquisa em Infecção Hospitalar, Instituto Oswaldo Cruz/Fundação Oswaldo Cruz (Fiocruz) (WDCM947; 39 CGEN022/2010) and its genome sequence is available in the GenBank database (accession CP021380).1313. Silveira M, Albano R, Asensi M, Assef APC. The draft genome sequence of multidrug-resistant Pseudomonas aeruginosa strain CCBH4851, a nosocomial isolate belonging to clone SP (ST277) that is prevalent in Brazil. Mem Inst Oswaldo Cruz. 2014; 109(8): 1086-7. For the orthology analysis, P. aeruginosa PAO1,66. Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature. 2000; 406(6799): 959-64.P. aeruginosa PA71414. Roy PH, Tetu SG, Larouche A, Elbourne L, Tremblay S, Ren Q, et al. Complete genome sequence of the multiresistant taxonomic outlier Pseudomonas aeruginosa PA7. PloS One. 2010; 5(1): e8842. and P. aeruginosa UCBPP-PA14 (PA14)1515. Lee DG, Urbach JM, Wu G, Liberati NT, Feinbaum RL, Miyata S, et al. Genomic analysis reveals that Pseudomonas aeruginosa virulence is combinatorial. Genome Biol. 2006; 7(10): R90. were chosen as reference strains.
Orthology-based model generation - Fitch1616. Fitch WM. Homology a personal view on some of the problems. Trends Genet TIG. 2000; 16(5): 227-31. defines orthologs as genes that diverged after a speciation event, but share a common ancestor. The most common approach to find orthologs is the reciprocal best hits (RBH) method.1717. Kristensen DM, Wolf YI, Mushegian AR, Koonin EV. Computational methods for Gene Orthology inference. Brief Bioinform. 2011; 12(5): 379-91. The regulatory interactions between TFs and TGs in P. aeruginosa PAO1, P. aeruginosa PA14 and P. aeruginosa PA7 strains were propagated to the reconstructed P. aeruginosa CCBH4851 network if both the TF and the TG formed RBH. The criterion used to define an orthologous relationship was the existence of RBH between the two genomes. Two genes, x and x’, of the genomes X and X’, respectively, were considered orthologs if they were also RBH, i.e., if upon aligning the sequence of x against the gene list of X’, we obtained x’ as the best alignment, and if upon aligning the sequence of x’ against the gene list of X, we obtained x as the best hit. Once the complete set of RBHs between X and X’ genomes was obtained, a regulatory interaction between a TF (gene x) and a TG (gene y) was propagated from the reference network to CCBH4851, if both the TF and the TG had their respective RBH in the CCBH4851 genome. The propagation of a regulatory interaction, x-y, from the reference genome X, held if the pair, x’-y’, existed in the genome X’, such that both (x, x’) and (y, y’) were RBH pairs. One disadvantage of the RBH method is its inability to detect multi-to-multi orthologous relationships. In this case, RBH only picks the hit with the best score alignment, resulting in false negatives. To prevent these false negatives, when a gene presented no ortholog in genome X’, manual curation was performed as follows: the protein sequence encoded by gene x of genome X was searched against genome X’ using the BLASTX algorithm. If the search returned two or more hits, the neighbouring region of each hit was assessed to determine which gene in genome X’ was orthologous to that specific protein, matching its genomic context. If the search returned no hits, the gene was considered to have no ortholog in genome X’. This test for the propagation of regulatory interactions was performed with all known interactions in PAO1, PA7 and PA14. All-against-all alignments were performed using the BLASTP program with the following stringent parameters: identity ≥ 90%, coverage ≥ 90% and E value cut-off of 1 e-5. Fig. 1 presents an overview of the reconstruction process.
overview of the general strategy for reconstruction of the Pseudomonas aeruginosa gene regulatory network (GRN). The process began with the alignment of the P. aeruginosa CCBH4851 genome and the three reference strains. Next, the reciprocal best hits (RBH) method was applied and the resulting genes were compared against the gene regulatory databases listed in the “Databases” box. Data obtained from these databases were then integrated and submitted to the curation process, which aims to solve network inconsistencies. Finally, the GRN was generated and its topology was analysed.
Identification of RBH - An algorithm was implemented using the Python programming language to automate and generate a list of RBH in a tabular format. Regulators and TGs were then identified and separated into a single table, thus extending the work done by Galán-Vásquez et al.1212. Galán-Vásquez E, Luna B, Martínez-Antonio A. The regulatory network of Pseudomonas aeruginosa. Microb Inform Exp. 2011; 14(1): 3.
Data integration - The data integration process brought together biological information from all strains with the aim of organising biological knowledge. The final network table is available as Supplementary data. This table is organised into six columns: Regulatory gene, Ortholog of the regulatory gene, Target gene, Ortholog of the target gene, Mode of regulation and Reference. The first column lists the regulatory genes of P. aeruginosa CCBH4851, the second column contains orthologs of regulatory genes in the reference strain (PAO1, PA7 or PA14), the third column refers to the target gene in CCBH4851, the fourth column lists orthologs of TGs in the reference strain, the fifth column describes the mode of regulation and the sixth column indicates the source of the corresponding data.
Curation process - Our group has developed a web application to support the curation of biological networks. This web application, called CurSystem1818. Ramos TG. Reconstrução da Rede Metabólica da Pseudomonas aeruginosa CCBH4851. 2018 [cited 2019 Mar 6]; Available from: https://www.arca.fiocruz.br/handle/icict/29528.
https://www.arca.fiocruz.br/handle/icict...
(available from: http://Pseudomonas.procc.fiocruz.br:8185/CurSystem) provides support for distributed, asynchronous interaction among specialists in the fields of biochemistry, chemistry, microbiology, molecular biology, computational and systems biology, computer and systems engineering and physics. This tool was used to select specific gene interactions, discuss their main peculiarities and determine whether they would be part of the network. This stage was fundamental to excluding doubtful biological information from the network.
Network generation and computational analysis - The R programming language and the open-source software, RStudio were used for network generation and computational analysis.1919. RStudio Team. RStudio: Integrated development environment for R [Internet]. 2012; Boston, MA. Available from: http://www.rstudio.com/.
http://www.rstudio.com/...
Analysis of degree, centrality, clustering coefficient, connectivity, cycles, paths and hierarchical levels was performed according to previously described methods.1212. Galán-Vásquez E, Luna B, Martínez-Antonio A. The regulatory network of Pseudomonas aeruginosa. Microb Inform Exp. 2011; 14(1): 3.,2020. Martínez-Antonio A, Janga SC, Thieffry D. Functional organisation of Escherichia coli transcriptional regulatory network. J Mol Biol. 2008; 381(1): 238-47. We used the dplyr, tibble, readr, igraph and scales packages for these analyses. The igraph package was used for the computation of feed-forward loop (FFL) motifs (function triad_census). Network degree-entropy was measured according to the method described by Breitkreutz et al.2121. Breitkreutz D, Hlatky L, Rietman E, Tuszynski JA. Molecular signaling network complexity is correlated with cancer patient survivability. Proc Natl Acad Sci. 2012; 109(23): 9209-12.
All data and code are available as Supplementary data.
RESULTS
General features of the GRN - The P. aeruginosa network reconstruction resulted in a total of 1,046 genes, of which 42 behaved as regulatory genes, 96 as both regulatory and TGs (i.e., a TF was influenced by another TF in the network) and 908 were TGs. We identified 1,576 regulatory interactions between regulators and their TGs. Altogether, these genes represented approximately 16.52% of the genome of P. aeruginosa CCBH4851, which was used as the model organism in this study. Despite the apparent low coverage, we included most of the TFs with known functions among the 138 regulators in the P. aeruginosa CCBH4851 network. The number of regulatory genes, TGs, and interactions represented an increase of 44.92, 34.69 and 35.27%, respectively, compared to previous study.1212. Galán-Vásquez E, Luna B, Martínez-Antonio A. The regulatory network of Pseudomonas aeruginosa. Microb Inform Exp. 2011; 14(1): 3. Network enrichment was not the only result observed in the P. aeruginosa CCBH4851 GRN reconstruction. As the reconstruction was based on the RBH method that involved comparing the CCBH4851 genome annotation with reference strains, it was not possible to infer an orthologous relationship for some genes, particularly oprD and mexZ, which are genes involved in antibiotic resistance mechanisms. The curation process revealed that these genes were either fully absent or annotated as pseudogenes in CCBH4851. A pseudogene is a DNA sequence that resembles a gene in the reference genome, but has modifications such as point mutations, insertions, deletions, premature stop codons or frameshifts, making it impossible to determine if its product is still functional in the target organism, without further experimentation. The lack of orthology resulted in the exclusion of these genes from the P. aeruginosa CCBH4851 GRN. In addition, certain regulatory genes and its interactions were kept as described in the previous network,1212. Galán-Vásquez E, Luna B, Martínez-Antonio A. The regulatory network of Pseudomonas aeruginosa. Microb Inform Exp. 2011; 14(1): 3. and in the databases and/or scientific literature used. For example, ihf (integration host factor) represents not a single gene, but a complex composed of the products of the himA and himD genes that act in combination as a TF for several TGs. However, regulatory systems such as quorum sensing or two-component systems are often formed by a pair of genes, but only one of them is able to bind to the promoter region of the target gene. However, both genes were listed as regulatory genes. Therefore, we were able to maintain equivalent notations to previous network.1212. Galán-Vásquez E, Luna B, Martínez-Antonio A. The regulatory network of Pseudomonas aeruginosa. Microb Inform Exp. 2011; 14(1): 3.
Basic network topological analysis: number of vertices, number of edges and density - We identified 1,576 edges in the CCBH4851 network. These interactions were classified into four types: activation (“+”), repression (“-”), dual (“d”, when the regulatory gene could act as an activator or a repressor, depending on the conditions), and unknown (“?”). An illustration of the CCBH4851 GRN is presented in Fig. 2. Network density is a measure of the interconnectivity between vertices. It is the ratio of the actual number of edges in the network and the maximum possible number of edges. The regulatory network of the CCBH4851 strain had a density of 1.44 e-03, which was slightly lower than the observed density of 2.12 e-03 for the PAO1 strain, but was of the same order of magnitude. A network diameter indicates the path length between the two most distant nodes. The CCBH4851 GRN had a diameter of 12 nodes, while the previous network had a diameter of 9 nodes. Another measure, the average path distance, also known as the average shortest path, is the average distance between two nodes. While the previous network showed an average path distance of 4.08, the CCBH5851 GRN had an average path distance of 4.80.1212. Galán-Vásquez E, Luna B, Martínez-Antonio A. The regulatory network of Pseudomonas aeruginosa. Microb Inform Exp. 2011; 14(1): 3.,2222. Bales ME, Johnson SB. Graph theoretic modeling of large-scale semantic networks. J Biomed Inform. 2006; 39(4): 451-64.
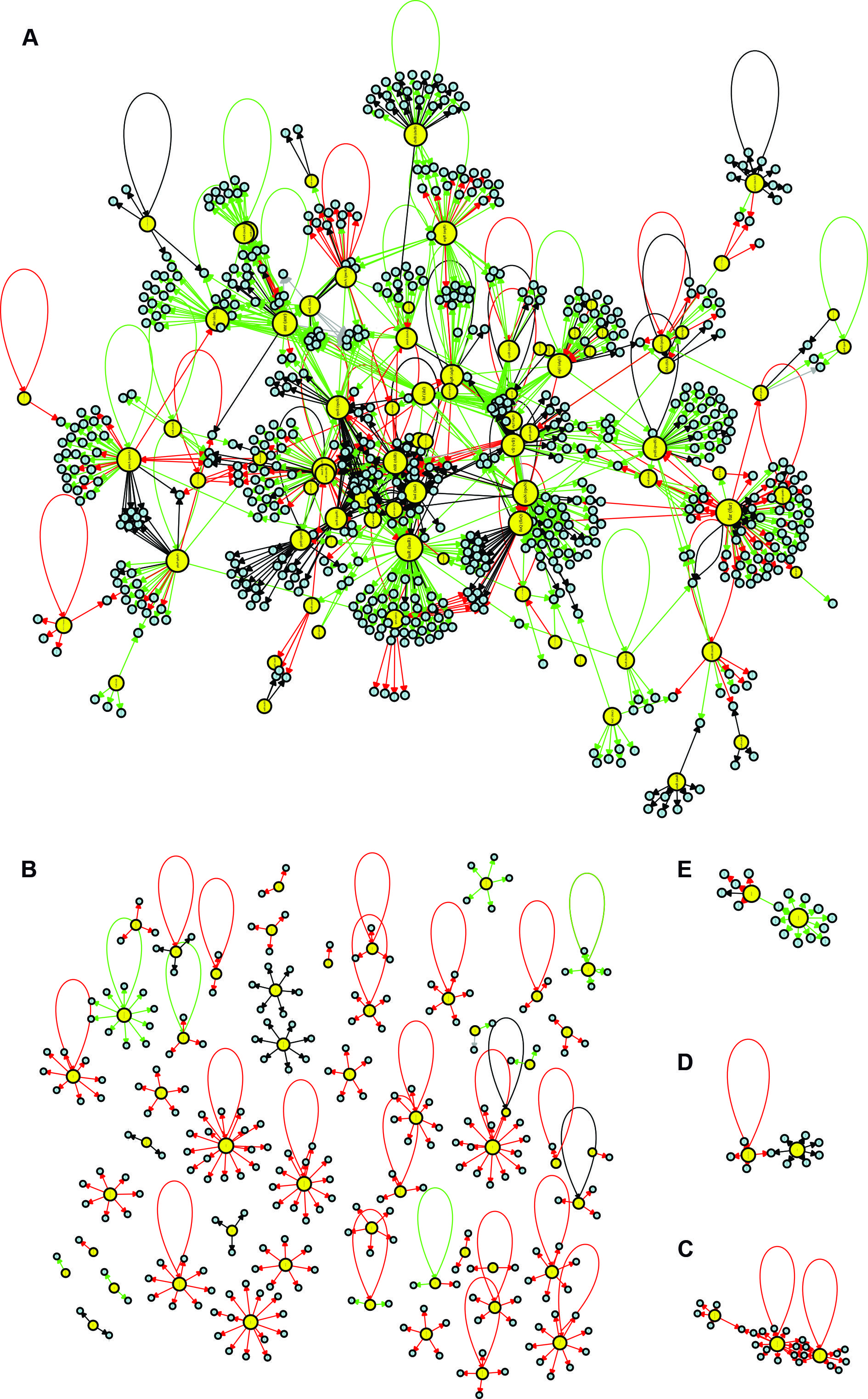
visualisation of the Pseudomonas aeruginosa CCBH4851 gene regulatory network (GRN). Yellow circles indicate regulatory genes, light blue circles indicate target genes (TGs), black lines indicate an unknown mode of regulation, green lines indicate activation, red lines indicate repression and grey lines indicate a dual mode of regulation. A: the GRN large highly connected network component; B: all regulatory and TGs that have no connections with the component depicted in A; C-E: clusters of lower connectivity compared to the component depicted in A. All figures are presented with higher resolution in the Supplementary data.
The degree k(i) of a vertex i is defined as its number of edges. Edges in directed networks can be of two types: they can “depart” from or “arrive” at node i, thus defining them as “incoming” (k-in) or “outgoing” (k-out) degrees, respectively. In the CCBH4851 GRN, each vertex was, on average, connected to three other vertices, which was the same as reported for the PAO1 GRN. Fig. 3 illustrates the incoming (3A, B) and outgoing (3C, D) degree distributions for the CCBH4851 GRN.
graphical representation of topological measurements of the Pseudomonas aeruginosa CCBH4851 gene regulatory network (GRN) (red) compared to the previously published network1212. Galán-Vásquez E, Luna B, Martínez-Antonio A. The regulatory network of Pseudomonas aeruginosa. Microb Inform Exp. 2011; 14(1): 3. (blue). A, B: incoming degree distribution of the P. aeruginosa CCBH4851 GRN; C, D: outgoing distribution of the P. aeruginosa CCBH4851 GRN. For clarity, the distributions are plotted both on a linear (A, C) and on a logarithmic scale (B, D); E: local clustering coefficient distribution; F: clustering coefficient by degree.
Scale-free is a common topology classification associated with biological networks, corresponding to complex networks in which the degree distribution follows a power law. In scale-free networks, most nodes (vertices) have few connections and few nodes have a large number of connections. Therefore, scale-free networks are dominated by a relatively small number of high-degree nodes, generally referred to as hubs.2323. Amaral LA, Scala A, Barthelemy M, Stanley HE. Classes of small-world networks. Proc Natl Acad Sci U S A. 2000; 97(21): 11149-52.
The degree distribution can be approximated by:
Equation 1 corresponds to a power-law distribution and the exponent, γ, is its degree exponent.2424. Barabási A-L, Bonabeau E. Scale-free networks. Sci Am. 2003; 288(5): 60-9. The degree distribution shown in Fig. 3B, D is on a double logarithmic axis and the straight line is consistent with a power-law distribution. For k-in, the estimated value for γ was 2.89, which is very close to the value reported for the reference network (γ =2.717).1212. Galán-Vásquez E, Luna B, Martínez-Antonio A. The regulatory network of Pseudomonas aeruginosa. Microb Inform Exp. 2011; 14(1): 3.
Clustering coefficient distribution - Given a node i with m(i) neighbours in a directed network, the maximum number of edges connecting the elements of this neighbourhood is given by m max (i) = m(i)(m(i)-1). The local clustering coefficient, C(i), is defined as the ratio of the actual number of edges N(i) occurring in node i neighbourhood and m max (i).2525. Watts DJ, Strogatz SH. Collective dynamics of "small-world" networks. Nature. 1998; 393(6684): 440-2. The local clustering coefficient is defined as C(i) = N(i)/m max (i). In GRNs, the local clustering coefficient C(i) is interpreted as the interaction between genes forming the regulatory groups. The distribution of local clustering coefficients can be seen in Fig. 3E.
The global clustering coefficient is proportional to the number of triangles present in the network, disregarding the directionality of the edges. A tringle is a set of three nodes with at least two connections between them. Triangles can be closed, with three connections within the set, or open triangles, with only two edges. The global clustering coefficient, C, is the ratio between the number of closed triangles and the total number of triangles (closed or open) in the network. The CCBH4851 network had a global clustering coefficient equal to 3.2 e-02.
Another interesting feature to observe is the correlation between the local clustering coefficient C(i) and the degree k(i), as shown in the scatter plot in Fig. 3F. The observed correlation was negative and Fig. 3 also shows that the vertices with high degree, k, corresponded to the same vertices with null clustering coefficients, while the vertices that formed clusters had low degrees. This observation confirmed that strongly cohesive groups are exceptions in the network and are formed by a small number of genes. These results were obtained for both the CCBH4851 GRN and the previously published P. aeruginosa GRN.1212. Galán-Vásquez E, Luna B, Martínez-Antonio A. The regulatory network of Pseudomonas aeruginosa. Microb Inform Exp. 2011; 14(1): 3.
Connectivity - Network connectivity is a concept that reflects the associations between every pair of genes. Nodes were considered a part of a connected component when they interacted through a direct or an indirect link (intermediate connections). In the connectivity analysis, network interactions were considered undirected. Similar to the reference GRN, the CCBH4851 network was disconnected. It showed one large connected component (including 751 nodes) and more than 48 small connected components, which is larger than the previously reported network.1212. Galán-Vásquez E, Luna B, Martínez-Antonio A. The regulatory network of Pseudomonas aeruginosa. Microb Inform Exp. 2011; 14(1): 3. However, there may be several reasons for the network being disconnected at specific points. Firstly, this may represent the natural behaviour of the organism, i.e., not all genes in a complex genome are linked, because cellular processes can be compartmentalised or global, constitutive or growth phase-dependent. Secondly, there may have been insufficient biological information to infer the interactions. Thirdly, P. aeruginosa genomes generally maintain a conserved core component, which accounts for the majority of the genome. However, additional strain-specific blocks of genes are acquired by horizontal gene transfer, as a result of evolutionary events, and these can result in a decreased degree of similarity with reference strains and an increased degree of similarity with newly reported strains. This process can result in the loss of existing interactions or a gain of interactions still not fully described, thus lacking a connection with other components in the network.
Dominant activity and autoregulation - The analysis of the frequency of the different modes of regulation indicated that activation is the predominant type of regulation mode in the CCBH4851 network, with frequency values very similar to those previously observed for the P. aeruginosa GRN. Overall, 48.92% of the interactions were of the activation mode, 28.8% were of the repression mode and 22.27% were of dual or unknown modes. Although the distribution pattern was maintained, a significant enrichment was observed in negative and unknown regulation modes. When considering autoregulation, i.e., a gene regulating its own expression, the CCBH5851 GRN showed a predominance of negative autoregulatory motifs, which differs from the findings of Galán-Vásquez et al.1212. Galán-Vásquez E, Luna B, Martínez-Antonio A. The regulatory network of Pseudomonas aeruginosa. Microb Inform Exp. 2011; 14(1): 3.
Motifs - The existence of cycles or motifs in biological networks is a necessary condition for the existence of multiple stationary states or attractors. In GRNs, the most common 3-genes motif is the FFL. The FFL motif comprises a gene A that regulates gene B. Then, both A and B regulate gene C. There are two types of FFL motifs: (i) coherent, where the regulatory effect of both paths, direct and indirect, are the same and (ii) incoherent, where the regulatory effects are different. In this study, we computed the total number of FFL motifs: the number of coherent type I FFL motifs (where all interactions are activations) and the number of incoherent type II motifs, (where all interactions are repressions).2626. Mangan S, Alon U. Structure and function of the feed-forward loop network motif. Proc Natl Acad Sci U S A. 2003; 100(21): 11980-5. The CCBH4851 GRN had a larger number of FFL motifs (when considering all variations), when compared to the GRN published by Galán-Vásquez et al. The coherent type I FFL motif was the most abundant in both networks, with 82 representatives in the PAO1 GRN and 79 in the CCBH4851 GRN. Meanwhile, there were four incoherent type II FFL motifs in the CCBH4851 GRN compared with three in the previously described network.1212. Galán-Vásquez E, Luna B, Martínez-Antonio A. The regulatory network of Pseudomonas aeruginosa. Microb Inform Exp. 2011; 14(1): 3.
Hubs - Identifying the most influential genes in a gene transcription network is a key step in determining therapeutic targets against an infectious agent. One way to identify possible targets is to identify so-called network hubs. Different definitions of a hub can be applied in the context of complex network theory. One method of identifying a hub is to determine which vertices have the highest k-out degrees in order to identify, in the case of a GRN, the genes with the greatest influence on target regulation. According to Vandereyken et al.,2727. Vandereyken K, Van Leene J, De Coninck B, Cammue BPA. Hub protein controversy: taking a closer look at plant stress response hubs. Front Plant Sci. 2018; 9: 1-24. the exact number of interactions that characterise a hub, also called the degree threshold, differs among different studies. Some studies have shown that the minimum number is five, whereas others have reported eight, 10, 20 or even 50 as the minimum number. In the present study, the degree threshold was defined as the average of the number of connections of all nodes having at least two edges. The application of this definition resulted in a cut-off value of 16 connections. Table I shows the 30 most influential hubs in the P. aeruginosa GRN. After pinpointing the hubs, an analysis was performed to determine whether they are interconnected (through direct or indirect interactions). Only two hubs were found to not be interconnected, np20 and PA4851_19380 (homologous to PA1520). The remaining hubs had a direct (when a hub affects the regulation of another hub) or indirect (when hubs affect the regulation of the same group of TGs) connection to other hubs (Fig. 4). Node interactions that were not common among hubs were hidden to improve visualisation in Fig. 4.
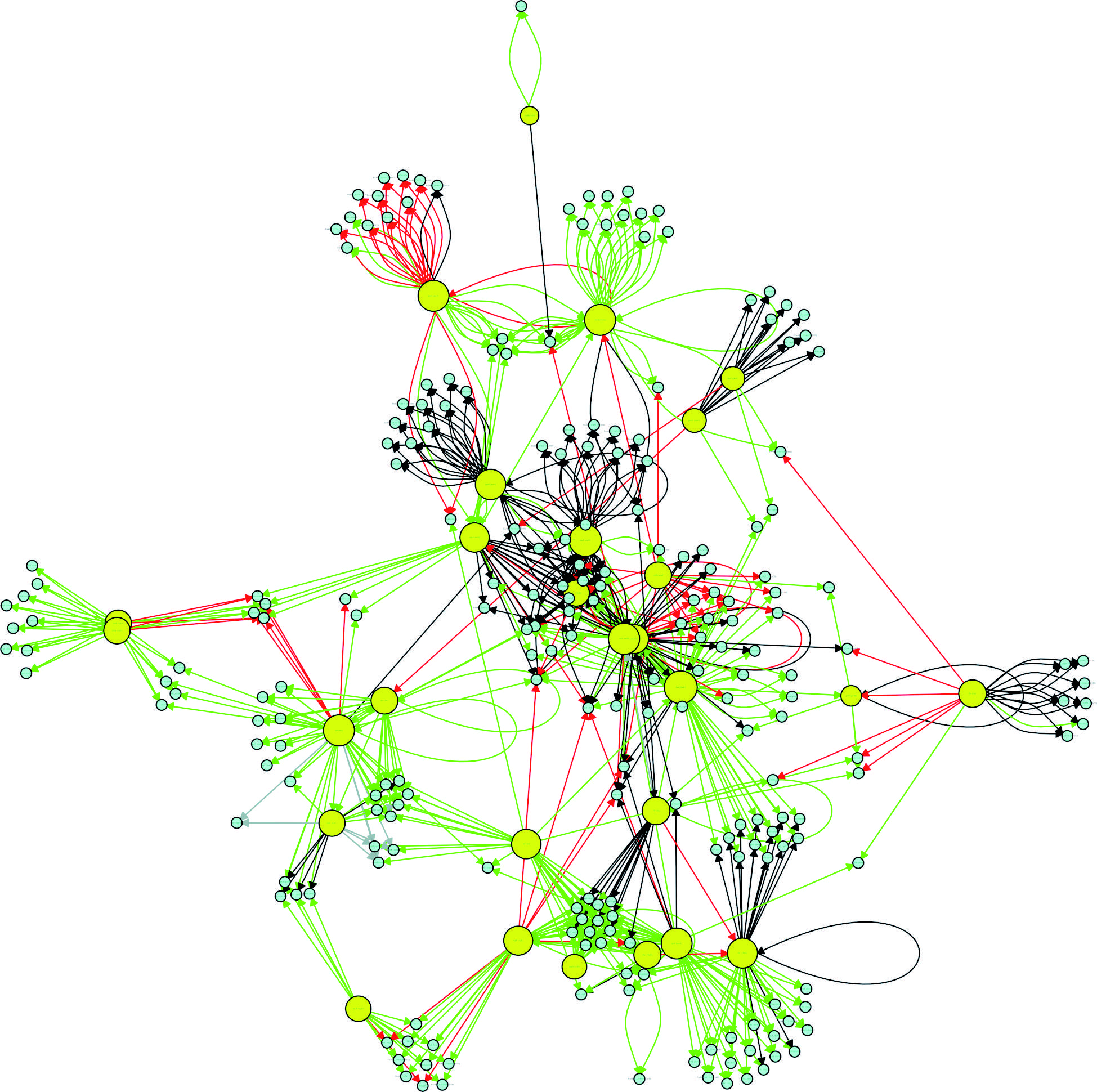
connectivity relationships among the 30 most influential hubs of the Pseudomonas aeruginosa CCBH4851 gene regulatory network. Yellow circles indicate regulatory genes considered hubs, light blue circles indicate target genes, black lines indicate an unknown mode of regulation, green lines indicate activation, red lines indicate repression and grey lines indicate a dual mode of regulation.
The summarised results comprising the network statistics are presented in Table II, which contains standard measures, such as the number of nodes, number of edges, number of autoregulatory motifs, diameter of the network and average path length. Other relevant measures were the number of coherent and incoherent feed-forward motifs, clustering coefficients and network entropy. Moreover, a comparison with data from the previous network1212. Galán-Vásquez E, Luna B, Martínez-Antonio A. The regulatory network of Pseudomonas aeruginosa. Microb Inform Exp. 2011; 14(1): 3. is included in Table II.
DISCUSSION
The importance of gene regulation on metabolic, adaptive, pathogenic and antibiotic resistance capabilities is well known. The GRN reconstruction and analysis of a versatile pathogen, such as P. aeruginosa, particularly when based on an MDR strain, contributes to an increase in the knowledge of related cellular processes. Multidrug resistance can be conferred by a combination of factors that vary according to the antimicrobial class. For instance, carbapenem resistance in P. aeruginosa is manly conferred by mutations in oprD and/or by the presence of MBLs. Mutations or differential expression of efflux system genes are also contributing factors for both carbapenem and aminoglycoside resistance. Multidrug resistance can also occur through other mechanisms, including the acquisition of genes through horizontal transfer and punctual mutations, in multiple combinations.2828. Kos VN, Déraspe M, McLaughlin RE, Whiteaker JD, Roy PH, Alm RA, et al. The resistome of Pseudomonas aeruginosa in relationship to phenotypic susceptibility. Antimicrob Agents Chemother. 2015; 59(1): 427-36. In addition, P. aeruginosa has the ability to form a biofilm, which can play a role in antibiotic penetration, antibiotic tolerance, the formation of persister cells and protection from the host immune system.44. Mulcahy LR, Isabella VM, Lewis K. Pseudomonas aeruginosa biofilms in disease. Microb Ecol. 2014; 68(1): 1-12. Due to the limitations of graphical representation, it is difficult to incorporate data such as gene expression variation and point mutations or genes lacking experimental evidence in a GRN graph. Overall, we excluded oprD, mexZ and pilA genes from our network. As oprD is a target gene, its exclusion had a minor impact on the network topology. However, this gene is extremely important for the cell because oprD encodes an outer membrane porin, which is important for the absorption of carbapenems. The lack of OprD leads to low outer membrane permeability. However, the exclusion of mexZ, a regulatory gene, resulted in the exclusion of its node from the network, as well as the exclusion of the interactions with its TGs. The mexZ product represses the transcription of mexX and mexY genes. MexXY proteins are part of an efflux pump system, whose overexpression leads to aminoglycoside resistance through the extrusion of this family of compounds. However, MexXY overexpression needs to be experimentally established and cannot be represented in a graph. PilA is a major pilin protein associated with bacterial adherence through the type VI pilus machinery. Therefore, PilA has a great importance in pathogenesis. The pilA gene is also a target, which only shows regulatory influence upon itself. The advantageous effect of PilA loss to an MDR strain is unclear. One can hypothesise that this loss may be compensated by newly acquired genes, since CCBH4851 has a chromosome approximately 600 kb larger than PAO1. However, these alterations are common among MDR strains2929. Quale J, Bratu S, Gupta J, Landman D. Interplay of efflux system, ampC, and oprD expression in carbapenem resistance of Pseudomonas aeruginosa clinical isolates. Antimicrob Agents Chemother. 2006; 50(5): 1633-41.,3030. Haghi F, Zeighami H, Monazami A, Toutouchi F, Nazaralian S, Naderi G. Diversity of virulence genes in multidrug resistant Pseudomonas aeruginosa isolated from burn wound infections. Microb Pathog. 2018;115: 251-6. and constructing a network comprising these features may impact the dynamics of simulations designed to assess MDR bacteria behaviour based on the P. aeruginosa CCBH4851 GRN. Overall, the reconstruction reported here included additional regulators, TGs and new interactions described in the literature or included in curated databases since the last P. aeruginosa GRN publication.1212. Galán-Vásquez E, Luna B, Martínez-Antonio A. The regulatory network of Pseudomonas aeruginosa. Microb Inform Exp. 2011; 14(1): 3. Several genes involved in virulence mechanisms were identified, such as those associated with the production of proteases and toxins, antimicrobial activity, iron uptake, antiphagocytosis, adherence and quorum sensing. In addition to the inclusion of new nodes and connections, previously identified nodes were excluded (by the curation process or the lack of homology) and interactions were revisited (due to genes whose regulatory effect had been recently elucidated). Two noteworthy examples of the included nodes and interactions are the regulatory effect of fleQ upon “psl” genes and the regulation of the efflux pump genes, mexA, mexE, and oprH, by brlR. The “psl” (polysaccharide synthesis locus) cluster comprises 15 exopolysaccharide biosynthesis-related genes organised in tandem. These genes are important for biofilm formation. The recently functionally characterised transcriptional regulator, BrlR, has biofilm-specific expression and plays a role in the antibiotic tolerance of biofilms by modulating the expression of efflux pump genes.44. Mulcahy LR, Isabella VM, Lewis K. Pseudomonas aeruginosa biofilms in disease. Microb Ecol. 2014; 68(1): 1-12. Altogether, these alterations directly influenced the topological characteristics of the network. However, topology measures of the P. aeruginosa CCBH4851 GRN, such as node degree distribution and clustering coefficient, remained consistent with a scale-free network type. The degree distribution followed the power-law distribution (Fig. 3B, D), meaning that a small number of nodes had many connections and a large number of nodes had few connections. Moreover, the correlation between local clustering coefficient and node degree (Fig. 3F) showed that nodes with lower degrees had larger local clustering coefficients than nodes with higher degrees. Indeed, construction of several networks representing biological processes showed similar topological characteristics.2424. Barabási A-L, Bonabeau E. Scale-free networks. Sci Am. 2003; 288(5): 60-9.,3131. Zhu X, Gerstein M, Snyder M. Getting connected: analysis and principles of biological networks. Genes Dev. 2007; 21(9): 1010-24. As with other mathematical aspects of the network, topology per se was consistent with the type of network obtained, but it is important to ensure that these measures are consistent with biological observations. The reconstructed network showed a low-density value, which was compatible with the fact that networks representing natural phenomena often have low density, due to their structural and dynamic flexibility.2222. Bales ME, Johnson SB. Graph theoretic modeling of large-scale semantic networks. J Biomed Inform. 2006; 39(4): 451-64. The low density observed in the CCBH4851 GRN indicated that the nodes were not all interconnected. Biologically, in an organism such as P. aeruginosa, which has an average of 6,000 coding sequences, it is not expected that all genes maintain an interaction, since they are related to distinct biological processes that are not all dependent on each other and are triggered during different growth phases, thus corroborating the observed low density. In the same way, the global clustering coefficient and connectivity parameters are affected by these biological behaviours, resulting in the large number of connected components found in the CCBH4851 GRN.
Although some nodes under positive regulation were lost (Table II), the most common regulatory activity found among CCBH4851 GRN interactions was activation. However, more than 50% of the autoregulation found was negative. This may be a consequence of the increase in negative regulation in the overall network interactions. A similar pattern is seen in the regulatory network of another member of the Gammaproteobacteria class, Escherichia coli, which exhibited the prevalence of negative autoregulation concurrently with the prevalence of positive regulation between TFs. The positive mode of regulation is important to ensure the continuity of biological processes. Adhesion, cell-to-cell signalling, production of virulence and resistance factors, biofilm formation, secretion of toxins and host-pathogen interaction factors are examples of processes that, once initiated, must reach a final stage in order to have the desired effect. In fact, we observed that genes such as lasR, rlhR, pvdS, anr, dnr and algU, involved in these types of processes mostly showed a positive mode of regulation in the CCBH4851 GRN. However, negative cycles are also important for life-sustaining cyclic processes, such as those involved in cellular homeostasis. This is the case for metabolic processes where genes such as lexA, hutC, iscR, desT and mvat (although involved in virulence factor biosynthesis, this gene regulates arginine metabolism), showed a negative mode of regulation as the predominant effect.2020. Martínez-Antonio A, Janga SC, Thieffry D. Functional organisation of Escherichia coli transcriptional regulatory network. J Mol Biol. 2008; 381(1): 238-47. Negative autoregulation is linked to cellular stability, providing a rapid response to variations in protein/toxin/metabolite concentrations, thus avoiding the energy cost of unneeded synthesis and avoiding undesired effects. Some examples of negative autoregulatory interactions included in the CCBH4851 network were algZ, lexA, metR, ptxR and rsaL. RsaL is a quorum-sensing repressor; LexA is involved in the SOS response; AlgZ is the transcriptional activator of AlgD, involved in alginate production; PtxR affects exotoxin A production; and MetR is involved in swarming motility and methionine synthesis. Overall, these autoregulatory genes tend to be further upstream in the regulatory chain.
The dominance of the activation mode was also observed when analysing network motifs. Motifs are patterns of topological structures statistically overrepresented in the network. The number of FFL motifs, considering all variations, was 218 for the CCBH4851 GRN, but 137 for the GRN published by Galán-Vásquez et al.1212. Galán-Vásquez E, Luna B, Martínez-Antonio A. The regulatory network of Pseudomonas aeruginosa. Microb Inform Exp. 2011; 14(1): 3. A common motif often related to transcriptional networks, the coherent FFL, was abundantly present in the CCBH4851 GRN (Table II).2626. Mangan S, Alon U. Structure and function of the feed-forward loop network motif. Proc Natl Acad Sci U S A. 2003; 100(21): 11980-5. In particular, the coherent type I FFL motif, where all interactions are positive, was common in both GRNs. These motifs act as sign-sensitive delays, i.e., a circuit that responds rapidly to step-like stimuli in one direction (ON to OFF), and as a delay to steps in the opposite direction (OFF to ON). While the temporary removal of the stimulus ceases transcription, the activation of expression requires a persistent signal to carry on. Although less represented, the incoherent type II FFL motif was also found in the CCBH4851 GRN. In contrast to the coherent FFL, these motifs act as sign-sensitive accelerators, i.e., a circuit that responds rapidly to step-like stimuli in one direction, but not in the other direction.2626. Mangan S, Alon U. Structure and function of the feed-forward loop network motif. Proc Natl Acad Sci U S A. 2003; 100(21): 11980-5. Overall, FFL motifs are important for the modulation of cellular processes according to environmental conditions.
One last characteristic revealed by the topological analysis was the presence of hubs. Hubs are nodes showing a large number of connections, a concept that is inherent to scale-free networks. As expected, analysis of the CCBH4851 GRN showed that genes known to have a great impact on the gene regulatory systems of P. aeruginosa, such as lasR, fur, anr, mexT and algU, were among the most influential hubs. These genes are involved in resistance, virulence, and pathogenicity mechanisms. LasR, for instance, directly activates the expression of 99 genes. The activity of LasR depends on the presence and binding of N-3-oxo-dodecanoyl-L-homoserine lactone (C12). Once bound, the LasR-C12 complex coordinates the expression of TGs, including many genes encoding factors involved in virulence and cell density.3232. Kiratisin P, Tucker KD, Passador L. LasR, a transcriptional activator of Pseudomonas aeruginosa virulence genes, functions as a multimer. J Bacteriol. 2002; 184(17): 4912-9. In addition, fur is a global regulator of iron uptake, rpoN is an alternative sigma factor, mexT regulates an efflux pump system and several virulence factors and anr is responsible for the regulation of anaerobic adaptation processes. All of these genes are known to control the expression of many other genes. We observed that, even though a few hubs remained unconnected, most of the influential genes belonged to the major connected component. This interaction can be direct, as in case of the positive effect of LasR on rlhR transcription, or indirect, when hubs are regulating the same targets, i.e., involved in the regulation of the same processes, such as fur and algU, both of which affect the expression of phuR, which encodes a member of a heme uptake system that facilitates host iron acquisition.3333. Firoved AM, Boucher JC, Deretic V. Global genomic analysis of AlgU (sE)-dependent promoters (sigmulon) in Pseudomonas aeruginosa and implications for inflammatory processes in cystic fibrosis. J Bacteriol. 2002; 184(4): 1057-64.,3434. Ochsner UA, Johnson Z, Vasil ML. Genetics and regulation of two distinct haem-uptake systems, phu and has, in Pseudomonas aeruginosa. Microbiol Read Engl. 2000; 146 (Pt 1): 185-98. Another example is the regulation of “alg” genes by algU, rpoN and cysB, which are not directly connected, but are related through their effect on alginate biosynthesis, which is important for the mucoid phenotype of P. aeruginosa colonies.3535. Ramsey DM, Wozniak DJ. Understanding the control of Pseudomonas aeruginosa alginate synthesis and the prospects for management of chronic infections in cystic fibrosis. Mol Microbiol. 2005; 56(2): 309-22. Direct and indirect interactions reflect the importance of influential genes, not only to their specific targets, but the effects of their targets on the regulation of subsequent processes, triggering a more pleiotropic effect. If a perturbation is required, a hub can affect more than one pathway, resulting in undesired effects. However, one of the interconnected nodes related to that hub may be a candidate for a perturbation that results in the impairment or improvement of a specific pathway. Nevertheless, isolated hubs are equally important. In fact, they are related to processes such as zinc uptake (np20) and purine metabolism (PA4851_19380), which are fundamental to bacterial survival, but can be considered somewhat independent of other processes, and are only triggered under specific conditions.
Table II shows a comparison of network statistics between the CCBH4851 GRN and the GRN network published by Galán-Vásquez et al.1212. Galán-Vásquez E, Luna B, Martínez-Antonio A. The regulatory network of Pseudomonas aeruginosa. Microb Inform Exp. 2011; 14(1): 3. One clear trend is that the CCBH4851 GRN represents a substantial improvement in terms of network completeness, since is includes more nodes, edges and network motifs. Other measures also reflect this improvement, such as the global clustering coefficient and the diameter of the network. Other comparisons between these networks are presented in Fig. 3. The charts and the data presented in Table II show an increase in the completeness and complexity of the CCBH4851 network, compared with the previous network, particularly when comparing clustering coefficients (Fig. 3E, F).
A concept addressed by Csermely,3636. Csermely P. The wisdom of networks: a general adaptation and learning mechanism of complex systems. The network core triggers fast responses to known stimuli; innovations require the slow network periphery and are encoded by core-remodeling. BioEssays News Rev Mol Cell Dev Biol. 2018; 40(1). is the plasticity of networks. Plastic networks have some interesting characteristics, such as a diffuse core, overlapping modules, fewer hierarchies/more loops, large network entropy and origin dominance, leading to many attractors. Csermely states that biologically plastic networks should be targeted by a “central impact” directed at their hubs, bridges and bottlenecks, because if they are attacked on their periphery, the effect of the drug will never reach the centre of the network, due its efficient dissipation. For this reason, topological characteristics, such as connected components, motifs and hubs are important to help determine the best approach to disturb a network in a way that promotes the desired phenotype in the cell. Indeed, it is noteworthy that the total network entropy of the CCBH4851 GRN was greater than the P. aeruginosa GRN published in 2011 (Table II). Therefore, the CCBH4851 GRN had greater plasticity than the GRN described by Galán-Vásquez et al.1212. Galán-Vásquez E, Luna B, Martínez-Antonio A. The regulatory network of Pseudomonas aeruginosa. Microb Inform Exp. 2011; 14(1): 3. This increased plasticity may be due to the increased size of the CCBH4851 GRN. Nevertheless, this observation may also be related to the fact that the CCBH4851 is an MDR strain, while the previous network is based mostly on P. aeruginosa PAO1.
This reconstruction of the P. aeruginosa GRN contributes to an increase in our understanding of the behaviour of this bacterium. In future studies, we intend to construct a dynamic model of this network, aimed at assisting researchers working on experimental drug design and screening. This will enable the prediction of dynamic behaviour in order to better understand these bacteria and allow the simulation of normal and stress conditions, eventually leading to the discovery of new therapeutic targets and the development of new drugs to combat P. aeruginosa infections.
ACKNOWLEDGEMENTS
To Inova-Fiocruz, Faperj and Capes, for financial support.
REFERENCES
-
1Agência Nacional de Vigilância Sanitária. Boletim Segurança do Paciente e Qualidade em Serviços de Saúde no 16. Avaliação dos indicadores nacionais das Infecções Relacionadas à Assistência à Saúde (IRAS) e Resistência microbiana do ano de 2016 [Internet]. Brasília, DF: Anvisa; 2017 [cited 2019 Jan 17]. Available from: http://portal.anvisa.gov.br/documents/33852/271855/Boletim+Seguran%C3%A7a+do+Paciente+e+Qualidade+em+Servi%C3%A7os+de+Sa%C3%BAde+n%C2%BA+16+-+Avalia%C3%A7%C3%A3o+dos+indicadores+nacionais+das+Infec%C3%A7%C3%B5es+Relacionadas+%C3%A0+Assist%C3%AAncia+%C3%A0+Sa%C3%BAde+%28IRAS%29+e+Resist%C3%AAncia+microbiana+do+ano+de+2016+%28REVISADO%29/e8ec4ea2-1832-489d-8354-0dbc7e3c2f7b
» http://portal.anvisa.gov.br/documents/33852/271855/Boletim+Seguran%C3%A7a+do+Paciente+e+Qualidade+em+Servi%C3%A7os+de+Sa%C3%BAde+n%C2%BA+16+-+Avalia%C3%A7%C3%A3o+dos+indicadores+nacionais+das+Infec%C3%A7%C3%B5es+Relacionadas+%C3%A0+Assist%C3%AAncia+%C3%A0+Sa%C3%BAde+%28IRAS%29+e+Resist%C3%AAncia+microbiana+do+ano+de+2016+%28REVISADO%29/e8ec4ea2-1832-489d-8354-0dbc7e3c2f7b -
2Matos ECO de, Andriolo RB, Rodrigues YC, Lima PDL de, Carneiro IC do RS, Lima KVB. Mortality in patients with multidrug-resistant Pseudomonas aeruginosa infections: a meta-analysis. Rev Soc Bras Med Trop. 2018; 51(4): 415-20.
-
3Lister PD, Wolter DJ, Hanson ND. Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin Microbiol Rev. 2009; 22(4): 582-610.
-
4Mulcahy LR, Isabella VM, Lewis K. Pseudomonas aeruginosa biofilms in disease. Microb Ecol. 2014; 68(1): 1-12.
-
5Neves PR, Mamizuka EM, Levy CE, Lincopan N. Pseudomonas aeruginosa multirresistente: um problema endêmico no Brasil. J Bras Patol e Med Lab. 2011; 47: 409-20.
-
6Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature. 2000; 406(6799): 959-64.
-
7Toleman MA, Simm AM, Murphy TA, Gales AC, Biedenbach DJ, Jones RN, et al. Molecular characterization of SPM-1, a novel metallo-beta-lactamase isolated in Latin America: report from the SENTRY antimicrobial surveillance programme. J Antimicrob Chemother. 2002; 50(5): 673-9.
-
8Carvalho APD, Albano RM, de Oliveira DN, Cidade DA de P, Teixeira LM, Marques E de A. Characterization of an epidemic carbapenem-resistant Pseudomonas aeruginosa producing SPM-1 metallo-beta-lactamase in a hospital located in Rio de Janeiro, Brazil. Microb Drug Resist Larchmt N. 2006; 12(2): 103-8.
-
9Fontes LC, Neves PR, Oliveira S, Silva KC, Hachich EM, Sato MIZ, et al. Isolation of Pseudomonas aeruginosa coproducing metallo-beta-lactamase SPM-1 and 16S rRNA methylase RmtD1 in an urban river. Antimicrob Agents Chemother. 2011; 55(6): 3063-4.
-
10Nascimento APB, Ortiz MF, Martins WMBS, Morais GL, Fehlberg LCC, Almeida LGP, et al. Intraclonal genome stability of the metallo-ß-lactamase SPM-1-producing Pseudomonas aeruginosa ST277, an endemic clone disseminated in Brazilian hospitals. Front Microbiol [Internet]. 2016. [cited 2019 Apr 10]. Available from: https://www.frontiersin.org/articles/10.3389/fmicb.2016.01946/full
» https://www.frontiersin.org/articles/10.3389/fmicb.2016.01946/full -
11Tatarinova TV, Nikolsky Y, editors. Biological networks and pathway analysis [Internet]. Humana Press; 2017 [cited 2019 Mar 6]. (Methods in Molecular Biology). Available from: https://www.springer.com/gb/book/9781493970254
» https://www.springer.com/gb/book/9781493970254 -
12Galán-Vásquez E, Luna B, Martínez-Antonio A. The regulatory network of Pseudomonas aeruginosa. Microb Inform Exp. 2011; 14(1): 3.
-
13Silveira M, Albano R, Asensi M, Assef APC. The draft genome sequence of multidrug-resistant Pseudomonas aeruginosa strain CCBH4851, a nosocomial isolate belonging to clone SP (ST277) that is prevalent in Brazil. Mem Inst Oswaldo Cruz. 2014; 109(8): 1086-7.
-
14Roy PH, Tetu SG, Larouche A, Elbourne L, Tremblay S, Ren Q, et al. Complete genome sequence of the multiresistant taxonomic outlier Pseudomonas aeruginosa PA7. PloS One. 2010; 5(1): e8842.
-
15Lee DG, Urbach JM, Wu G, Liberati NT, Feinbaum RL, Miyata S, et al. Genomic analysis reveals that Pseudomonas aeruginosa virulence is combinatorial. Genome Biol. 2006; 7(10): R90.
-
16Fitch WM. Homology a personal view on some of the problems. Trends Genet TIG. 2000; 16(5): 227-31.
-
17Kristensen DM, Wolf YI, Mushegian AR, Koonin EV. Computational methods for Gene Orthology inference. Brief Bioinform. 2011; 12(5): 379-91.
-
18Ramos TG. Reconstrução da Rede Metabólica da Pseudomonas aeruginosa CCBH4851. 2018 [cited 2019 Mar 6]; Available from: https://www.arca.fiocruz.br/handle/icict/29528
» https://www.arca.fiocruz.br/handle/icict/29528 -
19RStudio Team. RStudio: Integrated development environment for R [Internet]. 2012; Boston, MA. Available from: http://www.rstudio.com/
» http://www.rstudio.com/ -
20Martínez-Antonio A, Janga SC, Thieffry D. Functional organisation of Escherichia coli transcriptional regulatory network. J Mol Biol. 2008; 381(1): 238-47.
-
21Breitkreutz D, Hlatky L, Rietman E, Tuszynski JA. Molecular signaling network complexity is correlated with cancer patient survivability. Proc Natl Acad Sci. 2012; 109(23): 9209-12.
-
22Bales ME, Johnson SB. Graph theoretic modeling of large-scale semantic networks. J Biomed Inform. 2006; 39(4): 451-64.
-
23Amaral LA, Scala A, Barthelemy M, Stanley HE. Classes of small-world networks. Proc Natl Acad Sci U S A. 2000; 97(21): 11149-52.
-
24Barabási A-L, Bonabeau E. Scale-free networks. Sci Am. 2003; 288(5): 60-9.
-
25Watts DJ, Strogatz SH. Collective dynamics of "small-world" networks. Nature. 1998; 393(6684): 440-2.
-
26Mangan S, Alon U. Structure and function of the feed-forward loop network motif. Proc Natl Acad Sci U S A. 2003; 100(21): 11980-5.
-
27Vandereyken K, Van Leene J, De Coninck B, Cammue BPA. Hub protein controversy: taking a closer look at plant stress response hubs. Front Plant Sci. 2018; 9: 1-24.
-
28Kos VN, Déraspe M, McLaughlin RE, Whiteaker JD, Roy PH, Alm RA, et al. The resistome of Pseudomonas aeruginosa in relationship to phenotypic susceptibility. Antimicrob Agents Chemother. 2015; 59(1): 427-36.
-
29Quale J, Bratu S, Gupta J, Landman D. Interplay of efflux system, ampC, and oprD expression in carbapenem resistance of Pseudomonas aeruginosa clinical isolates. Antimicrob Agents Chemother. 2006; 50(5): 1633-41.
-
30Haghi F, Zeighami H, Monazami A, Toutouchi F, Nazaralian S, Naderi G. Diversity of virulence genes in multidrug resistant Pseudomonas aeruginosa isolated from burn wound infections. Microb Pathog. 2018;115: 251-6.
-
31Zhu X, Gerstein M, Snyder M. Getting connected: analysis and principles of biological networks. Genes Dev. 2007; 21(9): 1010-24.
-
32Kiratisin P, Tucker KD, Passador L. LasR, a transcriptional activator of Pseudomonas aeruginosa virulence genes, functions as a multimer. J Bacteriol. 2002; 184(17): 4912-9.
-
33Firoved AM, Boucher JC, Deretic V. Global genomic analysis of AlgU (sE)-dependent promoters (sigmulon) in Pseudomonas aeruginosa and implications for inflammatory processes in cystic fibrosis. J Bacteriol. 2002; 184(4): 1057-64.
-
34Ochsner UA, Johnson Z, Vasil ML. Genetics and regulation of two distinct haem-uptake systems, phu and has, in Pseudomonas aeruginosa. Microbiol Read Engl. 2000; 146 (Pt 1): 185-98.
-
35Ramsey DM, Wozniak DJ. Understanding the control of Pseudomonas aeruginosa alginate synthesis and the prospects for management of chronic infections in cystic fibrosis. Mol Microbiol. 2005; 56(2): 309-22.
-
36Csermely P. The wisdom of networks: a general adaptation and learning mechanism of complex systems. The network core triggers fast responses to known stimuli; innovations require the slow network periphery and are encoded by core-remodeling. BioEssays News Rev Mol Cell Dev Biol. 2018; 40(1).
-
Financial support: Inova-Fiocruz (grant VPPCB-007-FIO-18-2-29), Faperj, Capes
ERRATUM
-
0
In the article “Gene regulatory network inference and analysis of multidrug-resistant Pseudomonas aeruginosa”, DOI number: 10.1590/0074-02760190105, published in Mem Inst Oswaldo Cruz, Rio de Janeiro, Vol. 114: e190105, 2019: On page 7, Table I, where it reads: “PA4851_19380” It should read: “AL347_05155” On page 7, where it reads: “After pinpointing the hubs, an analysis was performed to determine whether they are interconnected (through direct or indirect interactions). Only two hubs were found to not be interconnected, np20 and PA4851_19380 (homologous to PA1520).” It should read: “After pinpointing the hubs, an analysis was performed to determine whether they are interconnected (through direct or indirect interactions). Only two hubs were found to not be interconnected, np20 and AL347_05155 (homologous to PA1520).” On page 10, where it reads: “In fact, they are related to processes such as zinc uptake (np20) and purine metabolism (PA4851_19380), which are fundamental to bacterial survival, but can be considered somewhat independent of other processes, and are only triggered under specific conditions.” It should read: “In fact, they are related to processes such as zinc uptake (np20) and purine metabolism (AL347_05155), which are fundamental to bacterial survival, but can be considered somewhat independent of other processes, and are only triggered under specific conditions.” On page 10, where it reads: “Table II shows a comparison of network statistics between the CCBH4851 GRN and the GRN network published by Galán-Vásquez et al. (12) One clear trend is that the CCBH4851 GRN represents a substantial improvement in terms of network completeness, since is includes more nodes, edges and network motifs.” It should read: “Table II shows a comparison of network statistics between the CCBH4851 GRN and the GRN network published by Galán-Vásquez et al. (12) One clear trend is that the CCBH4851 GRN represents a substantial improvement in terms of network completeness, since it includes more nodes, edges and network motifs.” On page 4,Fig. 2should be replaced by the figure below:On page 8,Fig. 4should be replaced by the figure below:Fig. 2:
visualisation of the Pseudomonas aeruginosa CCBH4851 gene regulatory network (GRN). Yellow circles indicate regulatory genes, light blue circles indicate target genes (TGs), black lines indicate an unknown mode of regulation, green lines indicate activation, red lines indicate repression and grey lines indicate a dual mode of regulation. A: the GRN large highly connected network component; B: all regulatory and TGs that have no connections with the component depicted in A; C-E: clusters of lower connectivity compared to the component depicted in A. All figures are presented with higher resolution in the Supplementary data.
The Supplementary material were corrected and attached in the link below:https://memorias.ioc.fiocruz.br/images/revistas/2020/115/0105er_sd.xlsxFig. 4:
connectivity relationships among the 30 most influential hubs of the Pseudomonas aeruginosa CCBH4851 gene regulatory network. Yellow circles indicate regulatory genes considered hubs, light blue circles indicate target genes, black lines indicate an unknown mode of regulation, green lines indicate activation, red lines indicate repression and grey lines indicate a dual mode of regulation.
Publication Dates
-
Publication in this collection
05 Aug 2019 -
Date of issue
2019
History
-
Received
26 Mar 2019 -
Accepted
26 June 2019