Resumo
This paper discuss the consequences of the equipartition principle when used to calculate the heat capacity of atoms and molecules, a discussion that appeared at the end of XIX century and beginning of the XX century. Classical molecular thermodynamics prediction of the heat capacity is introduced, followed by a presentation of the degrees of freedom of a system. The historical discussion that appeared at the time, by Dulong, Petit, Maxwell, Boltzmann, Rayleigh and Kelvin is discussed afterwards. The necessity of a new theory is also presented as a direct consequence of the equipartition principle collapse.
equipartition principle; degree of freedom; heat capacity
equipartition principle; degree of freedom; heat capacity
Educação
COLAPSO DA EQUIPARTIÇÃO DA ENERGIA
João Pedro Braga
Departamento de Química, ICEx, Universidade Federal de Minas Gerais, 31270-901 Belo Horizonte - MG
Recebido em 5/5/00; aceito em 19/2/01
THE COLLAPSE OF THE EQUIPARTITION PRINCIPLE. This paper discuss the consequences of the equipartition principle when used to calculate the heat capacity of atoms and molecules, a discussion that appeared at the end of XIX century and beginning of the XX century. Classical molecular thermodynamics prediction of the heat capacity is introduced, followed by a presentation of the degrees of freedom of a system. The historical discussion that appeared at the time, by Dulong, Petit, Maxwell, Boltzmann, Rayleigh and Kelvin is discussed afterwards. The necessity of a new theory is also presented as a direct consequence of the equipartition principle collapse.
Keywords: equipartition principle; degree of freedom; heat capacity.
INTRODUÇÃO
Ao calcularmos valores médios da energia cinética e da energia potencial, dentro de um contexto da termodinâmica molecular clássica, sempre usamos o princípio da equipartição da energia. Este princípio nos diz que a cada grau de liberdade translacional, vibracional ou rotacional deve ser associado o valor kT, onde k é a constante de Boltzmann e T a temperatura. A energia interna e consequentemente a capacidade calorífica do sistema em estudo podem ser facilmente calculadas com o uso direto deste princípio. Entretanto esta simplicidade nem sempre agradou aos cientistas que trabalhavam com termodinâmica. Este trabalho discute os pontos de vista de alguns cientistas, em torno de 1850, quando se deram conta de que havia alguma coisa de muito errado com o princípio da equipartição da energia. Uma discussão sobre o enfraquecimento deste princípio, ou seja, do colapso da equipartição da energia, e de suas conseqüências, teóricas, históricas e didáticas, é o principal objetivo deste trabalho.
O PRINCÍPIO DA EQUIPARTIÇÃO DA ENERGIA
O valor médio de uma função f(p), onde p é o momento da partícula, pode ser calculado na termodinâmica molecular como1,

com Ecsendo a energia cinética da partícula, b = 1/kT e dtpo elemento de volume no espaço dos momentos. Analogamente o valor médio de uma função f(R), onde R é a coordenada da partícula, é dado por,

onde U(R) é a energia potencial e dV o elemento de volume no espaço das coordenadas.
O cálculo destes dois valores médios assumem, implicitamente, que o número de estados acessíveis para o sistema é muito grande e adicionalmente a validade da aplicação da distribuição de Boltzmann. A validade, e uma simulação da distribuição de Boltzmann aparece, de forma didática, na referência [2]. O cálculo destes valores médios assumem também que a energia total, E, do sistema de N partículas, pode ser escrita como uma soma da energia cinética e da energia potencial.
Fazendo a =  teremos para o valor médio da energia cinética,
teremos para o valor médio da energia cinética,

Portanto o valor médio da energia cinética é simplesmente 1/2kT, um fato muito importante pois este valor médio não dependeu da massa do sistema em estudo. Em outras palavras, este valor médio é independente do sistema em questão. Átomos leves ou átomos pesados têm, dentro do contexto do princípio da equipartição da energia, a mesma energia cinética média.
O princípio que estabelece 1/2kT por grau de liberdade (acima fizemos a integral em uma dimensão, ou seja, um grau de liberdade) é conhecido como o princípio da equipartição da energia.
O cálculo da energia potencial média é feita de forma análoga. Tratando novamente o problema em uma dimensão e assumindo que o potencial é do tipo harmônico, isto é, U(R)=1/2 kfR2onde kfé a constante de força do sistema, podemos também calcular a energia potencial média, integrando sob o espaço das coordenadas. Entretanto esta integração é desnecessária se percebemos que o cálculo será semelhante. Para ver isto basta que associemos kfcom 1/m. Pelo mesmo motivo que o valor médio da energia cinética não dependeu da massa, o valor médio da energia potencial não irá também depender da constante de força. Portanto o valor médio de potenciais harmônicos é dado por,

Aqui também deve ser notado que o valor médio do potencial não leva em conta qualquer sistema em particular pois o resultado não dependeu da constante de força que é característica de cada sistema. Sistemas ligados por potenciais harmônicos com interações fortes (kfgrande) ou com interações fracas (kfpequeno) têm, dentro do princípio acima, a mesma energia potencial média.
GRAUS DE LIBERDADE E A EQUIPARTIÇÃO DA ENERGIA
Usando o princípio da equipartição da energia podemos calcular a energia total média do sistema,  . Para este cálculo precisamos saber o número de graus de liberdade do sistema em estudo. No que se segue o sistema será considerado ideal, isto é, o potencial intermolecular, Uinter, assumirá o valor igual a zero. Uma partícula ao mover-se em três dimensões terá, nestas condições, a energia total igual à energia translacional e podendo ser escrita como,
. Para este cálculo precisamos saber o número de graus de liberdade do sistema em estudo. No que se segue o sistema será considerado ideal, isto é, o potencial intermolecular, Uinter, assumirá o valor igual a zero. Uma partícula ao mover-se em três dimensões terá, nestas condições, a energia total igual à energia translacional e podendo ser escrita como,

donde, usando o princípio da equipartição, podemos calcular,

Portanto o número de graus de liberdade de um sistema de p átomos é igual a 3p.
O próximo grau de liberdade a ser considerado é o rotacional. A energia cinética rotacional de uma molécula não linear pode ser expressa por,

onde Lx, Lye Lzsão os momentos angulares nos eixos x, y e z com Ixx, Iyye Izzos seus respectivos momentos de inércia. A energia cinética acima, sendo da mesma forma que (5), terá também média igual a,

por molécula. Entretanto deve ser notado que se a molécula é linear o momento angular ao longo do eixo que contém os átomos é desprezível. Supondo que este eixo é o eixo z teremos para a energia rotacional,

e consequentemente,

por molécula linear. Ao considerarmos rotações em sistemas moleculares precisamos, portanto, distinguir entre moléculas não lineares e lineares.
Resta-nos considerar o grau de liberdade vibracional. Isto pode ser facilmente calculado pois do total de 3p graus de liberdade, 3 pertencem à rotação de moléculas não lineares e 2 à moléculas lineares. Portanto temos, por ora, (3p-3) ou (3p-2) graus de liberdade que podem pertencer à vibração. Entretanto precisamos retirar mais três graus de liberdade das expressões acima, que correspondem às translações da molécula como um todo. Ficamos então com (3p-6) graus de liberdade vibracional para a molécula não linear e (3p-5) graus de liberdade vibracional para a molécula linear.
Para ilustrar como o princípio da equipartição se aplica ao caso vibracional consideremos novamente o caso unidimensional. A energia vibracional neste caso será,

donde,

onde usamos os resultados obtidos em (3) e (4). Este resultado ilustra que cada grau de liberdade vibracional deve ser computado como kT, pois a vibração envolve a energia cinética e potencial.
CAPACIDADES CALORÍFICAS
Com o resultado do último item temos para a média da energia intramolecular,

para moléculas lineares, e
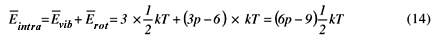
para moléculas não lineares. Se desejamos o resultado para N moléculas basta que multipliquemos os valores acima por este número.
Até agora só levamos em conta propriedades individuais de cada sub-sistema, no caso, átomos e moléculas, do sistema total em questão. Se queremos as propriedades do sistema como um todo temos também que levar em conta os graus de liberdade do centro de massa deste sistema. Para isto basta que adicionemos NkT aos valores encontrados acima. Este valor adicionado corresponde também à energia translacional média de sistemas atômicos. Portanto a energia média total, isto é, a energia interna, pode ser escrita na forma,

As correções da não idealidade podem ser feitas usando-se a expansão do virial, como por exemplo, nas referências [1] e [3].
Para o cálculo da capacidade calorífica a volume constante, CV, basta que tomemos a derivada primeira das energias acima,
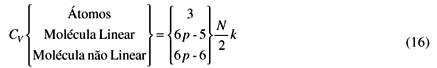
No que se segue usaremos o símbolo f para designar as três possibilidades acima, isto é,
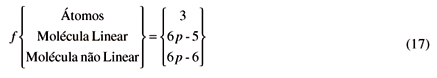
Note também que a capacidade calorífica à pressão constante, Cp, para forças intermoleculares iguais a zero, é igual a CP =  NkT.
NkT.
A LEI DE DULONG E PETIT
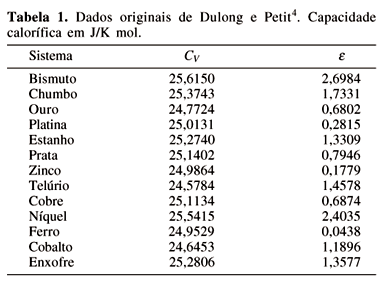
Baseando em medidas experimentais de capacidades caloríficas de treze substâncias, Petit e Dulong publicavam, em 1819, o seguinte resultado4,
Uma análise simples destes números nos permite perceber a relação que é tão formidável na sua simplicidade que nos leva a reconhecer de uma vez a existência de uma lei física, que pode ser extendida para todas substâncias elementares.
A tabela original apresentada no artigo de Dulong e Petit é reproduzida na Tabela 1, com os dados convertidos para J/K mol. Dulong e Petit usaram a massa molar normalizada para a massa molar do oxigênio. O valor de 16 g/mol foi usado para remover esta normalização conforme resultados apresentados na tabela abaixo. O erro percentual relativo, e =  x 100 , é também apresentado, sendo o erro percentual relativo médio, obtido desta tabela, de 1,1415. Devido a diversidade dos dados analisados e o baixo desvio apresentado, a conclusão de Petit e Dulong é bastante justificável. Apesar de estabelecerem uma lei o artigo acima termina com uma incerteza se este seria realmente o caso. Em homenagem a estes dois cientistas, o valor constante da capacidade calorífica, 3R por mol, para substâncias sólidas é conhecido como a lei de Dulong e Petit. A explicação teórica desta lei iria aparecer mais tarde.
x 100 , é também apresentado, sendo o erro percentual relativo médio, obtido desta tabela, de 1,1415. Devido a diversidade dos dados analisados e o baixo desvio apresentado, a conclusão de Petit e Dulong é bastante justificável. Apesar de estabelecerem uma lei o artigo acima termina com uma incerteza se este seria realmente o caso. Em homenagem a estes dois cientistas, o valor constante da capacidade calorífica, 3R por mol, para substâncias sólidas é conhecido como a lei de Dulong e Petit. A explicação teórica desta lei iria aparecer mais tarde.
UMA AULA DE MAXWELL
A explicação teórica da lei de Dulong e Petit teria que esperar um pouco. Nesta época existia muito pouco conhecimento sobre a estrutura interna de moléculas e sólidos. A interação intramolecular ainda não tinha sido levada em consideração e portanto não era conhecido o fato de que potenciais harmônicos teriam também valor médio igual ao valor da energia cinética. Apesar disto a necessidade de se levar em conta forças intramoleculares já era bem evidente.
Maxwell, numa aula na Chemical Society, em 18 de Fevereiro de 1875, discute teoricamente, entre outras coisas, o cálculo da capacidade calorífica5. Neste artigo fica bem claro que ele sabia da necessidade da inclusão da interação intramolecular apesar de, em nenhum ponto, mostrar o cálculo deste valor. Mas é impressionante a visão que tinha do problema, e qual direção deveria ser tomada. Maxwell simplesmente definiu os graus de liberdade de um sistema como (n+e), isto é, dois termos definindo e como,
onde e é uma quantidade positiva de valor desconhecido dependendo da lei da força que une os constituintes da molécula.
Isto equivale exatamente ao valor de (n+e) = f onde f foi deduzido anteriormente, equação (17). Maxwell prossegue deduzindo que CVteria que ser proporcional a (n+e) e que Cpteria também que ser proporcional a (n+e+2). Note que isto equivale precisamente ao raciocínio dado anteriormente.
Neste ponto Maxwell começa a comparar estes resultados teóricos com os resultados experimentais de CVda época donde conclui,
Mas se nós tomarmos os valores experimentais de capacidades caloríficas como encontrado por Regnault e compará-los com esta fórmula, achamos que n+e para o ar e vários outros gases não pode ser maior de 4,9.
Portanto Maxwell, ao tomar dados experimentais, verifica que o valor de f não podia ser maior do que 4,9 para o ar. Sabemos que o valor de f, fazendo o modelo de moléculas diatômicas, é igual a 7. O ponto para o qual Maxwell chama a atenção é bastante claro: a teoria estava em desacordo com os dados experimentais. Maxwell sabia da gravidade do problema, o que é bem claro na frase,
E aqui estamos face a face com a maior dificuldade que a teoria molecular jamais encontrou... .
ou ainda, mais abaixo no mesmo artigo, ao enfatizar o seu ponto de vista
Eu agora coloquei perante vós o que eu considero ser a maior dificuldade jamais encontrada pela teoria molecular.
Alguma coisa não estava correta com a teoria cinética. A estória do colapso da equipartição passa agora necessariamente por Boltzmann.
A EXPLICAÇÃO TEÓRICA DE BOLTZMANN PARA ÁTOMOS E MOLÉCULAS
No seu livro de teoria dos gases6, Boltzmann também fornece a sua explicação sobre os valores experimentais de capacidades caloríficas. Em algumas passagens do livro prefere discutir estes pontos referindo-se à razão das capacidades caloríficas à pressão e volume constante, que Boltzmann chamou de k. Dentro do contexto apresentado aqui esta quantidade equivale a(f+2)/f.
Considerando então o potencial intramolecular igual a zero e para sistemas monoatômicos Boltzmann obtém k=1, ou seja, o valor correto, pois neste caso f=3. Continua a discussão, agora falando de moléculas, mas supondo que a molécula seria rígida, com simetria esférica e ainda sem interação intramolecular. Não é nenhuma surpresa que tenha obtido o mesmo resultado do caso atômico. Boltzmann passa então a considerar uma molécula linear, como um corpo rígido, obtendo k = 1,4, um resultado novamente correto. Para este caso, sem estrutura interna, temos 2 graus de liberdade rotacional e três translacional, resultando em k =7/5.
No próximo exemplo tratado no livro, Boltzmann percebe que teria que levar em conta as forças intramoleculares. Tratando este potencial como harmônico, prova que o seu valor médio vale também kT, e usa este resultado para estabelecer o valor de k para uma molécula diatômica. Obtém então o valor de k =1,2857. É impressionante este valor pois coincide com o valor de 7 graus de liberdade para uma molécula diatômica, o resultado correto.
O trabalho de Boltzmann desta época é realmente fascinante. O caso diatômico, com estrutura interna é explicado por ele. Conseguiu ver que seria necessário levar em conta as forças intramoleculares para a descrição teórica da capacidade calorífica. Fez isto assumindo potenciais harmônicos e também mostra que o valor médio deste potencial equivaleria a kT. Com isto, o parâmetro e, uma quantidade positiva de valor desconhecido dependendo da lei da força que une os constituintes da molécula, previsto anteriormente por Maxwell, encontrava uma explicação. Exatamente por causa disto Boltzmann conseguia obter corretamente k =9/7 para uma molécula diatômica.
A LEI DE DULONG E PETIT ERA EXPLICADA
A lei de Dulong e Petit pedia uma explicação teórica e isto teria que ser feito usando-se o princípio da equipartição da energia, a única ferramenta teórica conhecida, por volta da metade do século XIX, para o cálculo da capacidade calorífica. Além disto esta explicação teria que vir de L. Boltzmann pois nesta época já sabia da importância, e do valor médio, da força intramolecular.
Numa nota de rodapé do seu livro teoria cinética de gases6, parte I de 1896 e parte II de 1898, encontra-se a seguinte dedução, colocada aqui na linguagem de hoje. Consideremos um sólido monoatômico onde a força intramolecular é do tipo harmônica. Se este sólido é composto de N átomos teremos que contar 3N graus de liberdade. Cada átomo deste sólido irá contribuir com1/2kTpara a energia cinética média e1/2kTpara a energia potencial média, já que estamos supondo que a interação é do tipo harmônica. Então teremos para a energia interna, E=3N(1/2kT+1/2kT)=3NkT donde CV= 3Nk. Por mol teremos CV=3R que é a lei de Dulong e Petit.
COMPARAÇÃO COM DADOS EXPERIMENTAIS
O conhecimento da estrutura interna de moléculas com mais de dois átomos era inexistente na época de Boltzmann e Maxwell. A explicação teórica sobre moléculas diatômicas havia sido feita, mas nada foi feito sobre a capacidade calorífica de moléculas poliatômicas, a não ser, como Boltzmann o fez, tratando estes sistemas como corpo rígido. Mas já existiam dados suficientes para desconfiar de que alguma coisa estava errada.
A capacidade calorífica do diamante a baixas temperaturas era mais um fato experimental que não se encaixava na termodinâmica clássica. A baixas temperaturas a capacidade calorífica a volume constante do diamante tendia para zero não correspondendo ao valor previsto pela equipartição da energia. Entretanto a altas temperaturas este valor da capacidade calorífica correspondia ao valor previsto pela termodinâmica, sendo igual a 3R. Na Figura 1 apresentamos os dados experimentais sobre a capacidade calorífica do diamante, como usados por Einstein7. Podemos ver claramente a validade da lei de Dulong e Petit para altas temperaturas, onde a capacidade calorífica vale 3R=24,942 J/Kmol. Entretanto fica também evidente o insucesso da lei de Dulong e Petit para baixas temperaturas.
Neste ponto é instrutivo apresentar também, junto com os dados do diamante acima, valores de capacidades caloríficas de átomos e moléculas, a altas e baixas temperaturas. A Tabela 2 mostra dados experimentais para alguns sistemas.
Note que para o argônio a temperatura de 298,15K já é alta suficiente para torná-lo um gás clássico. Os efeitos da quebra da equipartição da energia para o argônio podem ser vistos a 85K, na mesma pressão acima, quando Cp = 44,7 J/K mol 9.
A conclusão tirada das tabelas acima, usando dados recentes, é a mesma conclusão que foi tirada dos dados da capacidade calorífica do diamante por volta de 1880: alguma coisa estava faltando. A teoria molecular, a base teórica da termodinâmica, estava incompleta.
Entretanto é preciso deixar claro que o princípio da equipartição fornece resultados com boa precisão quando a temperatura é aumentada, o que está mostrado na Tabela 3.
Se a temperatura for aumentada ainda mais a concordância será melhor, mas esta região de temperatura não tem muito interesse num primeiro estudo de físico química. Mesmo a temperatura acima, que foi a mais alta encontrada na referência [8], é de pouco interesse.
O PROBLEMA ESTAVA LONGE DE SER RESOLVIDO
Tanto Maxwell quanto Boltzmann estavam conscientes de que ainda havia muito a ser feito. Na sua aula de 1875 Maxwell deixa isto bastante claro,
A lei da teoria molecular das capacidades caloríficas é imprecisa quando verificada experimentalmente, e a sua explicação completa depende de um conhecimento mais preciso da estrutura interna das moléculas do que o que temos agora.
Faz também uma previsão bastante interessante,
Um estudante inteligente, armado de cálculo e de um espectroscópio, dificilmente falhará na descoberta de alguns fatos importantes sobre a constituição interna das moléculas.
Apesar de ter esta visão do problema, Maxwell não especularia nada sobre as possíveis razões físicas que estavam levando a teoria cinética a não explicar as capacidades caloríficas experimentais.
Na parte II de seu livro6 Boltzmann é bastante claro quanto a sua posição,
Claro que o problema está longe de ser resolvido. Para muitos gases, k ainda tem valores menores ... .
e,
É fácil ver que muito ainda tem que ser feito na teoria de capacidades caloríficas.
Afirmava isto apesar de ter calculado os valores de k corretos para o caso atômico e diatômico. O não conhecimento da estrutura vibracional das moléculas era o grande impedimento para o cálculo correto para moléculas poliatômicas, como Maxwell já havia sentido. Boltzmann concordava com isto,
Naturalmente, por causa da obscuridade da natureza dos processos moleculares, todas as hipóteses sobre eles têm que ser expressas com grande cuidado.
Não é surpresa, portanto, notar o desespero de Boltzmann nesta altura, tentando explicar as capacidades caloríficas experimentais. Algumas passagens do seu livro mostram este desespero,
É justamente agora que os ataques na teoria dos gases começam a aumentar. Estou convencido de que estes ataques são meramente baseados numa falta de compreensão... . Na minha opinião será uma grande tragédia para a ciência se a teoria dos gases fosse esquecida... . Estou consciente de ser somente um indivíduo lutando fracamente contra a corrente do tempo. Mas ainda me resta energia... .
É evidente que Boltzmann não se dava por satisfeito e, mesmo sabendo da limitação do conhecimento da estrutura molecular propunha uma explicação para a discordância dos resultados teóricos com os experimentais. Boltzmann então conjecturou que isto ocorria porque alguns graus de liberdade, quando a temperatura era diminuída, não deviam ser contados,
Para aqueles corpos, cujas capacidades caloríficas é menor do que a metade prevista pela lei de Dulong e Petit(por exemplo, diamante) temos que assumir que o movimento relacionado com certos parâmetros entram em equilíbrio com outros tão devagar que eles não contribuem para a capacidade calorífica determinada por experimentos.
Esta era a solução que Boltzmann tentava dar para o problema, que como sabemos não pode estar correta. O tempo de relaxação de uma vibração ou rotação molecular é suficientemente pequeno para que este efeito não apareça nas medidas experimentais. A explicação teórica das discrepâncias acima teria que esperar um pouco mais.
LORD RAYLEIGH
Qualquer tentativa de interpretação de dados de capacidades caloríficas a baixas temperaturas usando a equipartição da energia resultará num insucesso. O princípio da equipartição foi obtido para altas temperaturas pois fizemos a integração ao invés de fazermos o somatório quando tratamos deste assunto. Os cientistas desta época sabiam que o problema estava com o princípio da equipartição mas não sabiam como resolver o problema.
Em 1900 Lord Rayleigh publicava um trabalho intitulado10, A lei da equipartição da energia cinética, em que discute o cálculo de valores médios e, como ele mesmo diz, aproveita a oportunidade para fazer algumas observações sobre a teoria cinética da matéria. Após discutir brevemente as dificuldades de reconciliar estrutura molecular com valores de capacidade calorífica enuncia,
O que parece ser necessário é alguma maneira de evitar a simplicidade destrutiva da conclusão geral ... . Tal escape tem que envolver uma repudiação do postulado fundamental de Maxwell aplicável praticamente para sistemas com um imenso número de graus de liberdade .
Em outras palavras Lord Rayleigh queria dizer que o princípio da equipartição era simples e ao mesmo tempo destrutivo pois trazia consigo conclusões demasiadamente gerais. Nós já sentimos o gosto disto. Atribuímos o valor kT para qualquer sistema, independente de suas características individuais. A discussão sobre a inconsistência da equipartição da energia passa agora por Lord Kelvin.
OUTRA AULA SOBRE A EQUIPARTIÇÃO
Os problemas relacionados com a equipartição da energia despertaram o interesse de Lord Kelvin, que numa aula dada em 27 de abril de 1900,
Nuvens do século dezenove sobre a teoria dinâmica do calor e da luz.
trata deste assunto. O título da aula, publicada em julho de 190111 é bastante interessante pois Kelvin achava que existia uma "nuvem" obscurecendo a teoria molecular do calor. Na parte em que discute a teoria do calor, começa mencionando o trabalho de Rayleigh10 e passa a discutir, como Boltzmann, o cálculo da quantidade k. Na verdade Kelvin preferiu discutir k - 1 mas isto não é relevante para a nossa discussão. As conclusões de Kelvin são as mesmas obtidas anteriormente: havia algo de incorreto na teoria cinética dos gases,
Ambos Boltzmann e Maxwell reconhecem a contradição experimental de sua doutrina representada pela teoria cinética dos gases, e sentem que uma explicação desta incompatibilidade é imperativamente necessária.
No parágrafo seguinte discute uma tabela, semelhante às Tabelas 2 e 3 apresentadas acima, e ao comparar com os dados experimentais conclui,
Mas, na realidade, a doutrina de Boltzmann-Maxwell erra enormemente mais do que mostrado na tabela precedente.
Esta é a mesma conclusão que tiramos ao analisarmos as Tabelas 2 e 3. Lord Kelvin termina o seu artigo com a frase,
A maneira mais simples de chegarmos aos resultados desejados é negar a conclusão; e então no começo do século vinte, perder de vista a nuvem que tem obscurecido o brilho da teoria molecular do calor ... .
Não havia mais espaço para a equipartição da energia, como formulada por Boltzmann e Maxwell.
A CORREÇÃO DE EINSTEIN
A necessidade de substituir o princípio da equipartição da energia por um princípio mais geral não é uma idéia nova e tem, no mínimo, 125 anos, como Maxwell já conseguia perceber. A pergunta óbvia, nesta altura, é então: O que deve ser feito para substituir a equipartição da energia?
Em abril de 1900 Kelvin dava a sua célebre aula e em dezembro deste mesmo ano Max Planck mostrava, baseando-se em dados experimentais, que a energia de osciladores harmônicos deveria ser quantizada. No mesmo caso de Kelvin, o artigo de Planck iria aparecer somente em 190112. Em 1905 A. Einstein usa estas idéias para explicar o efeito fotoelétrico13. Einstein usa também as idéias de Planck para investigar as anomalias que aconteciam com o princípio da equipartição explicando os dados apresentados na Figura 1 acima. Até 1907 três teorias haviam sido formuladas que abandonavam os conceitos clássicos: 1)A hipótese do quantum de energia, feito por Planck em 190012. 2)A teoria do efeito fotoelétrico, elaborada por Einstein em 190513. 3)A teoria das capacidades caloríficas, feita também por Einstein em 19077. Os conceitos usados nestes formalismos não tinham qualquer relação com a física clássica e culminaram com mecânica quântica. Para o envolvimento de Einstein com o assunto o leitor deve consultar a referência [14]. Esta referência, juntamente com as referências [15] e [16], tratam de aspectos do colapso da equipartição da energia e serviram de motivação inicial para o desenvolvimento do presente trabalho.
Tratando então um sólido como um conjunto de osciladores harmônicos, Einstein, como não podia fazer a integração como foi feito na equação (3), fez então o somatório para o cálculo da energia média de onde calculou a capacidade calorífica do modo vibracional como17,

A obtenção da equação acima é feita calculando-se primeiramente a energia média,

com a substituição de En=nhu. Este resultado nos fornece, para um mol e em três dimensões,

A equação (18) é obtida derivando-se, em relação à temperatura, a equação (20). A expressão da energia obtida por Einstein, como verificado mais tarde, estava incompleta pois um energia residual teria que ser levada em consideração. Esta energia residual, conhecida como energia de ponto zero, aparece como conseqüência do princípio da incerteza pois, como a partícula estará confinada numa região, haverá, como conseqüência deste princípio, uma incerteza no momento, gerando uma energia mínima. Entretanto a expressão (18) permanece inalterada pois esta energia de ponto zero independe da temperatura.
A correção de Einstein pode ser extendida para moléculas. Como a ordem de grandeza da energia translacional, Etrans, energia rotacional, Erot e energia vibracional, Evib, obedece a relação, Etrans << Erot << Evib,18 podemos manter a equipartição da energia para o modo translacional e rotacional, fazendo a correção de Einstein somente para o modo vibracional. Então,

para moléculas lineares e,
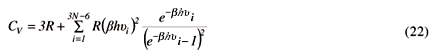
para moléculas não lineares. Definindo, Q =  , devidamente apresentados na Tabela 4, os valores de capacidade calorífica podem ser calculados.
, devidamente apresentados na Tabela 4, os valores de capacidade calorífica podem ser calculados.
Um exemplo de cálculo é dado para a molécula de N2a 298,15K. Calculando primeiramente a quantidade bhu  = 11,3869, e usando (21) com N=2, temos
= 11,3869, e usando (21) com N=2, temos
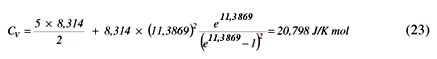
Ao adicionarmos o valor de R obtemos Cp= 29,111 J/K mol. Continuando com um raciocínio análogo para os outros sistemas da Tabela 2 obteremos os resultados apresentados na Tabela 5.
Para o argônio o resultado é o mesmo pois a correção é feita no modo intramolecular. O resultado para o diamante é semelhante. Para T=511K, CVexperimental (como na referência [7]) = 15,6 J/ K mol, e o teórico =14,710 J/ K mol, um resultado bem melhor do que o obtido pela equipartição que fornece CV=24,942 J/ K mol.
CONCLUSÃO E DISCUSSÃO
O presente trabalho discute as limitações do princípio da equipartição da energia onde argumentos históricos e teóricos são fornecidos. O insucesso da tentativa de conciliação de dados experimentais e dados teóricos de capacidades caloríficas foi um dos pontos que acelerou o desenvolvimento da mecânica quântica. É importante que o leitor reflita sobre a importância da termodinâmica, ou melhor, da falha do uso da termodinâmica clássica, no desenvolvimento da mecânica quântica.
Para que o princípio da equipartição forneça resultados em boa concordância com dados experimentais a temperatura tem que ser bastante alta. Para os sistemas estudados no presente trabalho esta temperatura equivale aproximadamente a 6000K. Esta temperatura tem certamente um interesse restrito para quem está iniciando os seus estudos em termodinâmica.
O conjunto de fatos, começando em 1819 com o trabalho de Petit e Dulong até 1900 com aula de Lord Kelvin, mostraram que o princípio da equipartição deveria ser substituído. O presente trabalho mostra que a imprecisão do princípio da equipartição da energia tem 125 anos, tomando como referência a aula de Maxwell em 1875. Usando as idéias de Planck, Einstein conseguiu explicar os dados experimentais do diamante. Estas idéias foram aqui usadas para moléculas diatômicas e poliatômicas deixando claro a importância de tal contribuição.
Se a imprecisão da equipartição da energia é um fato tão antigo e se as idéias de Einstein são tão simples, fica aqui o questionamento: As idéias apresentadas no presente artigo podem ser introduzidas num primeiro curso de físico química? Se estamos apresentando o material acima num primeiro curso de físico química temos que tomar o cuidado de apresentá-lo dentro deste contexto, isto é, obedecendo os pré-requisitos deste primeiro curso.
Convencer o estudante da necessidade da equipartição da energia ser substituída por uma nova teoria, não é uma tarefa difícil, após os resultados apresentados no presente trabalho. Mas é possível fazer a correção de Einstein num primeiro curso de físico química? A resposta é sim. Na verdade fazer o somatório é mais simples, matematicamente, do que fazer a integral em (3) e portanto não há quebra de pré-requisito neste ponto. A introdução da quantização da energia também não é um problema pois o aluno, neste estágio do curso, já foi apresentado às idéias de Planck. Isto aconteceu no primeiro semestre de seu curso, num curso de química geral, onde estudou, por exemplo, o átomo de Bohr.
AGRADECIMENTOS
Gostaria de agradecer ao CNPq, à CAPES e à FAPEMIG pelo apoio financeiro.
3. Braga, J. P.; Notas de termodinâmica, Não publicado. 1998.
- 1. Hirschfelder, J. O.; Curtiss, C. F.; Bird R. B.; Molecular Theory of Gases and Liquids; John Wiley; New York, 1954.
- 2. Plięgo Jr, J. R; Braga, J. P.; Quim. Nova 1994, 17, 496.
- 4. Petit, A. T.; Dulong, P. L.; Ann. Chim. Phys 1819, 10, 395.
- 5. Maxwell, J. C.; On the dynamical evidence of the molecular constitution of bodies, Aula dada na Chemical Society em 18 de fevereiro de 1875.
- 6. Boltzmann, L.; Lectures on gas theory; Dover Publication; New York, 1995.
- 7. Einstein, A.; Ann. d. Phys 1907, 22, 180.
- 8. Chase, M. W.; Janaf Thermochemical Tables, Springer Verlag, New York, 1986.
- 9. Lide, D. R.; Frederikse, H. P. R.; CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, 1996.
- 10. Lord Rayleigh; Phil. Mag 1900, 49, 98
- 11. Lord Kelvin; Phil. Mag (Sixth Series) 1901, 2, 1.
- 12. Planck, M.; Ann. d. Phys 1901, 4, 553.
- 13. Einstein, A.; Ann. d. Phys 1905, 17, 144.
- 14. Pais A.; Sutil é o senhor...", A cięncia e a vida de Albert Einstein, Editora Nova Fronteira, Rio de Janeiro, 1995.
- 15. Klein, M.; Science 1965, 148, 3667.
- 16. Hund, F; The history of quantum theory, Harrap, Londres, 1974.
- 17. Hill, T. L; An introduction to statistical thermodynamics, Dover Publication, New York, 1986.
- 18. Kittel, C.; Elementary statiscal physics, John Wiley, New York, 1967.
- 19. Herzberg, G.; Molecular Spectra and Molecular Structure, D. Van Nostrand, Princeton, 1950.
Datas de Publicação
-
Publicação nesta coleção
08 Nov 2001 -
Data do Fascículo
Out 2001
Histórico
-
Aceito
19 Fev 2001 -
Recebido
05 Maio 2000