Resumo
It has been carried out an investigation of ethanol electro-oxidation on Ti/IrO2 electrodes. The experimental results show a high selectivity towards acetaldehyde formation thus, offering potential advantages in cost and availability of raw material. It has been observed that the electrode is partially blocked by a film formed after the oxidation of the starting material which can be removed by pulse technique between RDO and RDH onset. The mechanism and the selectivity of the product formed is presented.
ethanol; electro-oxidation; iridium oxide
ethanol; electro-oxidation; iridium oxide
ARTIGO
Eletroxidação do etanol em eletrodos de Ti/IrO2
Carlos H.V. Fidelis, Paulo M. Donate e Adalgisa R. de Andrade*
Departamento de Química, Faculdade de Filosofia, Ciências e Letras de Ribeirão Preto, Universidade de São Paulo, 14040-901 Ribeirão Preto - SP
*e-mail: ardandra@ffclrp.usp.br
Recebido em 7/12/99; aceito em 15/6/00
Electro-oxidation of ethanol in Ti/IrO2. It has been carried out an investigation of ethanol electro-oxidation on Ti/IrO2 electrodes. The experimental results show a high selectivity towards acetaldehyde formation thus, offering potential advantages in cost and availability of raw material. It has been observed that the electrode is partially blocked by a film formed after the oxidation of the starting material which can be removed by pulse technique between RDO and RDH onset. The mechanism and the selectivity of the product formed is presented.
Keywords: ethanol; electro-oxidation; iridium oxide.
INTRODUÇÃO
O emprego de catalisadores para a transformação, economicamente viável, de produtos simples em outros de maior valor agregado, é uma busca freqüente nos mais diversos campos da química1,2. Um problema enfrentado nas sínteses em escala industrial é a coexistência do catalisador e do produto final na mistura reacional, exigindo assim a inclusão de uma etapa de separação. Fixar o catalisador na superfície de um eletrodo é uma das vantagens da indústria eletroquímica, uma vez que diminui o número de etapas de separação.
Na eletrossíntese orgânica o produto final é altamente dependente do material do ânodo e das condições experimentais empregadas, isto é, densidade de corrente aplicada, o tempo de eletrólise, solvente, acidez do meio, etc. A comparação direta dos resultados das eletrólises não é simples, no caso da a oxidação do etanol em eletrodos de Ti/RuO2 por exemplo, são relatados os seguintes produtos: em meio básico o aldol3, em meio ácido uma mistura contendo ácido acético e CO24. Recentemente, sob condições de eletrólise a potencial controlado obtivemos somente o acetaldeido5. Os DSAâ constituem de um suporte de titânio onde é depositado, por decomposição térmica, uma mistura apropriada de óxido [normalmente constituída de um óxido eletroquimicamente ativo (RuO2) e um óxido estabilizante (TiO2), cuja função principal é melhorar as propriedades mecânicas da mistura6]. Modificações na composição original têm sido investigadas visando as mais diferentes aplicações, tais como: eletrólise da água7, capacitores8 e aplicação em eletroquímica orgânica9. A utilização de ânodos de óxidos de metais nobres para realizar a oxidação de materiais orgânicos apresenta um crescimento constante nos últimos anos. Duas sub-divisões podem ser identificadas: (i) mineralização completa de substâncias orgânicas potencialmente tóxicas visando a aplicação em química ambiental10. Nesta área, o objetivo principal é a procura de materiais eletródicos resistentes à corrosão, que promovam a combustão completa e/ou quebrem as ligações químicas dessas substâncias, formando moléculas mais simples que posteriormente poderão ser degradadas por rotas já conhecidas11; (ii) eletrossíntese, onde aproveita-se de propriedades, tais como: o fácil preparo de misturas com características específicas para a reação de interesse, a utilização de O2 e Cl2 como oxidantes.
O etanol geralmente é utilizado como molécula modelo para a oxidação de álcoois saturados, pois, a sua estrutura simples e completamente solúvel em água, facilita a comparação dos resultados obtidos com outros materiais. Além disto, o Brasil detém um setor sucro-alcooleiro de grande dimensão e a investigação da transformação de etanol em produtos com maior valor agregado torna-se, portanto, bastante interessante. Neste contexto, temos investigado sistematicamente em nosso laboratório a oxidação do etanol utilizando-se eletrodos na forma de óxidos5. O objetivo deste trabalho foi estudar a oxidação do etanol em eletrodos de composição nominal Ti/IrO2, para verificar a seletividade da eletrossíntese em função do material empregado. O óxido de irídio apesar de apresentar menor atividade para a reação de oxigênio (maior sobrepotencial e maior coeficiente de Tafel, 60 mV contra 40 mV do óxido de rutênio) é apontado como um eventual substituto do óxido de rutênio por ser muito mais estável sob condições drásticas de eletrólise (altos sobrepotenciais e meio ácido). Portanto, a idéia de utilizar eletrodos de Ti/IrO2 em eletrólise de substâncias orgânicas está baseada na expectativa de se encontrar eletrodos com maior durabilidade, apesar do menor efeito catalítico. Neste trabalho, procurou-se estudar a relação entre o tipo de material eletródico, a adsorsão de intermediários na superfície dos óxidos e a razão dos produtos finais obtidos.
PARTE EXPERIMENTAL
Os eletrodos utilizados na oxidação do etanol foram preparados por decomposição térmica de 0,2 mol/L de IrCl3.3H2O (ALDRICH) dissolvido em HCl 1:1 (v/v). A solução percursora foi pincelada na superfície do titânio, previamente tratado conforme descrito na literatura12. Neste estudo utilizou-se as seguintes configurações de eletrodos: (a) placa de titânio (ALDRICH) de 10 x 10 x 0,127 mm para os estudos de voltametria cíclica, (b) disco de titânio (FAENQUIL-DEMAR) de 0,78 cm2 de diâmetro e 2 cm de altura para os estudos de eletrodo de disco rotatório (EDR), (c) rede de titânio (TRIKEM) de 300 x 350 x 0,5 mm (rede industrial cuja cobertura original foi removida com H2SO4 concentrado a quente) para as eletrólises.
Após a aplicação da solução percursora, evaporou-se o solvente com um jato de ar quente e calcinou-se o eletrodo a 400 ou 450 oC durante 5 minutos, sob um fluxo de oxigênio de 5 L/min. Ao atingir a massa equivalente a espessura de 2 µm o eletrodo foi calcinado por mais 60 minutos. A montagem dos eletrodos de placa seguiu procedimento análogo ao descrito anteriormente13. Os eletrodos de disco (Figura 1) foram usinados no próprio laboratório e adaptados ao rotor AFMSRX da Pine Instruments Company.
Utilizou-se produtos químicos de pureza analítica (Merck) sem nenhuma purificação prévia. A água utilizada era deionizada (equipamento Mili-Q plus da Millipore). Todos os estudos foram realizados usando uma solução 1,0 mol/L de HClO4 como eletrólito de suporte. As soluções foram desaeradas com nitrogênio durante os experimentos.
Para os estudos de voltametria cíclica utilizou-se uma célula de três compartimentos onde, no compartimento central foram introduzidos os eletrodos de trabalho e de referência (eletrodo calomelano saturado de sódio, ECSS) e nas laterais foram introduzidas duas espirais de platina platinizadas como eletrodos auxiliares. Quando a concentração de etanol era a variável experimental investigada esta foi mantida constante na faixa de 0,05 - 0,6 mol/L.
Os experimentos eletroquímicos foram realizados em um potenciostato EG&G PAR modelo 273 comandado pelo software M270. As eletrólises foram realizadas no potenciostato/galvanostato 173 da EG&G PAR. A célula de eletrólise, sem separação entre ânodo e cátodo, era termostatizada (30 oC) e continha um volume total de 150 ml de solução.
Após a eletrólise a mistura reacional foi neutralizada com uma solução 2,5 mol/L de KOH, filtrada e os produtos formados analisados em um equipamento de Cromatografia gasosa da HP-5890 II acoplado com um espectrômetro de massas HP-5988-A. Duas colunas de sílica fundida foram utilizadas: HP-FFAP (25 m x 0,32 mm x 0,25 µm) e HP-5 (25 m x 0,32 mm x 0,52 µm). A temperatura da coluna foi programada para ser elevada de 60 oC até 220 oC a uma taxa de 10 oC/min., utilizando-se He como gás de arraste.
RESULTADOS E DISCUSSÃO
Voltametria Cíclica na Ausência de Etanol
A Figura 2 mostra voltamogramas cíclicos representativos do eletrodo Ti/IrO2 em 1,0 mol/L de HClO4, preparados em duas temperaturas de calcinação diferentes. Os sítios ativos na superfície do eletrodo sofrem transições redox [Ir(III)/Ir(IV)] cuja carga é superior ao carregamento da dupla camada elétrica. Assim, a medida da carga fornece uma indicação da área eletroquimicamente ativa da superfície do eletrodo14. A análise da Figura 2 mostra que a área eletroquimicamente ativa obtida para os eletrodos de Ti/IrO2 preparados a 400 oC é aproximadamente o dobro da área obtida para os eletrodos preparados a 450 oC. Verifica-se que um aumento de 50 oC na temperatura de calcinação provoca uma diminuição significativa da área do eletrodo. Este comportamento pode ser explicado pelo aumento do grau de sinterização da superfície do eletrodo à medida que se aumenta a temperatura de calcinação e, conseqüentemente, o número de sítios ativos diminui com o aumento da temperatura.
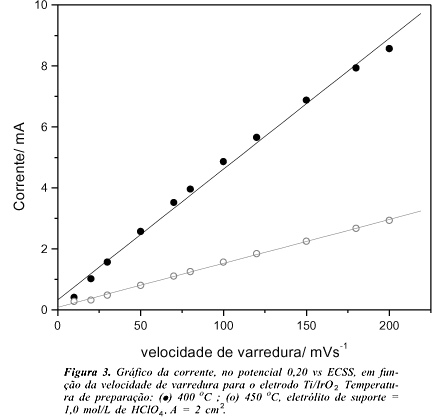
Um outro estudo que vem corroborar os resultados acima é a medida da rugosidade aparente dos eletrodos, obtida experimentalmente através do estudo de voltametria cíclica em várias velocidades de varredura (n < 200 mV s-1). A tangente da curva de corrente vs. velocidade de varredura (Figura 3) fornece a capacitância experimental, C. O potencial escolhido para a medida da corrente foi de 0,20 V vs ECSS. Embora se verifique a existência de bandas largas e mal definidas, a linearidade apresentada pela figura 3, é uma boa indicação de que no potencial escolhido a corrente é controlada pelo processo capacitivo. Assumindo-se que a capacitância teórica, C*, do eletrodo planar de IrO2 seja igual ao proposto para o RuO2 (0,080 mF cm-2)15 os fatores de rugosidade aparente, C/C*, obtidos foram de 540 e 180, respectivamente para eletrodos preparados a 400 e 450 oC.
Além de possuírem áreas eletroquímicamente ativa superiores, os eletrodos preparados a 400 oC apresentaram uma melhor definição das transições de estado sólido do par redox Ir(III)/Ir(IV)16, banda situada em 0,65 V vs ECSS. No entanto, estes eletrodos se mostraram instáveis, sofrendo acentuada perda de massa de óxido (> 50 %), durante os registros dos voltamogramas cíclicos no eletrólito de suporte ou ainda quando estocados em água por alguns dias. Esse comportamento inviabilizou os estudos com os eletrodos preparados a 400 oC na presença de material orgânico, visto ser necessário utilizar condições drásticas para remover o material orgânico adsorvido na superfície do eletrodo (ver discussão posterior). Portanto, em todos os estudos realizados na presença de material orgânico optou-se por utilizar os eletrodos preparados em 450 oC.
Voltametria Cíclica na Presença de Etanol
O voltamograma cíclico do eletrodo Ti/IrO2, em 1,0 mol/L de HClO4, na presença de etanol não apresenta nenhum pico anódico (Figura 4). Observa-se apenas um deslocamento da corrente, na direção catódica, em potenciais próximos a 1,1V vs. ECSS, região na qual a corrente de oxidação provavelmente é composta por Iet + IO2. Comparando-se com a oxidação de etanol em eletrodos de rutênio5, onde observou-se um pico bem definido situado em potenciais menos negativo que a RDO, pode-se afirmar que os eletrodos de óxido de rutênio são catalisadores mais eficientes para a oxidação de substratos orgânicos. No caso do óxido de irídio, verifica-se que a oxidação do etanol ocorre em sobrepotenciais maiores. Estudos prévios, realizados em nosso laboratório com outros substratos orgânicos16, mostraram resultados semelhantes, ou seja, que os eletrodos contendo óxido de irídio são menos eficientes para a oxidação de material orgânico do que os eletrodos contendo óxido de rutênio.
A reação de desprendimento de oxigênio, RDO, em eletrodo de Ti/IrO2 foi estudada por vários autores17,18 e pode ser simplificada através das seguintes etapas:
(2)
onde M é o sítio superficial ativo.
Visto que os resultados experimentais demonstram que a oxidação do etanol é simultânea com a RDO, pode-se concluir que na presença do etanol, além das etapas 1 e 2 especificadas acima, ocorre também a reação entre o sítio ativo do metal, MOx+1, com a molécula do álcool, através de um mecanismo similar ao proposto recentemente para o eletrodo de rutênio5 (etapa 3).
(3)
Na presença de etanol, os eletrodos de Ti/IrO2, após voltametria cíclica repetitiva do potencial entre 0,2 V e 1,3 V vs. ECSS, mostraram sinais de desativação, isto é, a corrente anódica cai acentuadamente à medida em que se realiza consecutivos ciclos de potencial. Após 5 ciclos observa-se uma perfeita concordância com os voltamogramas representativos do eletrólito de suporte. Este comportamento é idêntico ao apresentado para os eletrodos de óxido de rutênio5 e pode ser atribuído à formação de um filme insolúvel que bloqueia os sítios ativos da superfície do eletrodo. A formação desse filme ocorre após a transferência eletrônica, provavelmente devido à polimerização do acetaldeído formado como produto da oxidação do etanol. Para efetuar a remoção desse material polimérico da superfície, foi necessário retirar o eletrodo, colocando-o em um béquer contendo acetonitrila e mantendo-o sob agitação durante 10 minutos. Posteriormente, o eletrodo foi colocado em uma célula eletroquímica, contendo somente o eletrólito de suporte, e foi aplicado um programa de perturbação potenciostática por aproximadamente 10 minutos (E1 = 2,0 V, t = 4 s e E2 = -0,4 V, t = 2 s). Visto que, a reprodutibilidade do sistema é função da superfície ativa do eletrodo e esta se mostra bastante sensível e dependente da completa remoção do filme polimérico formado, todos os resultados apresentados foram obtidos em duplicata, adotando-se o procedimento de limpeza de pulso após o registro de cada voltamograma.
Para verificar o comportamento voltamétrico em função da concentração de etanol mediu-se a corrente, em um potencial fixo (E = 1,17 V), escolhido com o intuito de evitar a reação de desprendimento de oxigênio. Dessa forma, há uma maior probabilidade da corrente medida ser controlada somente pela reação de oxidação do etanol. Apesar dessa aproximação realizada, o gráfico de log I vs log Cet (figura 5) é linear. Portanto, em princípio, pode-se concluir que a eletroxidação de etanol é um processo controlado por difusão. A ordem da reação, obtida pelo coeficiente angular, é de 0,4. Ordens de reação fracionárias são freqüentes em eletrodos de óxidos metálicos e refletem a complexidade do mecanismo de oxidaçãoem função da heterogeneidade dos sítios ativos do eletrodo19.
Eletrólise
Experimentos de eletrólise a potencial controlado também foram realizados, utilizando um eletrodo de rede recoberto com IrO2, em uma célula termostatizada (30 oC). O potencial aplicado ao eletrodo de trabalho foi, continuamente, pulsado entre 1,4 V (10 s) e ¾ 0,6 (0,1 s) V vs. ECSS. A eletrólise era periodicamente interrompida e registrava-se voltamogramas cíclicos em um eletrodo especialmente colocado para este fim. O progresso da eletrólise é apresentado na Figura 6, onde pode ser verificado que a corrente diminui acentuadamente à medida que o reagente é consumido. Utilizando-se no sistema, um "trap" com KOH ou através das análises de cromatografia gasosa, não foi detectado a formação de CO2 durante a eletrólise. A análise do produto de reação por CG-MS mostrou a presença de um único pico, de massa molecular 44, correspondendo ao acetaldeído. Estes resultados demonstram que a oxidação do etanol, utilizando eletrodos de óxido de irídio é mais branda e portanto mais seletiva do que a oxidação em eletrodo de irídio metálico20, onde obtêm-se uma mistura contendo acetaldeído e ácido acético. O resultado obtido para o eletrodo de Ti/IrO2 é idêntico ao apresentado pelo eletrodo de RuO2 e está de acordo com o que foi sugerido por Comninellis e De Battisti21, que mostraram que eletrodos contendo óxido de irídio e/ou rutênio apresentam condições mais brandas de eletroxidação e, consequentemente, são bons materiais para realizar eletrossíntese orgânica, uma vez que uma maior seletividade pode ser obtida dependendo das condições experimentais utilizadas.
A obtenção do acetaldeído como único produto da reação (produto da transferência de 2 elétrons) já é, por si só, um indicativo da boa seletividade obtida, uma vez que a oxidação de álcoois primários a aldeídos é um processo bastante difícil de ser realizado em meio homogêneo, tendo em vista a posterior oxidação do aldeído ao ácido carboxílico correspondente22.
CONCLUSÕES
A eletroxidação de etanol utilizando eletrodos de Ti/IrO2 em meio ácido (1,0 mol/L de HClO4) apresentou alta seletividade, mostrando que, quando se escolhe condições brandas pode-se obter facilmente o produto intermediário da eletroxidação, o acetaldeído.
Um bloqueio da superfície do eletrodo, semelhante ao observado anteriormente para o eletrodo de Ti/RuO25,foi também observado para o eletrodo de Ti/IrO2. Uma vez que o bloqueio da superfície eletródica independe do óxido catalítico empregado pode-se inferir que a afinidade química entre os sítios ativos dos diferentes óxidos investigados (RuO2 e IrO2) e substância adsorvida não é o único fator responsável pela adsorsão.
A eletrooxidação realizada em eletrodo de óxido de irídio ocorre simultaneamente à reação de desprendimento de oxigênio e pode ser representada pelas etapas [1] ¾ [3] descritas previamente. Essa eletroxidação mostrou-se bem mais seletiva que a apresentada na literatura para irídio metálico20.
AGRADECIMENTOS
Os autores agradecem a FAPESP pelo apoio financeiro. C.H.V. de Fidellis agradece a bolsa IC-FAPESP (Processo 97/00339-3).
- 1. Couper, A. M.; Walsh, F. C.; Pletcher, D.; Chem. Rev. 1990, 90837.
- 2. Torii, S.; "Electroorganic Synteses", Kodansha Ltd., Tokio, 1985.
- 3. Taconi, N. R.; Lezna, R. O.; Beden, B.; Hahn, F.; Lamy, C.; J. Electrocanal. Chem. 1994, 379, 329.
- 4. Asokan, K.; Krishnan, V. B.; Electrochem 1988, 4, 827.
- 5. De Andrade, A. R.; Donate, P. M.; Alves, P. P. D.; Boodts, J. F. C.; Fidellis, C. H. V.; J. Electrochem. Soc. 1998, 145, 3839.
- 6. Trasatti, S.; Ed.; "Electrodes of Conductive Metallic Oxides", Elsevier, Amsterdam, Partes A e B, 1980 e 1981.
- 7. Wen, T-C.; Hu, C-C.; J. Electrochem. Soc. 1992, 139, 2158.
- 8. Lin, C.; Ritter, J. A.; Popov, B. N.; J. Electrochem. Soc. 1999, 146, 3155.
- 9. Cox, P.; Pletcher, D.; J. Appl. Electrochem. 1991, 21, 11.
- 10. Comninellis, C.; Electrochimica Acta 1994, 49, 1857.
- 11. Dong, R.; Wu, S.; Wat. Res. 1996, 30, 577.
- 12. Angelinetta, C.; Trasatti, S.; Atanasoska, Lj. D.; Atanasoski, R. T.; J. Electroanal. Chem 1986, 214, 535.
- 13. Garavaglia, R.; Mari, C. M.; Trasatti, S.; Surf. Technol 1984, 23, 41C.
- 14. Trasatti, S.; Petrii, O. A.; Pure Appl. Chem. 1991, 63, 711.
- 15. Da Silva, L. A.; Alves, V. A.; Da Silva, M. A. P.; Trasatti, S.; Boodts, J. F. C.; Can. J. Chem. 1997, 75, 1483.
- 16. Zanta, C. L. P. S.; De Andrade, A. R.; Boodts, J. F. C.; J. Appl. Electrochem. 2000, 30, 467.
- 17. Trasatti, S.; Electrochemical Hydrogen Tecnologies, Wendt, H., Ed., Elsevier, Amsterdam, 1990, p. 104.
- 18. Damjanovic, A.; Wong, M. K. Y.; J. Electrochem. Soc. 1967, 114, 596.
- 19. Anastasijevic, N. A.; Dimitrijevic, Z. M.; Adzic, R. R.; J. Electroanal. Chem 1986, 199, 251.
- 20. Tacconi, N. R.; Lezna, R. O.; Beden, B.; Hahn, F.; Lamy, C.; J. Electroanal. Chem. 1994, 379, 329.
- 21. Conminellis, C.; De Battisti; J. Chem. Phys. 1996, 93, 673.
- 22. House, H.O., Modern Synthetic Reactions, 2a Ediçăo, W. A. Benjamin, Inc., Menlo Park, 1972, p. 257-422.
Datas de Publicação
-
Publicação nesta coleção
09 Ago 2001 -
Data do Fascículo
Fev 2001
Histórico
-
Aceito
15 Jun 2000 -
Recebido
07 Dez 1999