Resumo
A number of ring-extended DNA adducts resulting from reaction of alpha,beta-unsaturated aldehydes, or their epoxides, with DNA bases have been characterized in recent years. These adducts can lead to miscoding during DNA replication which, if not repaired, result in mutations that can contribute to cancer development. Recently, the use of ultrasensitive methods allowed the detection of background levels of etheno DNA adducts in tissues of untreated animals and humans suggesting the existence of endogenous sources of reactive intermediates. In this review, we briefly summarize the recent advances in the chemistry of these DNA lesions.
exocyclic DNA adducts; etheno DNA adducts; lipid peroxidation
exocyclic DNA adducts; etheno DNA adducts; lipid peroxidation
Revisão
FORMAÇÃO DE ADUTOS EXOCÍCLICOS COM BASES DE DNA: IMPLICAÇÕES EM MUTAGÊNESE E CARCINOGÊNESE
Ana Paula M. Loureiro, Paolo Di Mascio e Marisa H. G. Medeiros*
Departamento de Bioquímica, Instituto de Química, Universidade de São Paulo, CP 26077, 05513-970 São Paulo - SP
*e-mail: mhgdmede@iq.usp.br
Recebido em 5/7/01; aceito em 28/11/01
EXOCYCLIC DNA ADDUCTS: IMPLICATIONS IN MUTAGENESIS AND CARCINOGENESIS. A number of ring-extended DNA adducts resulting from reaction of a,b-unsaturated aldehydes, or their epoxides, with DNA bases have been characterized in recent years. These adducts can lead to miscoding during DNA replication which, if not repaired, result in mutations that can contribute to cancer development. Recently, the use of ultrasensitive methods allowed the detection of background levels of etheno DNA adducts in tissues of untreated animals and humans suggesting the existence of endogenous sources of reactive intermediates. In this review, we briefly summarize the recent advances in the chemistry of these DNA lesions.
Keywords: exocyclic DNA adducts; etheno DNA adducts; lipid peroxidation.
INTRODUÇÃO
É muito antiga a observação de que a exposição dos seres humanos a determinadas substâncias presentes no meio ambiente ou no seu local de trabalho pode levar ao desenvolvimento de câncer. Já no final do século XVIII foi relatado que a ocorrência de câncer de escroto em limpadores de chaminés poderia estar relacionada com a deposição de fuligem e alcatrão nas pregas escrotais. Posteriormente foi mostrado que a aplicação repetida de alcatrão de hulha na pele de animais levava ao eventual desenvolvimento de tumores malignos no sítio da aplicação. Hoje sabemos que o alcatrão de hulha contém hidrocarbonetos aromáticos carcinogênicos, como o benzo[a]pireno. No século XIX foi percebida uma associação entre a exposição de pintores a aminas aromáticas, tal como benzidina, e o desenvolvimento de câncer na bexiga. Somente após 1930 foi demonstrada a associação entre a exposição de animais a diversas aminas aromáticas e o desenvolvimento desse tipo de câncer. Atualmente são conhecidas várias substâncias químicas carcinogênicas1.
Chama-se carcinogênese o processo de conversão de uma célula normal em uma célula maligna, sendo que os agentes indutores desse processo são denominados carcinógenos. Usualmente é necessária a exposição repetida aos carcinógenos para que haja o desenvolvimento de tumores malignos. Esse desenvolvimento ocorre muito lentamente devido à natureza complexa da carcinogênese, a qual pode ser dividida em três estágios: a iniciação, a promoção e a progressão, seqüência de eventos que foi esclarecida no decorrer dos últimos 20 anos2. Freqüentemente, após a alteração inicial, aparecem novas populações celulares que representam a evolução de células normais para células pré-neoplásicas, pré-malignas e malignas3,4.
A iniciação é causada por uma alteração irreversível do DNA, como a reação dessa molécula com substâncias carcinogênicas. Assim, mecanismos de detoxificação de carcinógenos, reparo do DNA e eliminação das células que tenham DNA modificado (por apoptose, por exemplo) são importantes para a proteção contra a iniciação do câncer. Para que a iniciação ocorra, é necessário que haja não só a modificação do DNA, mas também a sua replicação e a proliferação celular, de modo que a mutação inicial possa se fixar. A maioria dos cânceres humanos são originários de células epiteliais (carcinoma), pois as mesmas estão expostas aos carcinógenos (presentes no ar ou nos alimentos) e se proliferam rapidamente5.
Em geral, os carcinógenos são substâncias eletrofílicas ou são metabolizados para as mesmas durante o seu processo de detoxificação. Tais substâncias são atraídas por moléculas com alta densidade eletrônica, como é o caso das bases do DNA, às quais acabam se ligando e levando à formação de adutos6. A base do DNA mais suscetível a esse tipo de ataque é a guanina, mas já foram relatados adutos formados com todas as bases7. Sendo formados no DNA através de mecanismos químicos específicos, tais adutos podem levar a mutações em proto-oncogenes ou em genes supressores de tumor e iniciar o processo de carcinogênese8-10.
A elucidação das estruturas químicas de vários adutos formados com DNA foi de fundamental importância para os avanços feitos nesta área nas últimas duas décadas. Adutos formados tanto em reações in vitro como em sistemas experimentais de modelos biológicos foram caracterizados através de técnicas como espectrometria de massa, fluorescência e ressonância magnética nuclear (RMN)7,11-13.
A iniciação é seguida pela promoção, a qual envolve a seleção e expansão das células iniciadas, podendo levar ao desenvolvimento de um tumor benigno. Durante a promoção do tumor, o material genético alterado da célula iniciada altera a expressão dos genes que regulam a diferenciação e o crescimento celular. Quando administrados em altas doses, muitos carcinógenos são tanto iniciadores como promotores. Entretanto, uma dose baixa não indutora de tumor se torna efetiva se administrada juntamente com certas substâncias não carcinogênicas conhecidas como promotoras de tumor. Exemplos de promotores de tumor são o acetato de forbol miristato (PMA), um composto normalmente usado para induzir a explosão respiratória em células fagocíticas, e o 12-O-tetradecanoil-forbol-13-acetato (TPA). Muitos promotores de tumor são potentes indutores de inflamação, mas não há um princípio que explique a ação de todos. Eles podem levar a alterações reversíveis da proliferação celular e expressão fenotípica. A remoção do promotor pode resultar em retorno do tecido para um estado quase normal, embora ainda esteja iniciado14-16.
Na busca de uma correlação entre os níveis de adutos formados em DNA e a incidência de tumores, foi feito um estudo no qual foram compilados dados disponíveis a respeito dos níveis de diversos adutos após exposição crônica de ratos ou camundongos aos carcinógenos. Entre os diferentes carcinógenos investigados, foi observada grande variação nos níveis dos adutos que resultavam em 50% de incidência de tumor (53-5543 adutos/108 nucleotídeos), mostrando a existência de diferença na capacidade de os vários adutos de DNA levarem a mutações críticas1. Os níveis de adutos em DNA são um reflexo das velocidades de formação e remoção dos mesmos, o que depende da ativação do carcinógeno, da eficiência de reparo das lesões, da estabilidade do aduto e da velocidade de renovação do tecido17. Outros estudos também mostraram associações entre a formação de adutos em DNA e mutagênese e tumorigênese12,18-20, enquanto reduções dos níveis dos adutos foram associadas com quimioprevenção21. Os resultados obtidos nesses estudos mostraram que: a formação de adutos em DNA é tipicamente linear nas doses mais baixas de carcinógenos; esses adutos estão presentes na ausência de tumores, mas os tumores não se desenvolvem na ausência dos adutos; e outros eventos tecido-específicos (como proliferação celular) são necessários para que os tumores apareçam18-20.
O estágio final da carcinogênese consiste na progressão de uma lesão benigna ou pré-maligna para uma maligna, o que envolve alterações adicionais no DNA (por exemplo, nos genes supressores de tumor). A conversão de tumores benignos para malignos é acompanhada por perda no controle do crescimento, invasão de tecidos, metástase e instabilidade genética aumentada. Alguns exemplos são a progressão de pólipos benignos do cólon para adenocarcinoma e de nódulos hepáticos benignos para hepatoma3.
Estudos epidemiológicos moleculares com o potencial de elucidar as relações entre formação de adutos em DNA e risco de câncer em humanos provavelmente serão muito aplicados no futuro e poderão gerar hipóteses a respeito dos mecanismos biológicos básicos fundamentais que levam ao desenvolvimento de tumores. Grandes variações entre os indivíduos no que se refere à formação de adutos carcinógeno-DNA sugerem que determinantes genéticos do metabolismo de carcinógenos, reparo do DNA e controle do ciclo celular podem contribuir para as conseqüências dos danos ao DNA17.
DANOS BASAIS EM DNA: I. OXIDAÇÃO
Um grande avanço na área da carcinogênese ao longo dos últimos 20 anos foi a observação de que danos em DNA e mutações ocorrem mesmo na ausência de exposição a carcinógenos genotóxicos através de um tratamento específico ou um hábito particular. Os agentes que levam a esses danos incluem produtos normais do metabolismo celular, como espécies reativas de oxigênio, produtos da peroxidação lipídica, agentes alquilantes, estrógenos, espécies reativas de nitrogênio, agentes clorinantes e certos intermediários de algumas vias metabólicas22, e fontes exógenas inevitáveis, tais como radiação UV, radiação ionizante, radioisótopos de ocorrência natural e numerosas substâncias químicas genotóxicas presentes naturalmente ou como contaminantes na dieta e no ar23,24. O desenvolvimento de métodos analíticos suficientemente sensíveis para a detecção dos danos basais em DNA foi de fundamental importância para que pudesse ser avaliada a contribuição desses danos para a carcinogênese25-30.
Muitos dos danos endógenos observados no DNA correspondem a oxidações de bases31,32 (Figura 1). Entretanto, os níveis detectados de danos oxidativos no DNA de células normais variam de acordo com a metodologia utilizada (HPLC/EC, GC/MS ou 32P-postlabeling) e também entre laboratórios, o que tem sido motivo para grandes debates nesta área33. Níveis de 7,8-dihidro-8-oxo-2'-desoxiguanosina (8-oxodG) relatados em tecidos humanos variam de 1 aduto em 107 nucleotídeos a 1 aduto em 103 nucleotídeos34, e os níveis basais nos mesmos tecidos relatados por diferentes laboratórios variam em ordens de magnitude35. Esforços têm sido feitos no sentido de identificar e controlar as fontes de erros dessas medidas33,34,36,37. Mesmo assim, fazendo uma comparação com os níveis de adutos detectados em células expostas a carcinógenos conhecidos (o nível de adutos de benzo[a]pireno em DNA de carcinoma pulmonar de fumantes varia de 65 a 533 por 108 nucleotídeos), esses níveis de danos oxidativos são altos e podem ser importantes contribuintes para a etiologia de muitos cânceres humanos38,39.
A identidade dos oxidantes responsáveis pela oxidação de bases do DNA ainda é matéria de investigação40. O radical hidroxila (HO) é extremamente reativo, podendo se adicionar a bases do DNA ou abstrair átomos de hidrogênio das mesmas e originar muitos dos produtos observados no genoma41. É provável que o radical HO desempenhe um papel na oxidação endógena do DNA, mas a sua formação deve ser imediatamente adjacente à molécula de ácido nucleico para que os danos sejam gerados. Neste caso o peróxido de hidrogênio (H2O2) seria a espécie que se difundiria até o DNA, onde reagiria com íons metálicos gerando o oxidante42-44.
Outro oxidante que pode gerar muitos dos produtos de oxidação observados no DNA é o peroxinitrito (ONOO-), o qual pode se difundir intracelularmente e ser capturado por algumas células via canais aniônicos45-47. Uma série de complexos mecanismos levam à formação de diferentes espécies reativas de nitrogênio e oxigênio a partir do peroxinitrito, que devem ser responsáveis pelos danos observados48-59. O padrão de produtos gerados pela oxidação do DNA por peroxinitrito é complexo e reflete a diversidade de bases oxidadas detectadas nos tecidos60. Uma observação importante é a de que o peroxinitrito é bastante reativo com 8-oxodG (pelo menos 1000 vezes mais em relação à reatividade com 2'-desoxiguanosina) e, à medida que o nível dessa base oxidada aumenta, ela pode competir com as bases não modificadas no DNA pela reação com peroxinitrito, levando à formação de outros produtos60-62. Tal fato faz com que 8-oxodG não seja sempre um bom marcador para danos em DNA induzidos por estresse oxidativo. Óxido nítrico (NO) e ânion radical superóxido (O2-) são produzidos simultaneamente nos macrófagos e, portanto, níveis elevados de peroxinitrito (NO + O2-® ONOO-) serão produzidos em células inflamatórias ativadas. Condições patofisiológicas de infecções crônicas e inflamações são conhecidos fatores de risco para vários cânceres humanos e uma possível ligação entre inflamação e a indução de mutações é a habilidade de o peroxinitrito oxidar o DNA63. Recentemente foi demonstrada uma associação entre a ocorrência de transversões G®T no gene p53 de câncer colorretal humano e o nível de expressão da forma induzível da óxido nítrico sintase64.
O produto de oxidação do DNA mais extensivamente estudado, devido à facilidade com que pode ser detectado, é a 8-oxodG65-69. Suas propriedades biológicas foram amplamente investigadas, tendo sido verificado que é uma lesão mutagênica em bactérias e células de mamífero, levando principalmente a transversões G®T70-73. Transversões G®T são muito encontradas em genes supressores de tumor e proto-oncogenes mutados74. Outros estudos de mutagênese sítio-específica mostraram que 8-oxo-adenina, timina glicol, 5-hidroxiuracila, uracila glicol e 5-hidroxi-desoxicitidina também são lesões mutagênicas75,76.
Fortes evidências apontam no sentido de que a ocorrência de bases oxidadas no DNA leva ao surgimento de mutações espontâneas ao longo da vida de um indivíduo, podendo contribuir para o desenvolvimento de tumores. Níveis aumentados de vários produtos resultantes de danos oxidativos ao DNA foram encontrados em tecidos de câncer de mama em relação aos respectivos níveis presentes no DNA de áreas adjacentes. Aumentos semelhantes foram observados em cânceres de pulmão, cólon, estômago, ovário e cérebro77. Foi também observado que na hiperplasia prostática benigna, a qual freqüentemente se relaciona com o posterior desenvolvimento de câncer de próstata, há níveis elevados de bases oxidadas no DNA78.
Uma descoberta crucial que passou a sustentar a hipótese de que danos oxidativos no DNA são importantes contribuintes para a ocorrência de mutações espontâneas foi a de que existe um sistema elaborado de reparo em Escherichia coli para reduzir a mutagênese devida a 8-oxodG. Este sistema inclui uma glicosilase para remover 8-oxoguanina do DNA (glicosilase FAPY/produto do gene mutM), uma glicosilase que remove adenina no pareamento errado 8-oxodG:dA (produto do gene mutY) e uma hidrolase (8-oxodGTPase/produto do gene mutT) para eliminar 8-oxodGTP da reserva de nucleotídeos79-81. Linhagens de E. coli com mutações em qualquer um desses genes apresentam não só aumento dos níveis basais de 8-oxodG, como também aumento da freqüência de mutações espontâneas, principalmente tranversões G®T82-84. Existem também glicosilases para remoção de outras bases oxidadas do DNA85, sendo que as endonucleases III e VIII de E. coli participam da remoção de diversas pirimidinas oxidadas, tal como timina glicol, 6-hidroxi-5,6-dihidrocitosina e uracila glicol85-88. Homólogos dessas enzimas de reparo estão presentes em organismos eucariontes89-91.
As medidas dos níveis basais de danos oxidativos no DNA, a evidência molecular a respeito das propriedades mutagênicas dessas lesões e a verificação de que dietas ricas em antioxidantes são anticarcinogênicas sugerem fortemente que danos genéticos induzidos por oxidantes endógenos contribuem para a carcinogênese39,92,93.
PEROXIDAÇÃO LIPÍDICA E DANOS EM DNA
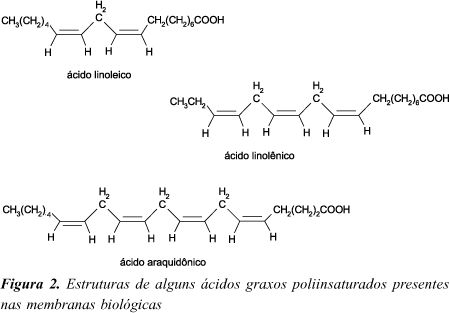
As espécies reativas de oxigênio e nitrogênio podem danificar não só o DNA, mas também outros componentes celulares. Devido à sua abundância nas células e susceptibilidade à oxidação pela presença de grupos metilênicos entre duplas ligações (Figura 2), os ácidos graxos poliinsaturados são, para os oxidantes, alvos mais prováveis do que o DNA94-97. É estimado que aproximadamente 60 moléculas de ácido linoleico (18:2 D9,12) e 200 de ácido araquidônico (20:4 D5,8,11,14) (os ácidos graxos poliinsaturados mais abundantes em nossas células) são consumidas por oxidante que reage com a bicamada lipídica98. Como essa oxidação desencadeia uma cascata autocatalítica que gera numerosas substâncias genotóxicas, tais danos aos lipídios têm grandes implicações para a integridade do DNA.
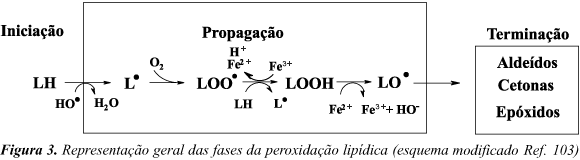
A peroxidação lipídica se inicia pelo ataque à bicamada lipídica de qualquer espécie suficientemente reativa para abstrair um átomo de hidrogênio bis-alílico de um ácido graxo poliinsaturado. Foi verificado que espécies tais como HO, HO2, NO2, RO, RO2 podem realizar essa oxidação99-101. Após iniciado, o processo se torna autocatalítico, levando à formação de hidroperóxidos e produtos secundários. As Figuras 3 e 4 resumem as três fases do processo. É importante mencionar que a oxidação enzimática do ácido araquidônico, que ocorre durante a síntese de eicosanóides, é uma importante fonte de espécies reativas de oxigênio. Além dos eicosanóides envolvidos na sinalização intra- e intercelular, radicais de oxigênio e hidroperóxidos lipídicos são gerados durante as reações catalisadas por ciclooxigenases ou lipoxigenases102.
Após a abstração do átomo de hidrogênio do ácido graxo poliinsaturado (LH) levando à formação de um radical lipídico centrado no carbono (L), este é estabilizado por um rearranjo molecular, adquirindo a estrutura de dieno conjugado. A adição extremamente rápida de uma molécula de oxigênio ao radical lipídico leva à formação do radical peroxil (LOO). Este é capaz de reagir com outro ácido graxo poliinsaturado, iniciando uma nova cadeia de oxidação a partir da formação de outro radical lipídico (L) (fase de propagação). O radical LOO combina-se com o átomo de hidrogênio abstraído e forma um hidroperóxido lipídico (LOOH)103. Alternativamente, os radicais LOO podem formar peróxidos cíclicos (Figura 4) pelo ataque a uma dupla ligação na mesma cadeia. Esses peróxidos cíclicos também podem propagar a peroxidação lipídica e, no caso da oxidação dos ácidos araquidônico, docosahexaenóico (22:6 D4,7,10,13,16,19) e eicosapentaenóico (20:5 D5,8,11,14,17), podem levar à formação de isoprostanos104. Os isoprostanos são uma classe de produtos tóxicos isômeros dos leucotrienos e prostaglandinas e são utilizados como biomarcadores de peroxidação lipídica. Estão presentes em plasma e urina de seres humanos sadios, o que indica que a peroxidação lipídica é um processo que ocorre continuamente105-109.
Os hidroperóxidos lipídicos são instáveis na presença de metais de transição, tais como ferro ou cobre, e formam radicais alcoxil (LO) e peroxil (LOO)110:

A reação de íons Fe2+ com hidroperóxidos lipídicos é freqüentemente mais rápida que a reação com H2O2 (k2 para Fe2+ + H2O2 é aproximadamente 76 M-1s-1; k2 para LOOH + Fe2+ é aproximadamente 1.5 x 103 M-1s-1). A reação dos hidroperóxidos com Fe2+ é mais rápida que a sua reação com Fe3+. Portanto, a velocidade de propagação da peroxidação lipídica na presença de Fe3+in vitro pode ser aumentada pela adição de agentes redutores em baixas concentrações, tal como ascorbato, de modo a ser atingida uma razão ótima Fe2+/Fe3+ que favoreça a propagação111. Os íons cobre são poderosos promotores da decomposição de peróxidos112.
Os radicais alcoxil podem abstrair átomos de hidrogênio de outros ácidos graxos poliinsaturados e hidroperóxidos lipídicos, formando outros radicais lipídicos (L) e radicais LOO, respectivamente. Assim, contribuem também para a propagação da peroxidação lipídica.
Além de contribuirem para a propagação da peroxidação lipídica, são também de interesse as reações dos radicais LOO com outras macromoléculas113,114. Seu tempo de vida é de aproximadamente 5 a 10 seg e, uma vez formados nas membranas nucleares, o DNA passa a ser um alvo importante115. Foi verificado, a partir da replicação em células humanas de vetores previamente modificados por LOO, que esses radicais induzem predominantemente substituições nos pares de base G:C, sendo que transversões G®T foram as principais substituições observadas116. Não são conhecidas as lesões pró-mutagênicas que levam a essas substituições22,117,118 e o esclarecimento dos eventos químicos responsáveis pelos efeitos mutagênicos dos radicais LOO é de fundamental importância para a avaliação da contribuição dessas lesões para o conjunto de danos basais presentes no DNA22.
Pode-se observar, portanto, que um único evento de iniciação leva à formação de várias moléculas de peróxidos como resultado da reação em cadeia. Um outro fator que aumenta a complexidade da peroxidação lipídica é que a abstração inicial de um átomo de hidrogênio pode ocorrer em diferentes pontos da cadeia de um ácido graxo poliinsaturado. Assim, a peroxidação do ácido linoleico dá origem a principalmente quatro diferentes hidroperóxidos (dienos trans,cis ou trans,trans substituídos em C-9 ou C-13), a do ácido linolênico (18:3 D9,12,15) também origina quatro (dienos trans,cis substituídos em C-9, C-12, C-13 ou C-16), a do ácido araquidônico seis (dienos trans,cis substituídos em C-5, C-8, C-9, C-11, C-12 ou C-15) e assim por diante104,119. Além disso, como mencionado anteriormente, os radicais peroxil podem reagir de modo a formar peróxidos cíclicos e endoperóxidos119. Outras variações surgem do fato de que os ácidos graxos poliinsaturados podem estar contidos em fosfatidilserina, fosfatidiletanolamina, fosfatidilcolina, glicolipídios ou esterificados ao colesterol e suas oxidações podem ter diferentes propriedades biológicas e patológicas120. A oxidação do colesterol para 5,6-epóxido e 5a-hidroperóxido é também de especial interesse biológico, uma vez que o colesterol epoxidado ocorre em altas concentrações em fluido de mama humana e foi identificado como mutagênico121,122.
A decomposição dos hidroperóxidos lipídicos é importante porque, além de gerar radicais que propagam a peroxidação lipídica, gera produtos não radicalares. Esses produtos secundários (aldeídos, cetonas, epóxidos, entre outros) são mais estáveis do que os radicais livres que iniciaram o processo e os radicais lipídicos formados durante a fase de propagação. Conseqüentemente, podem atingir pontos distantes do local em que se formaram103.
Os mecanismos pelos quais esses produtos secundários são gerados envolvem: cisão-b por clivagem homolítica entre o carbono do radical alcoxil e uma ligação C-C adjacente, originando um aldeído e um radical de hidrocarboneto que é estabilizado pela abstração de hidrogênio de uma outra molécula (esse mecanismo origina os gases etano e pentano, cujas medidas são utilizadas para a avaliação da peroxidação lipídica in vivo); estabilização do radical alcoxil para o respectivo ácido oxodienóico; ou ataque do radical alcoxil a uma dupla ligação adjacente, podendo formar epóxidos (Figura 5). Como os sistemas biológicos contêm uma mistura de diferentes ácidos graxos poliinsaturados com vários graus de insaturação, a peroxidação lipídica irá gerar uma mistura de hidroperóxidos lipídicos cujas quebras podem produzir uma variedade de produtos secundários diferentes, dentre os quais os aldeídos são os mais estudados119,123-125.
A Tabela 1 apresenta uma lista de aldeídos que foram identificados como produtos da peroxidação lipídica induzida em microssomos de fígado de ratos pelo sistema ADP-Fe2+/NADPH126-131.
A concentração gerada desses aldeídos depende das condições de oxidação e da composição de ácidos graxos poliinsaturados nas membranas. trans-4-Hidroxi-2-nonenal (HNE) é formado a partir da oxidação de ácidos graxos poliinsaturados w-6, tais como ácido araquidônico e linoleico, tendo sido detectado em vários tecidos de humanos, cachorros e roedores em níveis que variam de 0,3 a 8 nmol/g de tecido e 0,1 a 45 µM em plasma, dependendo do tecido e das condições patológicas120,132. A concentração intracelular de HNE medida em hepatócitos incubados com CCl4 (um indutor de peroxidação lipídica após ser metabolizado para CCl3 via citocromo P450) foi de aproximadamente 7 µM130 e, considerando que os aldeídos derivados de lipídios não se distribuem uniformemente entre as fases aquosa e lipídica, foi estimado que a concentração total local de aldeídos no interior das membranas de microssomos peroxidados pode atingir o nível de 100 mM127,128,130, sendo que essa concentração estimada de HNE fica em torno de 4,5 mM131.
Malonaldeído (MDA) é formado enzimaticamente durante o metabolismo de prostaglandinas102,133 e espermina134 e a partir da oxidação de ácidos graxos com mais de duas duplas ligações, tais como os ácidos linolênico, araquidônico, docosahexaenóico e eicosapentaenóico, sendo que os ácidos graxos w-3 (linolênico, docosahexaenóico e eicosapentaenóico) também originam 4-hidroxi-2-hexenal (HHE)120,135. Os níveis de MDA determinados em plasma de indivíduos sadios através de diferentes metodologias variam de zero a 47,2 µM, sendo que os valores mais baixos foram encontrados através da utilização de metodologias mais específicas, baseadas em HPLC ou cromatografia gasosa132. O nível de MDA encontrado em urina humana normal, determinado como derivado de pentafluorofenil-hidrazina por cromatografia gasosa acoplada à espectrometria de massa com ionização por captura de elétrons, situa-se na faixa de 0,2 a 0,8 µM136. MDA, HNE, HHE, juntamente com acroleína (2-propenal) e crotonaldeído (2-butenal), são os aldeídos mais largamente estudados com relação às suas propriedades químicas e biológicas132,137.
A peroxidação lipídica tem sido associada ao desenvolvimento de uma variedade de condições patológicas induzidas por exposição a agentes oxidantes. Alguns exemplos são a injúria devida à isquemia - reperfusão138-140, a hepatotoxicidade devida ao tetracloreto de carbono141-143, a carcinogênese16,144, a aterogênese145-151, a formação de pigmentos de lipofucsina associados ao envelhecimento152,153 e diabetes154-156. Considerando que os aldeídos a,b-insaturados (aqueles que apresentam uma dupla ligação C=C conjugada com o grupo carbonila), variantes estruturais dos mesmos (tais como 4-hidroxialcenais e epoxialdeídos) e malonaldeído são agentes alquilantes com capacidade de se ligarem covalentemente a grupos nucleofílicos presentes em DNA, peptídeos e proteínas, provocando alterações nas funções dessas moléculas, muitos trabalhos têm sugerido a implicação desses aldeídos em vários processos degenerativos associados ao estresse oxidativo16,151,153,157-161.
Recentemente foi descrita uma outra fonte endógena de aldeídos a,b-insaturados (Figura 6). A mieloperoxidase, na presença de H2O2 e Cl-, gera ácido hipocloroso (HOCl), o qual oxida aminoácidos para cloraminas. Estes últimos derivados perdem Cl- e CO2, formando iminas que são hidrolisadas para aldeídos. No caso da treonina, o hidroxialdeído resultante sofre desidratação e origina acroleína. Esta pode ser uma importante via de formação de alguns aldeídos em sítios de inflamação162.
DANOS BASAIS EM DNA: II. REAÇÃO COM MALONALDEÍDO
Malonaldeído (MDA) pode existir em diferentes formas em solução aquosa, de acordo com o pH (Figura 7). O valor de pK do grupo OH enólico é 4,5 e, portanto, em pH fisiológico a forma predominante é o ânion enolato, que tem baixa reatividade com a maioria dos grupos amino. A reatividade aumenta à medida em que o pH diminui e a forma enol não dissociada, b-hidroxiacroleína, se torna a espécie predominante, em equilíbrio com a forma ceto. Essa espécie é um eletrófilo que pode reagir com nucleófilos através de adição de Michael, de modo semelhante à reação de outros aldeídos a,b-insaturados132.
Diversos estudos mostraram que MDA é mutagênico em bactérias e células de mamífero163-168 e carcinogênico em ratos169. Tais características podem ser explicadas pela capacidade de reação deste aldeído com proteínas e DNA132,170.
Reiss e colaboradores descreveram a formação de produtos fluorescentes com emissão máxima a 460 nm quando excitados a 390 nm após a reação de DNA com MDA171. Foi sugerido que essa fluorescência era devida à formação de uma estrutura amino-iminopropeno conjugada, R-N=C-C=C-N-R', entre os grupos amino das bases do DNA e malonaldeído171. Alguns anos mais tarde foram caracterizados adutos resultantes da reação de dG, dA e dC com MDA (Figura 7)172-176. Apenas o aduto cíclico pirimidopurinona formado com dG (M1dG) é fluorescente, sendo também o de maior rendimento na reação de MDA com DNA (presente em concentração cinco vezes maior que N6-(3-oxopropenil)desoxiadenosina (M1dA)). O N4-(3-oxopropenil)desoxicitidina (M1dC) é formado em quantidades muito pequenas170.
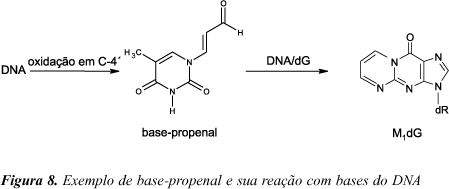
Uma via alternativa para formação de M1dG e, provavelmente, M1dA e M1dC é a reação das respectivas bases com base-propenais177. A oxidação direta do DNA por agentes que abstraem o átomo de hidrogênio 4' da desoxirribose inicia uma série de reações que levam à formação de base-propenais. Um desses agentes oxidantes é o antibiótico antitumoral Fe(II) bleomicina que induz quebras no DNA178-180. Base-propenais são derivados oxopropenil das bases do DNA, estruturalmente análogos a acroleínas substituídas na posição b (Figura 8). Esses compostos transferem o seu grupo oxopropenil para dG, formando M1dG. O tratamento de DNA com bleomicina na ausência completa de lipídios leva à formação de M1dG. Esse aduto também é formado pela reação direta de adenina-propenal com DNA177.
Para obtenção de informação a respeito da presença do aduto M1dG em DNA in vivo, foram desenvolvidas metodologias altamente sensíveis e muito específicas para a detecção de danos basais. A metodologia utilizada deve permitir a verificação inequívoca do aduto, o que não é uma tarefa fácil para qualquer aduto de DNA170.
Primeiramente foi utilizada pós-marcação com 32P (32P-postlabeling) para detecção e quantificação do aduto M1dG em DNA. Foi detectado um nível de 6 adutos/106 nucleotídeos em DNA de fígado de camundongos tratados com MDA (20 µmol/kg peso corpóreo)181. Em estudos subseqüentes, o tratamento de camundongos com MDA na concentração de 200 µmol/kg peso corpóreo estimulou a formação de adutos MDA-hemoglobina em sangue e M1dG em fígado. Os níveis de M1dG diminuiram lentamente após o tratamento com MDA, apresentando uma meia-vida de 12,5 dias182. Em outro estudo foi demonstrado um aumento de duas a três vezes no nível de M1dG em rins de hamsters Syrian tratados com doses de dietilestilbestrol ou oestradiol indutoras de câncer nesse órgão183. Um nível de 2,6 ± 1,2 adutos por 107 nucleotídeos foi detectado em leucócitos humanos de 26 voluntários e uma análise paralela em sete voluntárias indicou um nível de 3,0 ± 1,3 M1dG por 107 nucleotídeos em tecido de mama normal184. Um outro estudo mostrou que o nível de adutos em tecido de mama normal de mulheres com câncer de mama é duas a três vezes maior que no tecido de mama de mulheres sem câncer. Além disso, o nível de adutos encontrado no tumor foi menor que o presente no tecido normal circundante185. Foi ainda verificado um grande aumento (aproximadamente 20 vezes) do nível de M1dG em leucócitos de mulheres (mas não de homens) alimentadas com uma dieta rica em ácidos graxos poliinsaturados (1,2 a 28 adutos/107 nucleotídeos) em relação a um grupo que recebeu uma dieta rica em ácidos graxos monoinsaturados (0,2 a 2,5 adutos/107 nucleotídeos)186.
O uso da cromatografia gasosa acoplada à espectrometria de massa (GC-MS) com ionização química negativa permitiu a detecção e quantificação de M1dG em fígado, células brancas do sangue e pâncreas humanos em níveis que variam desde abaixo do nível de detecção (1 aduto/108 nucleotídeos) até 1,2 adutos por 106 nucleotídeos (aproximadamente 6500 adutos por célula)30,187,188. A especificidade do método é dada pela combinação de cromatografia por imunoafinidade para separar o aduto dos outros nucleosídeos presentes no DNA, derivatização química, coeluição cromatográfica com um padrão interno adicionado à amostra de DNA antes da hidrólise e monitoramento de um único íon no espectrômetro de massa170. A identidade de M1dG nas amostras de DNA de fígado humano e células brancas do sangue foi independentemente verificada por cromatografia líquida acoplada à espectrometria de massa em tandem187,189. É importante notar que os níveis de M1dG encontrados in vivo se assemelham aos níveis de 8-oxodG.
O potencial mutagênico dos adutos MDA-DNA foi avaliado por diferentes abordagens. A reação de MDA, em pH neutro, com um genoma simples-fita de bacteriófago, seguida por transformação de linhagens de E. coli induzidas para resposta SOS, provocou mutações por substituição de pares de bases (76%) e deslocamento da seqüência de bases (devido a adições) no gene lacZa contido no vetor. Aumento na concentração de MDA levou a aumento no número de mutações, coincidente com o aumento no nível de M1dG. As principais substituições observadas foram transversões G®T (35%) e transições C®T (32%) e A®G (25%), provavelmente decorrentes dos adutos correspondentes formados com MDA168. Experimentos de mutagênese sítio-específica utilizando vetores dupla-fita confirmaram que o aduto M1dG é mutagênico em E. coli, induzindo transversões para T e transições para A190,191. A freqüência total de mutações foi de aproximadamente 18% 191.
Transformações de linhagens de E. coli deficientes em enzimas de reparo com vetores contendo M1dG sugeriram que o reparo deste aduto se dá por meio de excisão de nucleotídeos. Linhagens deficientes em formamidopirimidina (FAPY) glicosilase ou 3-metiladenina glicosilase, enzimas conhecidas por participarem do reparo de 8-oxodG e adutos exocíclicos, não tiveram as freqüências de mutações afetadas, enquanto que um grande aumento na freqüência de mutações foi observado em células deficientes em reparo por excisão de nucleotídeos (uvrA- ou uvrB-)191,192.
Recentemente foi verificado, através de análises de espectros de ressonância magnética nuclear, que M1dG sofre rápida e quantitativa abertura do anel exocíclico para N2-oxopropenil-dG quando está presente em DNA dupla-fita, mas não quando em DNA simples-fita (Figura 9). A desnaturação térmica do DNA leva à rápida ciclização de N2-oxopropenil-dG para M1dG e o inverso ocorre quando o DNA é renaturado. A presença do grupo amino exocíclico da dC oposta ao aduto M1dG parece ser importante para a abertura do anel, uma vez que tal abertura não ocorre quando M1dG está oposta à timina193. Essa interconversão resulta em diferenças significativas no potencial mutagênico e susceptibilidade para o reparo, além de fornecer grupos funcionais reativos no interior da molécula de DNA que podem levar à formação de ligações cruzadas entre as cadeias do DNA e entre proteínas e DNA193,194. Foi demonstrado que MDA induz tais ligações cruzadas, mas os eventos moleculares responsáveis pelas mesmas ainda não são bem compreendidos195-198.
Apesar de os estudos de mutagenicidade indicarem um envolvimento direto do aduto M1dG na indução de mutações, mais investigações são necessárias para que seja esclarecida a importância desse aduto na carcinogênese humana e outras doenças crônicas. Para isso, torna-se importante avaliar a distribuição de M1dG em diferentes tecidos ou tipos celulares e se os níveis deste aduto podem ser modulados pela dieta ou outros fatores ambientais199.
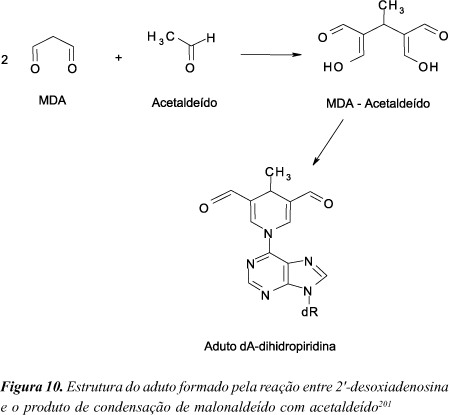
Uma outra possível fonte da fluorescência observada por Reis e colaboradores em DNA incubado com MDA171 pode ser o aduto 1,4-dihidropiridina altamente fluorescente resultante da reação do grupo amino exocíclico de 2'-desoxiadenosina com um produto da condensação de duas moléculas de malonaldeído com uma de acetaldeído (Figura 10)200. Acetaldeído pode ser formado a partir de malonaldeído via oxidação deste para o semialdeído ácido malônico e descarboxilação132. Esse aduto pode ser biologicamente importante, uma vez que o acetaldeído é um composto formado endogenamente a partir da oxidação enzimática do etanol no fígado, além de também estar presente na fumaça de cigarro e emissões de automóveis. Em fígado de ratos que ingeriram etanol foi encontrado o aduto dihidropiridina correspondente em proteínas, indicando que o produto da condensação de MDA com acetaldeído é formado in vivo201,202. Trabalhos anteriores haviam mostrado que derivados dihidropiridínicos são formados em reações de aminas primárias com MDA em misturas contendo aldeídos saturados203,204. Foi sugerido que a condensação de aldeídos alifáticos com malonaldeído é uma reação geral, o que significa que outros aldeídos, além de acetaldeído, presentes em fluidos fisiológicos podem formar com malonaldeído produtos de condensação reativos205.
DANOS BASAIS EM DNA: III. PROPANOADUTOS
Além de malonaldeído, outros aldeídos gerados endogenamente podem contribuir para o conjunto de danos basais presentes no DNA. Dentre eles, os mais estudados são os trans-4-hidroxi-2-alcenais e trans-2-alcenais, os quais, em pH fisiológico, são mais eletrofílicos que o MDA206,207.
O trans-4-hidroxi-2-nonenal (HNE), principal hidroxialcenal resultante do processo de peroxidação lipídica, exibe, de acordo com a sua concentração, uma série de atividades biológicas. Estas incluem inibição de enzimas, inibição da síntese de proteínas, DNA e RNA, indução de proteínas de choque térmico, bloqueio da proliferação de várias linhagens celulares, redução de comunicação intercelular via junções comunicantes, estímulo da migração de neutrófilos e modulação de vários genes131,132,208-212. Tem sido verificado que proteínas modificadas por HNE ocorrem em lesões ateroscleróticas e que existem anticorpos contra as mesmas em plasma de coelhos e humanos151,213,214 . HNE também apresenta efeitos genotóxicos e mutagênicos em células132,215.
Outro 4-hidroxialcenal, o trans-4-hidroxi-2-hexenal (HHE), recebeu atenção após ter sido verificado que, além de ser um produto de peroxidação lipídica132,135, é o produto final formado no fígado durante o metabolismo de alcalóides pirrolizidínicos, tal como senecionina, podendo desempenhar um papel na toxicidade devida a esses alcalóides216. Tais alcalóides são produzidos por uma variedade de plantas e são responsáveis por muitas mortes de animais e envenenamentos humanos217. Muitos são hepatotóxicos218, genotóxicos219,220 e carcinogênicos221.
A reatividade dos 4-hidroxialcenais é devida à presença de três grupos funcionais principais em suas estruturas: o grupo aldeído, a dupla ligação e o grupo hidroxila (Figura 11)132. A sua habilidade em reagir rapidamente com nucleófilos em pH fisiológico contribui significativamente para sua toxicidade. Assim, reagem com grupos tióis (-SH) presentes em GSH e proteínas e grupos amino presentes em bases de DNA, proteínas (como resíduos de lisina em lipoproteínas) e nos fosfolipídios fosfatidiletanolamina e fosfatidilserina132.
O padrão de danos induzidos em DNA por esses aldeídos é bem diversificado, uma vez que os adutos podem ser formados tanto através da reação direta dos aldeídos com as bases do DNA, como via seus derivados epoxidados137. No caso da reação direta dos aldeídos com bases do DNA (adição de Michael do grupo amino exocíclico da base ao C-3 do aldeído, seguida por ciclização), os principais produtos são os adutos cíclicos formados com dG, os quais são análogos saturados, 8-hidroxi propano C-6 substituídos, do aduto derivado da reação de dG com malonaldeído (Figura 11). Recebem a denominação geral de propanoadutos. A cadeia lateral no C-6 do novo anel revela qual enal gerou o aduto. Uma vez que os adutos contêm dois novos centros quirais, os produtos compreendem uma mistura de diastereoisômeros222,223.
Dentre os trans-2-alcenais, os mais estudados são acroleína [H2C=CH-HC=O] e crotonaldeído [H3C-CH=CH-HC=O]. Eles são formados endogenamente como resultado da oxidação de lipídios132, aminoácidos162 e poliaminas224 e ocorrem no ambiente como componentes da fumaça de cigarro (acroleína: 10-140 µg/cigarro) e exaustão de automóveis, sendo também produzidos quando alimentos contendo gordura são cozidos132,225,226; ocorrem também como produtos do metabolismo de drogas224,227-229, carcinógenos (ex.: N-nitrosopirrolidina origina crotonaldeído) e agentes quimioterapêuticos (ex.: ciclofosfamida e ifosfamida originam acroleína)230-232. Outro trans-2-alcenal, o hexenal, é um importante aromatizante que ocorre naturalmente em muitas frutas e vegetais, sendo, por causa do seu aroma, utilizado como aditivo alimentar, além de também ser gerado por processos de autoxidação233-236. Diversos estudos mostram que esses aldeídos são mutagênicos em bactérias e células de mamífero167,237-241, tendo sido verificado que crotonaldeído leva à formação de tumores em fígado de ratos após administração oral crônica (42 mg/L de água)242 e acroleína inicia a carcinogênese em bexiga urinária de ratos243.
Acroleína, crotonaldeído e hexenal também reagem, via adição de Michael, com bases do DNA, levando à formação dos respectivos propanoadutos236,244,245. Como no caso dos trans-4-hidroxi-2-alcenais, os principais adutos observados são os resultantes da reação com dG, embora adutos cíclicos também sejam formados a partir da reação de acroleína com desoxiadenosina e desoxicitidina238,239,246,247.
Adutos 1,N2-propano-2'-desoxiguanosina derivados da reação de dG com acroleína ou crotonaldeído foram detectados em tecidos de fígado, pele, glândulas mamárias, rins, cérebro, pulmão, próstata, leucócitos e bexiga urinária de roedores não submetidos a qualquer tratamento (nível total: 1,1 a 16,9 adutos por 107 dG) e em fígado, leucócitos, gengiva e mama humanos (nível total: 0,1 a 75,3 adutos por 107 dG)232,248,249. O aduto derivado da reação de dG com HNE foi recentemente detectado em DNA de fígado e cólon de ratos não tratados e humanos250. Diversos estudos realizados in vitro e in vivo mostraram que a oxidação dos ácidos graxos poliinsaturados é uma importante fonte desses níveis basais de propanoadutos251.
A presença de níveis basais de adutos 1,N2-propano-2'-desoxiguanosina em tecidos de roedores e humanos levanta questões a respeito da participação dessas lesões nos processos de mutagênese e carcinogênese. Entretanto, as propriedades mutagênicas desses adutos ainda não foram avaliadas por experimentos de mutagênese sítio-específica167,252 devido ao fato de a instabilidade dos mesmos impedir a sua incorporação em oligonucleotídeos e vetores ponte253. Uma nova estratégia para formação do aduto 1,N2-propano-2'-desoxiguanosina derivado de acroleína após a síntese do oligonucleotídeo foi recentemente descrita e deverá tornar possível a construção dos vetores necessários para esses estudos253. Um modelo do aduto 1,N2-propano-2'-desoxiguanosina sem o grupo hidroxila no anel propano, o qual não ocorre naturalmente, foi usado como substituto nos estudos de mutagênese sítio-específica realizados. Esses estudos mostraram que esse aduto modelo induz substituições G®A e G®T com alta eficiência em E. coli, além de alteração na seqüência de bases254-256. Com a utilização de vetores ponte replicados em células de mamífero foi determinado o espectro de mutações induzidas por crotonaldeído, consistindo em 86% das substituições envolvendo pares de base G:C257. As alterações mais comuns foram transversões G®T seguidas por transições G®A257.
Pouco também é conhecido a respeito do reparo desses adutos. O aduto modelo não hidroxilado é substrato para o complexo de reparo por excisão de nucleotídeos de E. coli e células de mamífero, sendo também reconhecido e reparado pelo sistema de reparo de pareamento errado (reparo de "mismatch")192,258.
Os dados existentes até o momento sugerem que os propanoadutos são lesões continuamente geradas em nosso DNA e, provavelmente, os adutos resultantes das reações de dG com crotonaldeído, acroleína ou HNE representam apenas uma pequena parcela dessas lesões. O entendimento do papel desses adutos cíclicos no desenvolvimento do câncer é uma importante e desafiante área para investigação futura, bem como a observação da forma com que os níveis basais e padrões dessas lesões pró-mutagênicas em tecidos humanos variam de acordo com a dieta e fatores genéticos e ambientais251.
DANOS BASAIS EM DNA: IV. ETENOADUTOS
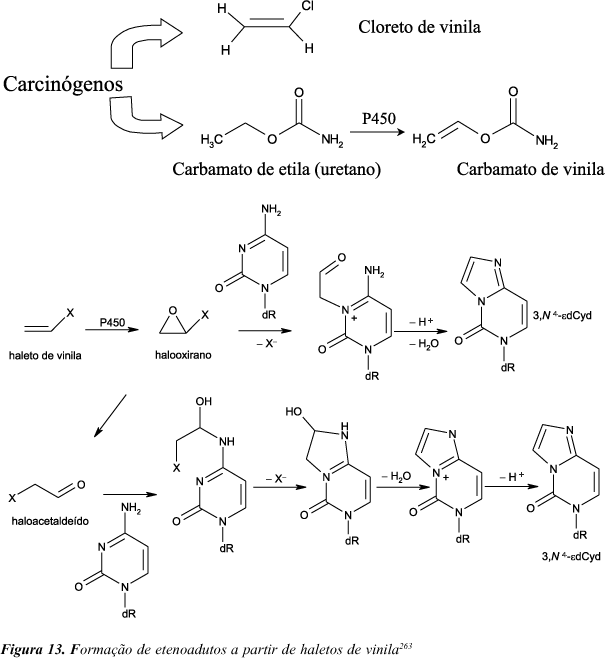
Dentre os adutos exocíclicos de DNA, os etenoadutos (Figura 12) têm sido bastante estudados ao longo dos últimos 25 anos259. Um grande interesse no estudo dessas lesões teve início com a verificação de que elas são induzidas em DNA por conhecidos carcinógenos ocupacionais e ambientais, tais como cloreto de vinila e carbamato de etila (uretano), após metabolização para os respectivos oxiranos (Figura 13)260-262. Os derivados exocíclicos 1,N6-eteno-2'-desoxiadenosina (edAdo), 3,N4-eteno-2'-desoxicitidina (edCyd) e N2,3-eteno-2'-desoxiguanosina (N2,3-edG) foram detectados em diferentes tecidos de ratos expostos ao cloreto de vinila (600 ppm, 4 h/dia, 5 dias) em níveis que variam de 2,9 a 9,8 edCyd/107dC, 0,4 a 2,1 edAdo/107dA e 2,1 a 18,1 N2,3-edG/107dG28,29,263-265 e levam à incorporação errada de bases após a replicação ou a transcrição266-270. Os estudos de fidelidade de replicação ou transcrição foram feitos inicialmente in vitro com a utilização de oligonucleotídeos modificados em um sítio específico e as especificidades mutagênicas encontradas foram subseqüentemente confirmadas por estudos de mutagênese sítio-específica através da replicação de vetores simples-fita em E. coli e células de mamífero. Nesses estudos foi verificado que edAdo induz transições A®G e transversões A®T e A®C270,271, edCyd induz transversões C®A e transições C®T255,272, e N2,3-edG induz transições G®A267, sendo que as eficiências mutagênicas de edAdo e edCyd foram maiores em células de mamífero (70% para edAdo e 81% para edCyd) que em E. coli (edAdo: 0,53% em células pré-irradiadas com UV e menos que 0,18% em células não irradiadas; edCyd: 32% em células pré-irradiadas com UV e 2% em células não irradiadas)255,271,273. Os resultados desses estudos foram consistentes com os resultados de dois estudos separados referentes às mutações em tumores de ratos e humanos expostos ao cloreto de vinila. Em um desses estudos, cinco de seis angiosarcomas de fígado humano associados com exposição ao cloreto de vinila mostraram transições G:C®A:T no codon 13 do gene c-Ki-ras-2, o que está de acordo com as propriedades mutagênicas de edCyd e N2,3-edG274. No outro estudo, tumores de ratos expostos ao cloreto de vinila mostraram transições G®A, transversões A®C e transições C®T em genes N-ras, também consistentes com as propriedades pró-mutagênicas dos etenoadutos275.
Com os avanços tecnológicos que permitiram o desenvolvimento de métodos analíticos ultra-sensíveis para detecção de adutos em DNA, tais como GC-MS, LC-ESI/MS, HPLC/radioimunoensaio, imunoafinidade/32P-postlabeling, níveis basais de vários adutos exocíclicos, incluindo os etenoadutos edAdo, edCyd e N2,3-edG, passaram a ser observados em DNA de roedores e humanos não expostos a carcinógenos137,262,276. Os níveis basais de edAdo/107dA detectados em tecidos humanos variam de <0,005 a 2,5 e em tecidos de roedores variam de 0,002 a 2,1; os valores correspondentes para edCyd/107dC variam de <0,005 a 1,1 e 0,003 a 2,4 respectivamente276. Tais observações estimularam a busca por substâncias geradas endogenamente que poderiam levar à formação dessas lesões. Há, atualmente, muitas evidências de que níveis basais dos adutos exocíclicos são derivados da reação de produtos da peroxidação lipídica com o DNA.
Um número crescente de trabalhos têm mostrado que os aldeídos interagem diretamente com o DNA ou são metabolizados a epóxidos, compostos conhecidos por serem agentes alquilantes do DNA com alta atividade mutagênica277,278. Acroleína, crotonaldeído, trans-2-hexenal e trans-4-hidroxi-2-nonenal (HNE) reagem com bases do DNA originando adutos cíclicos do tipo propano que puderam ser detectados in vivo251. Alternativamente esses aldeídos podem ser convertidos para os correspondentes derivados epoxidados tanto enzimaticamente como via reação com hidroperóxidos de ácidos graxos endógenos ou H2O2279. A reação dos epoxi-aldeídos com bases do DNA leva à formação dos etenoadutos.
Muito esforço tem sido feito no sentido de elucidar as fontes, o significado toxicológico e as conseqüências genéticas dessas lesões basais em DNA; os tipos de fatores exógenos ou condições patofisiológicas que levam ao seu aumento; e se esses adutos, quando gerados através de reações mediadas por estresse oxidativo, desempenham um papel na carcinogênese humana ou em outras doenças degenerativas259.
Estudos in vitro mostraram que 1,N6-etenoadenina (eA) e 3,N4-etenocitosina (eC) são formados quando microssomos de fígado de ratos são submetidos à peroxidação lipídica mediada por ferro na presença de nucleosídeos ou nucleotídeos280 e que aldeídos a,b-insaturados, tais como HNE, acroleína e crotonaldeído, reagem com bases de DNA, via seus epóxidos, originando os etenoadutos edAdo, edCyd e um outro etenoaduto de dG, 1,N2-eteno-2'-desoxiguanosina (1,N2-edG)137,281-283.
Apesar de ter sido sugerida a possibilidade de 1,N2-edG ser um bom marcador para danos em DNA induzidos por produtos da peroxidação lipídica283, a sua formação em sistemas biológicos ainda não foi demonstrada22,284. Apenas recentemente foram publicados os primeiros dados referentes a mutações induzidas por esse aduto em E. coli285 e células de mamífero286 com a utilização de vetores dupla-fita. No caso do estudo feito em bactérias, houve incorporação errada de bases em cerca de 3% da progênie viral, consistindo principalmente em transições G®A, seguidas por transversões G®T e G®C 285. No estudo feito em células de mamífero, foi utilizado um sistema de mutagênese sítio-específica intracromossômico, tendo sido observada uma freqüência aparente de mutações de 4,6%. A análise da seqüência de 21 clones derivados da fração mutante revelou cinco que correspondiam a mutações diretamente no sítio de incorporação de 1,N2-edG (principalmente transições G®A). Os outros mutantes diferiam dos gerados a partir do oligonucleotídeo não modificado e incluíam deleções, rearranjos, duplos-mutantes e substituições de pares de base em sítios próximos ao sítio do aduto286.
Em estudos in vivo, o tratamento de ratos com indutores de peroxidação lipídica, tais como CCl430, ferro + CCl4 ou ferro + etanol276 resulta em aumento dos níveis de etenoadutos nos fígados desses animais. Condições que induzem estresse oxidativo ou peroxidação lipídica em animais ou humanos, como produção excessiva de óxido nítrico (NO)287 e certas doenças genéticas de estoque de metais, tais como doença de Wilson e hemocromatose primária288, aumentam os níveis de etenoadutos nos tecidos afetados. A produção excessiva de NO ocorre sob condições patofisiológicas de infecções crônicas e inflamações, conhecidos fatores de risco para vários cânceres humanos. Recentemente foi demonstrado em um modelo de rato que a formação de etenoadutos em DNA ocorre como conseqüência da superprodução de NO in vivo276,287. Esta observação sugere que os etenoadutos podem contribuir para o desenvolvimento de cânceres humanos associados com processos inflamatórios crônicos causados por infecções virais, bacterianas e parasitárias persistentes289. De forma interessante, não foi detectado aumento concomitante de 8-oxodG nas mesmas amostras de DNA analisadas para verificação da presença de etenoadutos. O aumento no nível de 8-oxodG seria esperado, uma vez que o peroxinitrito (ONOO-) é um forte agente oxidante. Entretanto, a velocidade de degradação de 8-oxodG por peroxinitrito parece ser mais rápida que a sua velocidade de formação, o que faz com que essa base oxidada não seja sempre um bom marcador para danos ao DNA induzidos por estresse oxidativo. Neste caso, produtos de oxidação secundários mais estáveis, como os adutos exocíclicos, parecem ser marcadores mais úteis259. Outra evidência de que a peroxidação lipídica está envolvida na formação de etenoadutos em DNA vem da observação da presença de níveis elevados desses adutos exocíclicos em DNA de leucócitos de mulheres com dieta rica em ácidos graxos poliinsaturados w-6 290.
Em um estudo recente foi observada correlação entre o acúmulo de metabólitos do ácido araquidônico (ácidos 8-hidroxieicosatetraenóico (8-HETE) e 12-hidroxieicosatetraenóico (12-HETE)) e a formação dos adutos edAdo e edCyd no modelo de carcinogênese induzida por 7,12-dimetilbenzantraceno (DMBA) e promovida por 12-O-tetradecanoilforbol-13-acetato (TPA) em pele de camundongos16. Em papilomas reversíveis obtidos após 20 semanas de tratamento com TPA foram observados aumentos de 15 e 68 vezes nos níveis de 8-HETE e 12-HETE, respectivamente, em relação ao controle. Paralelamente, os níveis de edAdo e edCyd aumentaram, respectivamente, em 12 e 9 vezes16. Alguns estudos mostraram que tumores de pele induzidos experimentalmente acumulam altos níveis de prostaglandinas291 e ácidos 8-HETE e 12-HETE (precursores dos leucotrienos)292-294 e têm expressão aumentada das ciclooxigenases e lipoxigenases envolvidas na síntese dessas substâncias. A inibição dessas enzimas inibe significativamente o desenvolvimento de tumor em pele de camundongos295. Foi também demonstrado um aumento significativo dos níveis de etenoadutos em DNA de pólipos de pacientes com polipose adenomatosa familiar (FAP) nos quais havia superexpressão da ciclooxigenase-2, em comparação com o epitélio de cólon normal296. Esses dados sugerem uma correlação entre a formação de etenoadutos em DNA e a síntese de eicosanóides, além de indicarem o envolvimento dos etenoadutos no processo de carcinogênese16.
A verificação de que os etenoadutos são reconhecidos por enzimas de reparo mostra a existência de mecanismos de defesa celular contra essas lesões genotóxicas259. O aduto eA é substrato para a enzima de reparo alquilpurina-DNA-N-glicosilase de células de mamífero, mesma enzima que efetua o reparo de 3-metiladenina. O aduto eC é reparado eficientemente por uma outra DNA-glicosilase, a enzima timina-DNA glicosilase que repara T no pareamento errado G:T 297. Os etenoadutos 1,N2-eG e N2,3-eG são liberados por extratos de células HeLa a uma velocidade menor que eA ou eC, mas não são conhecidas as enzimas que efetuam esse reparo297. A inibição dessas enzimas de reparo ou a falta de expressão das mesmas em certos tipos de células podem levar a um acúmulo ou persistência dos etenoadutos em seu DNA265,298 e a todas as conseqüentes mutações. Apesar de serem bons substratos para o reparo por glicosilases, não pode ser descartada a possibilidade de que outras vias também estejam envolvidas no reparo de etenoadutos. Por exemplo, a mutagenicidade de 1,N2-edG aumenta quando presente em vetores que são transfectados para linhagens de E. coli deficientes em reparo por excisão de nucleotídeos285.
Todos esses estudos nos levam a acreditar que ao se utilizarem métodos específicos e sensíveis para analisar amostras biológicas facilmente disponíveis, como leucócitos e urina, vários fatores que influenciam a peroxidação lipídica, tais como dietas ricas em lipídios, baixos níveis de antioxidantes e processos inflamatórios, podem ser avaliados com a utilização dos etenoadutos como marcadores de exposição às espécies reativas formadas a partir do processo de lipoperoxidação299.
ESTUDOS REALIZADOS COM trans,trans-2,4- decadienal
Sabemos que muitos aldeídos são formados na fase de terminação da peroxidação lipídica. Em sistemas de peroxidação não enzimáticos, aproximadamente 30 a 40 moles de compostos carbonílicos são formados por mol de MDA103.
Em uma série de estudos foi caracterizada a formação de produtos fluorescentes a partir da interação de lipídios oxidados com o DNA. Esses produtos fluorescentes apresentavam um espectro com excitação máxima a 315 nm e emissão máxima a 420 nm300,301, sendo, portanto, diferente do obtido a partir da reação de MDA com DNA171. Esses resultados foram indicativos de que outros compostos carbonílicos formados durante a peroxidação lipídica poderiam reagir com o DNA e originar produtos caracterizados por diferentes espectros de fluorescência301.
Foram testados hidroperóxidos resultantes da oxidação do metil linolato e linolenato. Os testes foram feitos na presença de metais e agentes redutores (cisteína e ácido ascórbico), pois, nessa condição, os hidroperóxidos são facilmente decompostos, resultando na formação de uma grande variedade de compostos carbonílicos e espécies reativas de oxigênio que podem reagir com o DNA301.
Foi verificado que epidióxidos de linolenato autoxidado são precursores de 2,4-alcadienais302. Os hidroperoxi-epidióxidos do linolenato estão entre os mais ativos dos produtos secundários da oxidação do linolenato, reagindo com DNA na presença de metais e agentes redutores e levando à formação de produtos fluorescentes com emissão máxima a 420 nm (lexc.= 315 nm)301. Diversos aldeídos poliinsaturados foram testados, sendo que os 2,4-alcadienais e o 2,4,7-decatrienal foram os mais eficientes na formação dos adutos fluorescentes com o DNA303.
Hasegawa e colaboradores investigaram as estruturas químicas dos produtos formados a partir da reação de adenina com hidroperóxidos do metil linolato ou com aldeídos resultantes da decomposição desses hidroperóxidos na presença de Fe2+ e ácido ascórbico. O 2,4-decadienal (DDE) e o 2-octenal reagiram com adenina, levando à formação de produtos fluorescentes304.
Nosso laboratório tem se dedicado à caracterização da estrutura química de etenoadutos resultantes da reação de bases do DNA com derivados epoxidados de trans,trans-2,4-decadienal (DDE). DDE é um aldeído bastante difundido no meio ambiente, sendo encontrado na água305, em alimentos como peixe306-308, carne309,310, frango311,312 e pão313, estando presente como aromatizante natural em plantas314,315, sendo parte de secreções de defesa de alguns insetos316 e também gerado a partir de ácidos graxos poliinsaturados pela ação de lipoxigenases de plantas317,318. Além disso, é gerado endogenamente como produto da peroxidação lipídica131. Estudos de citotoxicidade mostraram que este aldeído é um dos produtos da quebra de hidroperóxidos lipídicos mais tóxicos para células319,320, inibe o crescimento celular, altera o nível de glutationa celular e induz fragmentação do DNA321. Recentemente foram caracterizados em nosso laboratório cinco etenoadutos fluorescentes resultantes da reação de epóxidos do DDE com 2'-desoxiadenosina in vitro322-324 e três etenoadutos não fluorescentes resultantes da reação de epóxidos do DDE com 2'-desoxiguanosina in vitro325. Todos os etenoadutos foram também detectados em DNA incubado com DDE in vitro. Duas possíveis vias para a formação dos etenoadutos de 2'-desoxiguanosina estão esquematizadas nas Figuras 14 e 15.
Estudos in vitro mostraram que outros produtos da peroxidação lipídica, tais como epóxidos de acroleína, crotonaldeído e 4-hidroxi-2-nonenal, reagem com 2'-desoxiguanosina (dGuo) originando o aduto 1,N2-eteno-2'-desoxiguanosina 281,282. Nossos resultados mostraram que DDE oxidado reage com dGuo levando à formação de vários adutos, dentre os quais três foram isolados e identificados com base em extensiva análise espectroscópica: aduto A1 (1,N2-eteno-2'-desoxiguanosina) e dois novos produtos diastereoisoméricos denominados adutos A2-1 e A2-2 (1-{[3-(2'-deoxy-b-D-erythro-pentafuranosyl)-5,9-dihydro-9 H-imidazo[2,1-i]purin-9-hydroxy]-7-yl}-2-one-3-octanol) 325.
Uma das possíveis vias para formação desses adutos é o mecanismo já descrito para reações de compostos carbonílicos epoxidados com bases de ácidos nucleicos282,283,324. Para a formação do aduto A1 poderia haver a epoxidação inicial das duas duplas ligações do aldeído ou apenas da dupla ligação em C2. Para a formação dos adutos A2-1 e A2-2 seria necessária a epoxidação inicial das duas duplas ligações com subseqüente hidrólise do grupo epóxido em C4. O ataque do grupo amino exocíclico do nucleosídeo ao carbono da carbonila do epoxi-aldeído, seguido por ciclização via ataque nucleofílico ao carbono interno do grupo epóxido em C2 pelo N-1 e eliminação de água, através do rearranjo de pinacol envolvendo os átomos ligados aos carbonos 10 e 11, levaria à formação dos etenoadutos A2-1 e A2-2. A eliminação adicional da cadeia lateral levaria à formação do aduto A1 (Figura 14)325.
Recentemente foi identificado um novo produto da peroxidação lipídica: 4-oxo-2-nonenal326. A reação desse produto diretamente com 2'-desoxiguanosina326 e 2'-desoxiadenosina327 levou à formação de etenoadutos substituídos contendo um grupo carbonila na cadeia lateral na posição correspondente a que encontramos nos adutos A2-1 e A2-2. Uma reação análoga ocorre entre 4-oxo-2-pentenal (produto da degradação atmosférica de tolueno, da solvólise de a-acetoxi-N-nitrosopiperidina e metabólito de 2-metilfurano) e 2'-desoxiguanosina, levando à formação do etenoaduto correspondente328,329. Em um trabalho anteriormente realizado, em que foi investigada a formação de adutos entre proteínas e produtos da peroxidação lipídica, utilizando n-butilamina e 4-hidroxi-2-nonenal, foi descrita a formação de um produto que parecia surgir a partir da oxidação de 4-hidroxi-2-nonenal para o seu análogo 4-oxo-2-nonenal. A formação do produto era acelerada quando 4-hidroxi-2-nonenal e n-butilamina reagiam na presença de Cu2+ e não detectada na ausência de oxigênio. A reação foi reproduzida a partir da incubação de n-butilamina com 4-oxo-2-nonenal330. Rindgen e colaboradores326 consideraram que, além da possível formação de 4-oxo-2-nonenal a partir da oxidação de 4-hidroxi-2-nonenal, mediada por metais de transição ou hidroperóxidos presentes no solvente, esse produto poderia também ser formado diretamente a partir de um intermediário da decomposição de hidroperóxidos lipídicos. Essa via para a formação de 4-hidroxi-2-nonenal a partir de ácidos graxos poliinsaturados w-6 foi originalmente proposta por Pryor e Porter331.
Um outro aldeído identificado como produto da peroxidação de lipídios microssomais de fígado induzida por NADPH-Fe2+ é o 4,5-dihidroxi-2-decenal128, podendo ser formado a partir da hidrólise do 4,5-epoxi-2-decenal detectado em diferentes sistemas332 e identificado como um dos produtos da decomposição do ácido 12,13(E)-epoxi-9-hidroperoxi-10-octadecenóico, um intermediário da oxidação do ácido linoleico (w-6)333. Analogamente, 4,5-epoxi-2-heptenal foi identificado como produto da oxidação de ácidos graxos poliinsaturados w-3334. Existem trabalhos mostrando que os 4,5-epoxi-2-alcenais são capazes de modificar aminoácidos rapidamente e que essas reações podem contribuir para a produção in vivo de pigmentos macromoleculares marrons com características fluorescentes do tipo lipofucsina153,335,336.
Considerando os dados acima apresentados, nos parece também concebível uma outra via para formação dos etenoadutos que caracterizamos. Segundo esse outro mecanismo (Figura 15), teríamos inicialmente apenas a epoxidação, catalisada por peróxidos, da dupla ligação em C4, obtendo-se o 4,5-epoxi-2-decenal. Dependendo das condições utilizadas, a monoepoxidação pode ocorrer especificamente apenas na dupla ligação envolvendo o carbono C4336. A subseqüente hidrólise desse epóxido levaria à formação do 4,5-dihidroxi-2-decenal. Como ocorre com o 4-hidroxi-2-nonenal326,330, teríamos a oxidação do álcool em C4 para cetona, obtendo-se um novo produto de oxidação do DDE, que seria o 4-oxo-5-hidroxi-2-decenal. Este produto reagiria então com 2'-desoxiguanosina segundo o mecanismo descrito para a reação de 4-oxo-2-nonenal com dGuo 326. O ataque do grupo amino exocíclico do nucleosídeo ao carbono da carbonila do 4-oxo-5-hidroxi-2-decenal, seguido por ciclização via ataque nucleofílico do N-1 ao C2 e eliminação de água levaria à formação dos etenoadutos A2-1 e A2-2. A eliminação adicional da cadeia lateral levaria à formação do aduto A1 (Figura 15). Investigações para a comprovação desse mecanismo podem ser feitas incubando-se diretamente 2'-desoxiguanosina com 4,5-dihidroxi-2-decenal e analisando-se os produtos formados.
Para investigarmos a formação desses etenoadutos em DNA incubado com DDE in vitro e, subseqüentemente, em DNA de células incubadas com o aldeído, em DNA de fígado de ratos e em DNA de leucócitos humanos, desenvolvemos uma metodologia seletiva e sensível que consiste em pré-purificação das amostras de DNA por HPLC de fase reversa e detecção utilizando o monitoramento de reação múltipla (MRM) do espectrômetro de massa. Este método nos permitiu um limite de sensibilidade para a detecção de 4 adutos A1/108 dGuo e 5 adutos A2/107 dGuo (relação sinal/ruído = 3). Para as suas quantificações nas amostras de DNA utilizamos padrões isotópicos [15N5] dos mesmos adutos325. Nas amostras de DNA incubado com DDE in vitro encontramos os níveis de 1110,2 ± 57,3 1,N2-edGuo/107 dGuo e 854,3 ± 24,2 A2/107 dGuo325. Estamos investigando a ocorrência dos mesmos em amostras biológicas, uma vez que a verificação da ocorrência dessas lesões em DNA in vivo é o primeiro passo para que se possa considerá-las marcadores de exposição a produtos de peroxidação lipídica tanto de origem endógena como exógena. Além disso, investigar as possíveis propriedades mutagênicas dessas lesões é passo determinante para a avaliação do seu papel na etiologia de diversos tipos de câncer.
AGRADECIMENTOS
Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), Conselho Nacional para o Desenvolvimento Científico e Tecnológico (CNPq) e Programa de Apoio aos Núcleos de Excelência, PRONEX/FINEP.
REFERÊNCIAS
313. Schieberle, P.; Grosch, W.; Zeitschrift fur Lebensmittel-Untersuchung Und-Forschung 1991, 192, 130.
- 1. Otteneder, M.; Lutz, W. K.; Mutat. Res. 1999, 424, 237.
- 2. Balmain, A.; Harris, C. C.; Carcinogenesis 2000, 21, 371.
- 3. Sun, Y.; Free Radical Biol. Med. 1990, 8, 583.
- 4. DiGiovanni, J.; Pharmacol. Ther. 1992, 54, 63.
- 5. Brown, K.; Kemp, C.; Burns, P.; Balmain, A.; Arch. Toxicol. 1994, 16 (Suppl.), 253.
- 6. Bartsch, H.; Mutat. Res. 1996, 340, 67.
- 7. Dipple, A.; Carcinogenesis 1995, 16, 437.
- 8. Lehman, T. A.; Harris, C. C.; IARC Sci. Publ. 1994, 399.
- 9. Kinzler, K. W.; Vogelstein, B.; Nature 1996, 379, 19.
- 10. Kinzler, K. W.; Vogelstein, B.; Cell 1996, 87, 159.
- 11. Weston, A.; Mutat. Res. 1993, 288, 19.
- 12. Hemminki, K.; Carcinogenesis 1993, 14, 2007.
- 13. Hemminki, K.; Dipple, A.; Shuker, D. E. G.; Kadlubar, F. F.; Segerback, D.; Bartsch, H.; IARC Sci. Publ. 1994, 125, 361.
- 14. Kensler, T. W.; Taffe, B. G.; Advances in Free Radical Biology and Medicine 1986, 2, 347.
- 15. Wei, L.; Wei, H.; Frenkel, K.; Carcinogenesis 1993, 14, 841.
- 16. Nair, J.; Furstenberger, G.; Burger, F.; Marks, F.; Bartsch, H.; Chem. Res. Toxicol. 2000, 13, 703.
- 17. Poirier, M. C.; Santella, R. M.; Weston, A.; Carcinogenesis 2000, 21, 353.
- 18. Poirier, M. C.; Beland, F. A.; Chem. Res. Toxicol. 1992, 5, 749.
- 19. Verna, L.; Whysner, J.; Williams, G. M.; Pharmacol. Ther. 1996, 71, 83.
- 20. Culp, S. J.; Gaylor, D. W.; Sheldon, W. G.; Goldstein, L. S.; Beland, F. A.; Carcinogenesis 1998, 19, 117.
- 21. Kensler, T. W.; Groopman, J. D.; Wogan, G. N.; IARC Sci. Publ. 1996, 237.
- 22. Burcham, P. C.; Mutat. Res. 1999, 443, 11.
- 23. Ames, B. N.; Mutat. Res. 1989, 214, 41.
- 24. Gupta, R. C.; Lutz, W. K.; Mutat. Res. 1999, 424, 1.
- 25. Gupta, R. C.; Reddy, M. V.; Randerath, K.; Carcinogenesis 1982, 3, 1081.
- 26. Randerath, K.; Reddy, M. V.; Disher, R. M.; Carcinogenesis 1986, 7, 1615.
- 27. Dizdaroglu, M.; Gajewski, E.; Methods Enzymol. 1990, 186, 530.
- 28. Fedtke, N.; Boucheron, J. A.; Turner, M. J.; Swenberg, J. A.; Carcinogenesis 1990, 11, 1279.
- 29. Fedtke, N.; Boucheron, J. A.; Walker, V. E.; Swenberg, J. A.; Carcinogenesis 1990, 11, 1287.
- 30. Chaudhary, A. K.; Nokubo, M.; Reddy, G. R.; Yeola, S. N.; Morrow, J. D.; Blair, I. A.; Marnett, L. J.; Science 1994, 265, 1580.
- 31. Douki, T.; Onuki, J.; Medeiros, M. H. G.; Bechara, E. J.; Cadet, J.; Di Mascio, P.; FEBS Lett. 1998, 428, 93.
- 32. Cadet, J.; Douki, T.; Pouget, J. P.; Ravanat, J. L.; Sauvaigo, S.; Teixeira, P. C.; Onuki, J.; Medeiros, M. H. G.; Bechara, E. J. H.; Di Mascio, P. Em New Aspects of Trace Element Research; Abdulla, M.; Bost, M.; Gamon, S.; Arnaud, P.; Chazot, G., eds.; Smith-Gordon: London, 1999, p. 163.
- 33. Ravanat, J. L.; Turesky, R. J.; Gremaud, E.; Trudel, L. J.; Stadler, R. H.; Chem. Res. Toxicol. 1995, 8, 1039.
- 34. Cadet, J.; Douki, T.; Ravanat, J. L.; Environ. Health Perspect. 1997, 105, 1034.
- 35. Beckman, K. B.; Ames, B. N.; Mutat. Res. 1999, 424, 51.
- 36. Douki, T.; Delatour, T.; Bianchini, F.; Cadet, J.; Carcinogenesis 1996, 17, 347.
- 37. Jenner, A.; England, T. G.; Aruoma, O. I.; Halliwell, B.; Biochem. J. 1998, 331 ( Pt 2), 365.
- 38. Totter, J. R.; Proc. Natl. Acad. Sci. U. S. A. - Biological Sciences 1980, 77, 1763.
- 39. Loft, S.; Poulsen, H. E.; J. Mol. Med. 1996, 74, 297.
- 40. Ravanat, J. L.; Di Mascio, P.; Martinez, G. R.; Medeiros, M. H. G.; Cadet, J.; J. Biol. Chem. 2000, 275, 40601.
- 41. Ward, J. F.; Prog. Nucleic Acid Res. Mol. Biol. 1988, 35, 95.
- 42. Pryor, W. A.; Annu. Rev. Physiol. 1986, 48, 657.
- 43. Meneghini, R.; Benfato, M. S.; Bertoncini, C. R.; Carvalho, H.; Gurgueira, S. A.; Robalinho, R. L.; Teixeira, H. D.; Wendel, C. M. A.; Nascimento, A. L. T. O.; Cancer Journal 1995, 8, 109.
- 44. Meneghini, R.; Free Radical Biol. Med. 1997, 23, 783.
- 45. Beckman, J. S.; Beckman, T. W.; Chen, J.; Marshall, P. A.; Freeman, B. A.; Proc. Natl. Acad. Sci. U. S. A. 1990, 87, 1620.
- 46. Koppenol, W. H.; Moreno, J. J.; Pryor, W. A.; Ischiropoulos, H.; Beckman, J. S.; Chem. Res. Toxicol. 1992, 5, 834.
- 47. Radi, R.; Chem. Res. Toxicol. 1998, 11, 720.
- 48. Santos, C. X.; Bonini, M. G.; Augusto, O.; Arch. Biochem. Biophys. 2000, 377, 146.
- 49. Augusto, O.; Gatti, R. M.; Radi, R.; Arch. Biochem. Biophys. 1994, 310, 118.
- 50. Gatti, R. M.; Alvarez, B.; Vasquez-Vivar, J.; Radi, R.; Augusto, O.; Arch. Biochem. Biophys. 1998, 349, 36.
- 51. Nakao, L. S.; Ouchi, D.; Augusto, O.; Chem. Res. Toxicol. 1999, 12, 1010.
- 52. Bonini, M. G.; Radi, R.; Ferrer-Sueta, G.; Ferreira, A. M.; Augusto, O.; J. Biol. Chem. 1999, 274, 10802.
- 53. Santos, C. X.; Anjos, E. I.; Augusto, O.; Arch. Biochem. Biophys. 1999, 372, 285.
- 54. Martinez, G. R.; Di Mascio, P.; Bonini, M. G.; Augusto, O.; Briviba, K.; Sies, H.; Maurer, P.; Rothlisberger, U.; Herold, S.; Koppenol, W. H.; Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 10307.
- 55. Kissner, R.; Nauser, T.; Bugnon, P.; Lye, P. G.; Koppenol, W. H.; Chem. Res. Toxicol. 1997, 10, 1285.
- 56. Richeson, C. E.; Mulder, P.; Bowry, V. W.; Ingold, K. U.; J. Am. Chem. Soc. 1998, 120, 7211.
- 57. Uppu, R. M.; Squadrito, G. L.; Pryor, W. A.; Arch. Biochem. Biophys. 1996, 327, 335.
- 58. Lymar, S. V.; Jiang, Q.; Hurst, J. K.; Biochemistry 1996, 35, 7855.
- 59. Lymar, S. V.; Hurst, J. K.; Inorg. Chem. 1998, 37, 294.
- 60. Burney, S.; Niles, J. C.; Dedon, P. C.; Tannenbaum, S. R.; Chem. Res. Toxicol. 1999, 12, 513.
- 61. Uppu, R. M.; Cueto, R.; Squadrito, G. L.; Salgo, M. G.; Pryor, W. A.; Free Radical Biol. Med. 1996, 21, 407.
- 62. Tretyakova, N. Y.; Wishnok, J. S.; Tannenbaum, S. R.; Chem. Res. Toxicol. 2000, 13, 658.
- 63. De Flora, S.; Izzotti, A.; Randerath, K.; Randerath, E.; Bartsch, H.; Nair, J.; Balansky, R.; van Schooten, F.; Degan, P.; Fronza, G.; Walsh, D.; Lewtas, J.; Mutat. Res. 1996, 366, 197.
- 64. Ambs, S.; Bennett, W. P.; Merriam, W. G.; Ogunfusika, M. O.; Oser, S. M.; Harrington, A. M.; Shields, P. G.; Felley-Bosco, E.; Hussain, S. P.; Harris, C. C.; J. Natl. Cancer Inst. 1999, 91, 86.
- 65. Floyd, R. A.; Watson, J. J.; Wong, P. K.; Altmiller, D. H.; Rickard, R. C.; Free Radical Res. Commun. 1986, 1, 163.
- 66. Di Mascio, P.; Teixeira, P. C.; Onuki, J.; Medeiros, M. H. G.; Ornemann, D.; Douki, T.; Cadet, J.; Arch. Biochem. Biophys. 2000, 373, 368.
- 67. Fraga, C. G.; Onuki, J.; Lucesoli, F.; Bechara, E. J.; Di Mascio, P.; Carcinogenesis 1994, 15, 2241.
- 68. Onuki, J.; Ribas, A. V.; Medeiros, M. H. G.; Araki, K.; Toma, H. E.; Catalani, L. H.; Di Mascio, P.; Photochem. Photobiol. 1996, 63, 272.
- 69. Teixeira, P. C.; Onuki, J.; Medeiros, M. H. G.; Dörnemann, D.; Di Mascio, P.; Biol. Chem. 2001, 382, 913.
- 70. Shibutani, S.; Takeshita, M.; Grollman, A. P.; Nature 1991, 349, 431.
- 71. Cheng, K. C.; Cahill, D. S.; Kasai, H.; Nishimura, S.; Loeb, L. A.; J. Biol. Chem. 1992, 267, 166.
- 72. Moriya, M.; Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 1122.
- 73. Le Page, F.; Margot, A.; Grollman, A. P.; Sarasin, A.; Gentil, A.; Carcinogenesis 1995, 16, 2779.
- 74. Hussain, S. P.; Harris, C. C.; Cancer Res. 1998, 58, 4023.
- 75. Wang, D.; Kreutzer, D. A.; Essigmann, J. M.; Mutat. Res. 1998, 400, 99.
- 76. Kreutzer, D. A.; Essigmann, J. M.; Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 3578.
- 77. Malins, D. C.; Polissar, N. L.; Gunselman, S. J.; Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 2557.
- 78. Olinski, R.; Zastawny, T. H.; Foksinski, M.; Barecki, A.; Dizdaroglu, M.; Free Radical Biol. Med. 1995, 18, 807.
- 79. Tchou, J.; Kasai, H.; Shibutani, S.; Chung, M. H.; Laval, J.; Grollman, A. P.; Nishimura, S.; Proc. Natl. Acad. Sci. U. S. A. 1991, 88, 4690.
- 80. Michaels, M. L.; Tchou, J.; Grollman, A. P.; Miller, J. H.; Biochemistry 1992, 31, 10964.
- 81. Mo, J. Y.; Maki, H.; Sekiguchi, M.; Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 11021.
- 82. Michaels, M. L.; Miller, J. H.; J. Bacteriol. 1992, 174, 6321.
- 83. Tajiri, T.; Maki, H.; Sekiguchi, M.; Mutat. Res. 1995, 336, 257.
- 84. Glassner, B. J.; Posnick, L. M.; Samson, L. D.; Mutat. Res. 1998, 400, 33.
- 85. Singer, B.; Hang, B.; Chem. Res. Toxicol. 1997, 10, 713.
- 86. Katcher, H. L.; Wallace, S. S.; Biochemistry 1983, 22, 4071.
- 87. Breimer, L. H.; Lindahl, T.; J. Biol. Chem. 1984, 259, 5543.
- 88. Purmal, A. A.; Lampman, G. W.; Bond, J. P.; Hatahet, Z.; Wallace, S. S.; J. Biol. Chem. 1998, 273, 10026.
- 89. Demple, B.; Harrison, L.; Annu. Rev. Biochem. 1994, 63, 915.
- 90. Girard, P. M.; Boiteux, S.; Biochimie 1997, 79, 559.
- 91. Mitra, S.; Hazra, T. K.; Roy, R.; Ikeda, S.; Biswas, T.; Lock, J.; Boldogh, I.; Izumi, T.; Mol. Cells 1997, 7, 305.
- 92. Potter, J. D.; Steinmetz, K.; IARC Sci. Publ. 1996, 61.
- 93. Tanaka, T.; Crit. Rev. Oncol. Hematol. 1997, 25, 73.
- 94. Cerutti, P. A.; Science 1985, 227, 375.
- 95. Porter, N. A.; Acc. Chem. Res. 1986, 19, 262.
- 96. Dix, T. A.; Aikens, J.; Chem. Res. Toxicol. 1993, 6, 2.
- 97. Wagner, B. A.; Buettner, G. R.; Burns, C. P.; Biochemistry 1994, 33, 4449.
- 98. Howard, J. A.; Free Radicals, Wiley-Interscience: New York, 1973, p. 3.
- 99. Barber, D. J. W.; Thomas, J. K.; Radiat. Res. 1978, 74, 51.
- 100. Pryor, W. A.; Lightsey, J. W.; Science 1981, 214, 435.
- 101. Aikens, J.; Dix, T. A.; J. Biol. Chem. 1991, 266, 15091.
- 102. Marnett, L. J.; Cancer Metastasis Rev. 1994, 13, 303.
- 103. Vaca, C. E.; Wilhelm, J.; Harms-Ringdahl, M.; Mutat. Res. 1988, 195, 137.
- 104. Gardner, H. W.; Free Radical Biol. Med. 1989, 7, 65.
- 105. Awad, J. A.; Morrow, J. D.; Takahashi, K.; Roberts, L. J.; J. Biol. Chem. 1993, 268, 4161.
- 106. Morrow, J. D.; Roberts, L. J.; Biochem. Pharmacol. 1996, 51, 1.
- 107. Roberts, L. J.; Morrow, J. D.; Biochim. Biophys. Acta 1997, 1345, 121.
- 108. Basu, S.; FEBS Lett. 1998, 428, 32.
- 109. Lawson, J. A.; Rokach, J.; FitzGerald, G. A.; J. Biol. Chem. 1999, 274, 24441.
- 110. Halliwell, B.; Gutteridge, J. M. C.; Free Radicals in Biology and Medicine, Oxford University Press: New York, 1999, v. 3.
- 111. Minotti, G.; Aust, S. D.; Lipids 1992, 27, 219.
- 112. Gutteridge, J. M.; Stocks, J.; Crit. Rev. Clin. Lab. Sci. 1981, 14, 257.
- 113. Viner, R. I.; Ferrington, D. A.; Aced, G. I.; Miller-Schlyer, M.; Bigelow, D. J.; Schoneich, C.; Biochim. Biophys. Acta 1997, 1329, 321.
- 114. Garner, B.; Witting, P. K.; Waldeck, A. R.; Christison, J. K.; Raftery, M.; Stocker, R.; J. Biol. Chem. 1998, 273, 6080.
- 115. Greenley, T. L.; Davies, M. J.; Biochim. Biophys. Acta 1994, 1226, 56.
- 116. Burcham, P. C.; Harkin, L. A.; Mutagenesis 1999, 14, 135.
- 117. Harkin, L. A.; Burcham, P. C.; Biochem. Biophys. Res. Commun. 1997, 237, 1.
- 118. Valentine, M. R.; Rodriguez, H.; Termini, J.; Biochemistry 1998, 37, 7030.
- 119. Porter, N. A.; Methods Enzymol. 1984, 105, 273.
- 120. Esterbauer, H.; Eckl, P.; Ortner, A.; Mutat. Res. 1990, 238, 223.
- 121. Petrakis, N. L.; Gruenke, L. D.; Craig, J. C.; Cancer Res. 1981, 41, 2563.
- 122. Sevanian, A.; Peterson, A. R.; Proc. Natl. Acad. Sci. U. S. A. 1984, 81, 4198.
- 123. Mlakar, A.; Spiteller, G.; Biochim. Biophys. Acta 1994, 1214, 209.
- 124. Loidl-Stahlhofen, A.; Hannemann, K.; Spiteller, G.; Biochim. Biophys. Acta 1994, 1213, 140.
- 125. Mlakar, A.; Spiteller, G.; Chem. Phys. Lipids 1996, 79, 47.
- 126. Benedetti, A.; Comporti, M.; Esterbauer, H.; Biochim. Biophys. Acta 1980, 620, 281.
- 127. Esterbauer, H.; Cheeseman, K. H.; Dianzani, M. U.; Poli, G.; Slater, T. F.; Biochem. J. 1982, 208, 129.
- 128. Benedetti, A.; Comporti, M.; Fulceri, R.; Esterbauer, H.; Biochim. Biophys. Acta 1984, 792, 172.
- 129. Benedetti, A.; Fulceri, R.; Comporti, M.; Biochim. Biophys. Acta 1984, 793, 489.
- 130. Poli, G.; Dianzani, M. U.; Cheeseman, K. H.; Slater, T. F.; Lang, J.; Esterbauer, H.; Biochem. J. 1985, 227, 629.
- 131. Esterbauer, H.; Free Radicals in Liver Injury, IRL Press: Oxford, 1985, p. 29.
- 132. Esterbauer, H.; Schaur, R. J.; Zollner, H.; Free Radical Biol. Med. 1991, 11, 81.
- 133. Hecker, M.; Ullrich, V.; J. Biol. Chem. 1989, 264, 141.
- 134. Quash, G.; Ripoll, H.; Gazzolo, L.; Doutheau, A.; Saba, A.; Gore, J.; Biochimie 1987, 69, 101.
- 135. van Kuijk, F. J.; Holte, L. L.; Dratz, E. A.; Biochim. Biophys. Acta 1990, 1043, 116.
- 136. Tomita, M.; Okuyama, T.; Hatta, Y.; Kawai, S.; J. Chromatogr. 1990, 526, 174.
- 137. Chung, F. L.; Chen, H. J.; Nath, R. G.; Carcinogenesis 1996, 17, 2105.
- 138. Grune, T.; Siems, W. G.; Schneider, W.; Free Radical Biol. Med. 1993, 15, 125.
- 139. Grune, T.; Siems, W. G.; Kowalewski, J.; Esterbauer, H.; Life Sci. 1994, 55, 693.
- 140. Siems, W. G.; Grune, T.; Esterbauer, H.; Life Sci. 1995, 57, 785.
- 141. Morrow, J. D.; Awad, J. A.; Kato, T.; Takahashi, K.; Badr, K. F.; Roberts, L. J.; Burk, R. F.; J. Clin. Invest. 1992, 90, 2502.
- 142. Brent, J. A.; Rumack, B. H.; J. Toxicol. Clin. Toxicol. 1993, 31, 173.
- 143. McGregor, D.; Lang, M.; Mutat. Res. 1996, 366, 181.
- 144. Cheeseman, K. H.; Mol. Aspects Med. 1993, 14, 191.
- 145. Esterbauer, H.; Jurgens, G.; Quehenberger, O.; Koller, E.; J. Lipid Res. 1987, 28, 495.
- 146. Salmon, S.; Maziere, C.; Theron, L.; Beucler, I.; Ayrault-Jarrier, M.; Goldstein, S.; Polonovski, J.; Biochim. Biophys. Acta 1987, 920, 215.
- 147. Yla-Herttuala, S.; Palinski, W.; Rosenfeld, M. E.; Parthasarathy, S.; Carew, T. E.; Butler, S.; Witztum, J. L.; Steinberg, D.; J. Clin. Invest. 1989, 84, 1086.
- 148. Witztum, J. L.; Steinberg, D.; J. Clin. Invest. 1991, 88, 1785.
- 149. Kotani, K.; Maekawa, M.; Kanno, T.; Kondo, A.; Toda, N.; Manabe, M.; Biochim. Biophys. Acta 1994, 1215, 121.
- 150. Holvoet, P.; Perez, G.; Zhao, Z.; Brouwers, E.; Bernar, H.; Collen, D.; J. Clin. Invest. 1995, 95, 2611.
- 151. Salomon, R. G.; Kaur, K.; Podrez, E.; Hoff, H. F.; Krushinsky, A. V.; Sayre, L. M.; Chem. Res. Toxicol. 2000, 13, 557.
- 152. Yin, D.; Free Radical Biol. Med. 1996, 21, 871.
- 153. Hidalgo, F. J.; Zamora, R.; Chem. Res. Toxicol. 2000, 13 , 501.
- 154. Sato, Y.; Hotta, N.; Sakamoto, N.; Matsuoka, S.; Ohishi, N.; Yagi, K.; Biochem. Med. 1979, 21, 104.
- 155. Suzuki, Y. J.; Tsuchiya, M.; Packer, L.; Free Radical Res. Commun. 1992, 17, 211.
- 156. Reagan, L. P.; Magarinos, A. M.; Yee, D. K.; Swzeda, L. I.; Van Bueren, A.; McCall, A. L.; McEwen, B. S.; Brain Res. 2000, 862, 292.
- 157. Brambilla, G.; Bassi, A. M.; Faggin, P.; Ferro, M.; Finollo, R.; Martelli, A.; Sciaba, L.; Marinari, U. M.; Free Radicals in Liver Injury, IRL Press: Oxford, 1985, p. 59.
- 158. Witz, G.; Free Radical Biol. Med. 1989, 7, 333.
- 159. Eder, E.; Scheckenbach, S.; Deininger, C.; Hoffman, C.; Toxicol. Lett. 1993, 67, 87.
- 160. Szweda, L. I.; Uchida, K.; Tsai, L.; Stadtman, E. R.; J. Biol. Chem. 1993, 268, 3342.
- 161. Requena, J. R.; Fu, M. X.; Ahmed, M. U.; Jenkins, A. J.; Lyons, T. J.; Baynes, J. W.; Thorpe, S. R.; Biochem. J. 1997, 322 ( Pt 1), 317.
- 162. Hazen, S. L.; Hsu, F. F.; d'Avignon, A.; Heinecke, J. W.; Biochemistry 1998, 37, 6864.
- 163. Shamberger, R. J.; Corlett, C. L.; Beaman, K. D.; Kasten, B. L.; Mutat. Res. 1979, 66, 349.
- 164. Marnett, L. J.; Tuttle, M. A.; Cancer Res. 1980, 40, 276.
- 165. Yonei, S.; Furui, H.; Mutat. Res. 1981, 88, 23.
- 166. Basu, A. K.; Marnett, L. J.; Carcinogenesis 1983, 4, 331.
- 167. Marnett, L. J.; Hurd, H. K.; Hollstein, M. C.; Levin, D. E.; Esterbauer, H.; Ames, B. N.; Mutat. Res. 1985, 148, 25.
- 168. Benamira, M.; Johnson, K.; Chaudhary, A.; Bruner, K.; Tibbetts, C.; Marnett, L. J.; Carcinogenesis 1995, 16, 93.
- 169. Spalding, J. W.; National Toxicology Program, Washington: DC, 1988, chap. NTP Tech. Rep. 331, p. 5.
- 170. Marnett, L. J.; IARC Sci. Publ. 1999, 17.
- 171. Reiss, U.; Tappel, A. L.; Chio, K. S.; Biochem. Biophys. Res. Commun. 1972, 48, 921.
- 172. Seto, H.; Okuda, T.; Takesue, T.; Ikemura, T.; Bull. Chem. Soc. Jpn. 1983, 56, 1799.
- 173. Nair, V.; Turner, G. A.; Offerman, R. J.; J. Am. Chem. Soc. 1984, 106, 3370.
- 174. Marnett, L. J.; Basu, A. K.; Ohara, S. M.; Weller, P. E.; Rahman, A. F. M. M.; Oliver, J. P.; J. Am. Chem. Soc. 1986, 108, 1348.
- 175. Stone, K.; Ksebati, M. B.; Marnett, L. J.; Chem. Res. Toxicol. 1990, 3, 33.
- 176. Stone, K.; Uzieblo, A.; Marnett, L. J.; Chem. Res. Toxicol. 1990, 3, 467.
- 177. Dedon, P. C.; Plastaras, J. P.; Rouzer, C. A.; Marnett, L. J.; Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 11113.
- 178. Burger, R. M.; Berkowitz, A. R.; Peisach, J.; Horwitz, S. B.; J. Biol. Chem. 1980, 255, 11832.
- 179. Giloni, L.; Takeshita, M.; Johnson, F.; Iden, C.; Grollman, A. P.; J. Biol. Chem. 1981, 256, 8608.
- 180. Grollman, A. P.; Takeshita, M.; Pillai, K. M.; Johnson, F.; Cancer Res. 1985, 45, 1127.
- 181. Vaca, C. E.; Vodicka, P.; Hemminki, K.; Carcinogenesis 1992, 13, 593.
- 182. Kautiainen, A.; Vaca, C. E.; Granath, F.; Carcinogenesis 1993, 14, 705.
- 183. Wang, M. Y.; Liehr, J. G.; Carcinogenesis 1995, 16, 1941.
- 184. Vaca, C. E.; Fang, J. L.; Mutanen, M.; Valsta, L.; Carcinogenesis 1995, 16, 1847.
- 185. Wang, M.; Dhingra, K.; Hittelman, W. N.; Liehr, J. G.; de Andrade, M.; Li, D.; Cancer Epidemiol. Biomarkers Prev. 1996, 5, 705.
- 186. Fang, J. L.; Vaca, C. E.; Valsta, L. M.; Mutanen, M.; Carcinogenesis 1996, 17, 1035.
- 187. Rouzer, C. A.; Chaudhary, A. K.; Nokubo, M.; Ferguson, D. M.; Reddy, G. R.; Blair, I. A.; Marnett, L. J.; Chem. Res. Toxicol. 1997, 10, 181.
- 188. Kadlubar, F. F.; Anderson, K.; Lang, N.; Thompson, P.; MacLeod, S.; Mikhailova, M.; Chou, M.; Plastaras, J. P.; Marnett, L. J.; Haussermann, S.; Bartsch, H.; Proc. Am. Assoc. Cancer Res. 1998, 39, 286.
- 189. Chaudhary, A. K.; Nokubo, M.; Oglesby, T. D.; Marnett, L. J.; Blair, I. A.; J. Mass Spectrom. 1995, 30, 1157.
- 190. Reddy, G. R.; Marnett, L. J.; J. Am. Chem. Soc. 1995, 117, 5007.
- 191. Fink, S. P.; Reddy, G. R.; Marnett, L. J.; Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 8652.
- 192. Johnson, K. A.; Fink, S. P.; Marnett, L. J.; J. Biol. Chem. 1997, 272, 11434.
- 193. Mao, H.; Schnetz-Boutaud, N. C.; Weisenseel, J. P.; Marnett, L. J.; Stone, M. P.; Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 6615.
- 194. Niedernhofer, L. J.; Riley, M.; Schnetz-Boutaud, N.; Sanduwaran, G.; Chaudhary, A. K.; Reddy, G. R.; Marnett, L. J.; Chem. Res. Toxicol. 1997, 10, 556.
- 195. Summerfield, F. W.; Tappel, A. L.; Biochim. Biophys. Acta 1983, 740, 185.
- 196. Summerfield, F. W.; Tappel, A. L.; Anal. Biochem. 1984, 143, 265.
- 197. Summerfield, F. W.; Tappel, A. L.; Arch. Biochem. Biophys. 1984, 233, 408.
- 198. Voitkun, V.; Zhitkovich, A.; Mutat. Res. 1999, 424, 97.
- 199. Leuratti, C.; Singh, R.; Lagneau, C.; Farmer, P. B.; Plastaras, J. P.; Marnett, L. J.; Shuker, D. E.; Carcinogenesis 1998, 19, 1919.
- 200. Le Curieux, F.; Pluskota, D.; Munter, T.; Sjoholm, R.; Kronberg, L.; Chem. Res. Toxicol. 1998, 11, 989.
- 201. Tuma, D. J.; Thiele, G. M.; Xu, D.; Klassen, L. W.; Sorrell, M. F.; Hepatology 1996, 23, 872.
- 202. Xu, D.; Thiele, G. M.; Kearley, M. L.; Haugen, M. D.; Klassen, L. W.; Sorrell, M. F.; Tuma, D. J.; Chem. Res. Toxicol. 1997, 10, 978.
- 203. Kikugawa, K.; Ido, Y.; Lipids 1984, 19, 600.
- 204. Nair, V.; Offerman, R. J.; Turner, G. A.; Pryor, A. N.; Baenziger, N. C.; Tetrahedron 1988, 44, 2793.
- 205. GomezSanchez, A.; Hermosin, I.; Lassaletta, J. M.; Maya, I.; Tetrahedron 1993, 49, 1237.
- 206. Burcham, P. C.; Mutagenesis 1998, 13, 287.
- 207. Toyokuni, S.; Luo, X. P.; Tanaka, T.; Uchida, K.; Hiai, H.; Lehotay, D. C.; Free Radical Biol. Med. 1997, 22, 1019.
- 208. Fazio, V. M.; Barrera, G.; Martinotti, S.; Farace, M. G.; Giglioni, B.; Frati, L.; Manzari, V.; Dianzani, M. U.; Cancer Res. 1992, 52, 4866.
- 209. Parola, M.; Pinzani, M.; Casini, A.; Albano, E.; Poli, G.; Gentilini, A.; Gentilini, P.; Dianzani, M. U.; Biochem. Biophys. Res. Commun. 1993, 194, 1044.
- 210. Spycher, S.; Tabataba-Vakili, S.; O'Donnell, V. B.; Palomba, L.; Azzi, A.; Biochem. Biophys. Res. Commun. 1996, 226, 512.
- 211. Barrera, G.; Pizzimenti, S.; Serra, A.; Ferretti, C.; Fazio, V. M.; Saglio, G.; Dianzani, M. U.; Biochem. Biophys. Res. Commun. 1996, 227, 589.
- 212. Leonarduzzi, G.; Scavazza, A.; Biasi, F.; Chiarpotto, E.; Camandola, S.; Vogel, S.; Dargel, R.; Poli, G.; FASEB J. 1997, 11, 851.
- 213. Steinberg, D.; Parthasarathy, S.; Carew, T. E.; Khoo, J. C.; Witztum, J. L.; N. Engl. J. Med. 1989, 320, 915.
- 214. Palinski, W.; Yla-Herttuala, S.; Rosenfeld, M. E.; Butler, S. W.; Socher, S. A.; Parthasarathy, S.; Curtiss, L. K.; Witztum, J. L.; Arteriosclerosis 1990, 10, 325.
- 215. Brambilla, G.; Sciaba, L.; Faggin, P.; Maura, A.; Marinari, U. M.; Ferro, M.; Esterbauer, H.; Mutat. Res. 1986, 171, 169.
- 216. Segall, H. J.; Wilson, D. W.; Dallas, J. L.; Haddon, W. F.; Science 1985, 229, 472.
- 217. Bull, L. B.; Culvenor, C. C. J.; Dick, A. T.; Front. Biol. 1968, 9, 1.
- 218. McLean, E. K.; Pharmacol. Rev. 1970, 22, 429.
- 219. Williams, G. M.; Mori, H.; Hirono, I.; Nagao, M.; Mutat. Res. 1980, 79, 1.
- 220. Green, C. E.; Segall, J. H.; Byard, J. L.; Toxicol. Appl. Pharmacol. 1981, 60, 176.
- 221. Schoental, R.; Cancer Res. 1968, 28, 2237.
- 222. Winter, C. K.; Segall, H. J.; Haddon, W. F.; Cancer Res. 1986, 46, 5682.
- 223. Yi, P.; Zhan, D.; Samokyszyn, V. M.; Doerge, D. R.; Fu, P. P.; Chem. Res. Toxicol. 1997, 10, 1259.
- 224. Alarcon, R. A.; Arch. Biochem. Biophys. 1970, 137, 365.
- 225. Wynder, E. L.; Goodman, D. A.; Hoffmann, D.; Cancer 1965, 18, 505.
- 226. Wilson, V. L.; Foiles, P. G.; Chung, F. L.; Povey, A. C.; Frank, A. A.; Harris, C. C.; Carcinogenesis 1991, 12, 1483.
- 227. Rees, K. R.; Tarlow, M. J.; Biochem. J. 1967, 104, 757.
- 228. Patel, J. M.; Wood, J. C.; Leibman, K. C.; Drug Metab. Dispos. 1980, 8, 305.
- 229. Nelson, T. J.; Boor, P. J.; Biochem. Pharmacol. 1982, 31 , 509.
- 230. Izard, C.; Libermann, C.; Mutat. Res. 1978, 47, 115.
- 231. Hales, B. F.; Cancer Res. 1982, 42, 3016.
- 232. Nath, R. G.; Chung, F. L.; Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 7491.
- 233. Gotzschmidt, E. M.; Schreier, P.; J. Agric. Food Chem. 1986, 34, 212.
- 234. Feron, V. J.; Til, H. P.; de Vrijer, F.; Woutersen, R. A.; Cassee, F. R.; van Bladeren, P. J.; Mutat. Res. 1991, 259, 363.
- 235. VanRuth, S. M.; Roozen, J. P.; Cozijnsen, J. L.; Food Chem. 1995, 53, 15.
- 236. Golzer, P.; Janzowski, C.; Pool-Zobel, B. L.; Eisenbrand, G.; Chem. Res. Toxicol. 1996, 9, 1207.
- 237. Curren, R. D.; Yang, L. L.; Conklin, P. M.; Grafstrom, R. C.; Harris, C. C.; Mutat. Res. 1988, 209, 17.
- 238. Smith, R. A.; Cohen, S. M.; Lawson, T. A.; Carcinogenesis 1990, 11, 497.
- 239. Smith, R. A.; Williamson, D. S.; Cerny, R. L.; Cohen, S. M.; Cancer Res. 1990, 50, 3005.
- 240. Canonero, R.; Martelli, A.; Marinari, U. M.; Brambilla, G.; Mutat. Res. 1990, 244, 153.
- 241. Eder, E.; Deininger, C.; Neudecker, T.; Deininger, D.; Environ. Mol. Mutagen. 1992 , 19, 338.
- 242. Chung, F. L.; Tanaka, T.; Hecht, S. S.; Cancer Res. 1986, 46, 1285.
- 243. Cohen, S. M.; Garland, E. M.; St John, M.; Okamura, T.; Smith, R. A.; Cancer Res. 1992, 52, 3577.
- 244. Chung, F. L.; Hecht, S. S.; Cancer Res. 1983, 43, 1230.
- 245. Chung, F. L.; Young, R.; Hecht, S. S.; Cancer Res. 1984, 44, 990.
- 246. Sodum, R. S.; Shapiro, R.; Bioorg. Chem. 1988, 16 , 272.
- 247. Smith, R. A.; Williamson, D. S.; Cohen, S. M.; Chem. Res. Toxicol. 1989, 2, 267.
- 248. Nath, R. G.; Ocando, J. E.; Chung, F. L.; Cancer Res. 1996, 56, 452.
- 249. Nath, R. G.; Ocando, J. E.; Guttenplan, J. B.; Chung, F. L.; Cancer Res. 1998, 58, 581.
- 250. Chung, F. L.; Nath, R. G.; Ocando, J.; Nishikawa, A.; Zhang, L.; Cancer Res. 2000, 60, 1507.
- 251. Chung, F. L.; Zhang, L.; Ocando, J. E.; Nath, R. G.; IARC Sci. Publ. 1999, 45.
- 252. Cajelli, E.; Ferraris, A.; Brambilla, G.; Mutat. Res. 1987, 190, 169.
- 253. Khullar, S.; Varaprasad, C. V.; Johnson, F.; J. Med. Chem. 1999, 42, 947.
- 254. Benamira, M.; Singh, U.; Marnett, L. J.; J. Biol. Chem. 1992, 267, 22392.
- 255. Moriya, M.; Zhang, W.; Johnson, F.; Grollman, A. P.; Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 11899.
- 256. Burcham, P. C.; Marnett, L. J.; J. Biol. Chem. 1994, 269 , 28844.
- 257. Kawanishi, M.; Matsuda, T.; Sasaki, G.; Yagi, T.; Matsui, S.; Takebe, H.; Carcinogenesis 1998, 19, 69.
- 258. Johnson, K. A.; Mierzwa, M. L.; Fink, S. P.; Marnett, L. J.; J. Biol. Chem. 1999, 274, 27112.
- 259. Bartsch, H.; IARC Sci. Publ. 1999, 1.
- 260. Barbin, A.; Bresil, H.; Croisy, A.; Jacquignon, P.; Malaveille, C.; Montesano, R.; Bartsch, H.; Biochem. Biophys. Res. Commun. 1975, 67, 596.
- 261. Foiles, P. G.; Miglietta, L. M.; Nishikawa, A.; Kusmierek, J. T.; Singer, B.; Chung, F. L.; Carcinogenesis 1993, 14, 113.
- 262. Bartsch, H.; Barbin, A.; Marion, M. J.; Nair, J.; Guichard, Y.; Drug Metab. Rev. 1994, 26, 349.
- 263. Eberle, G.; Barbin, A.; Laib, R. J.; Ciroussel, F.; Thomale, J.; Bartsch, H.; Rajewsky, M. F.; Carcinogenesis 1989, 10, 209.
- 264. Ciroussel, F.; Barbin, A.; Eberle, G.; Bartsch, H.; Biochem. Pharmacol. 1990, 39, 1109.
- 265. Swenberg, J. A.; Fedtke, N.; Ciroussel, F.; Barbin, A.; Bartsch, H.; Carcinogenesis 1992, 13, 727.
- 266. Singer, B.; Spengler, S. J.; Chavez, F.; Kusmierek, J. T.; Carcinogenesis 1987, 8, 745.
- 267. Cheng, K. C.; Preston, B. D.; Cahill, D. S.; Dosanjh, M. K.; Singer, B.; Loeb, L. A.; Proc. Natl. Acad. Sci. U. S. A. 1991, 88, 9974.
- 268. Mroczkowska, M. M.; Kusmierek, J. T.; Mutagenesis 1991, 6, 385.
- 269. Singer, B.; Kusmierek, J. T.; Folkman, W.; Chavez, F.; Dosanjh, M. K.; Carcinogenesis 1991, 12, 745.
- 270. Basu, A. K.; Wood, M. L.; Niedernhofer, L. J.; Ramos, L. A.; Essigmann, J. M.; Biochemistry 1993, 32, 12793.
- 271. Pandya, G. A.; Moriya, M.; Biochemistry 1996, 35, 11487.
- 272. Palejwala, V. A.; Rzepka, R. W.; Simha, D.; Humayun, M. Z.; Biochemistry 1993, 32, 4105.
- 273. Shibutani, S.; Suzuki, N.; Matsumoto, Y.; Grollman, A. P.; Biochemistry 1996, 35, 14992.
- 274. Marion, M. J.; Froment, O.; Trepo, C.; Mol. Carcinog. 1991, 4, 450.
- 275. Froment, O.; Boivin, S.; Barbin, A.; Bancel, B.; Trepo, C.; Marion, M. J.; Cancer Res. 1994, 54, 5340.
- 276. Nair, J.; Barbin, A.; Velic, I.; Bartsch, H.; Mutat. Res. 1999, 424, 59.
- 277. Ehrenberg, L.; Osterman-Golkar, S.; Segerbck, D.; Svensson, K.; Calleman, C. J.; Mutat. Res. 1977, 45, 175.
- 278. Segerback, D.; Chem. Biol. Interact. 1983, 45, 139.
- 279. Chen, H. J.; Chung, F. L.; Chem. Res. Toxicol. 1996, 9, 306.
- 280. el Ghissassi, F.; Barbin, A.; Nair, J.; Bartsch, H.; Chem. Res. Toxicol. 1995, 8, 278.
- 281. Goldschmidt, B. M.; Blazej, T. P.; Van Duuren, B. L.; Tetrahedron Lett. 1968, 13, 1583.
- 282. Nair, V.; Offerman, R. J.; J. Org. Chem. 1985, 50, 5627.
- 283. Sodum, R. S.; Chung, F. L.; Cancer Res. 1988, 48, 320.
- 284. Moller, P.; Wallin, H.; Mutat. Res. 1998, 410, 271.
- 285. Langouet, S.; Mican, A. N.; Muller, M.; Fink, S. P.; Marnett, L. J.; Muhle, S. A.; Guengerich, F. P.; Biochemistry 1998, 37, 5184.
- 286. Akasaka, S.; Guengerich, F. P.; Chem. Res. Toxicol. 1999, 12, 501.
- 287. Nair, J.; Gal, A.; Tamir, S.; Tannenbaum, S. R.; Wogan, G. N.; Bartsch, H.; Carcinogenesis 1998, 19, 2081.
- 288. Nair, J.; Carmichael, P. L.; Fernando, R. C.; Phillips, D. H.; Strain, A. J.; Bartsch, H.; Cancer Epidemiol. Biomarkers Prev. 1998, 7, 435.
- 289. Ohshima, H.; Bartsch, H.; Mutat. Res. 1994, 305, 253.
- 290. Nair, J.; Vaca, C. E.; Velic, I.; Mutanen, M.; Valsta, L. M.; Bartsch, H.; Cancer Epidemiol. Biomarkers Prev. 1997, 6, 597.
- 291. Muller-Decker, K.; Scholz, K.; Marks, F.; Furstenberger, G.; Mol. Carcinog. 1995, 12, 31.
- 292. Lehmann, W. D.; Stephan, M.; Furstenberger, G.; Anal. Biochem. 1992, 204, 158.
- 293. Krieg, P.; Kinzig, A.; Ress-Loschke, M.; Vogel, S.; Vanlandingham, B.; Stephan, M.; Lehmann, W. D.; Marks, F.; Furstenberger, G.; Mol. Carcinog. 1995, 14, 118.
- 294. Burger, F.; Krieg, P.; Kinzig, A.; Schurich, B.; Marks, F.; Furstenberger, G.; Mol. Carcinog. 1999, 24, 108.
- 295. Muller-Decker, K.; Kopp-Schneider, A.; Marks, F.; Seibert, K.; Furstenberger, G.; Mol. Carcinog. 1998, 23, 36.
- 296. Schmid, K.; Nair, J.; Winde, G.; Velic, I.; Bartsch, H.; Int. J. Cancer 2000, 87, 1.
- 297. Singer, B.; Hang, B.; IARC Sci. Publ. 1999, 233.
- 298. Guichard, Y.; el Ghissassi, F.; Nair, J.; Bartsch, H.; Barbin, A.; Carcinogenesis 1996, 17, 1553.
- 299. Nair, J.; IARC Sci. Publ. 1999, 55.
- 300. Reiss, U.; Tappel, A. L.; Lipids 1973, 8, 199.
- 301. Fujimoto, K.; Neff, W. E.; Frankel, E. N.; Biochim. Biophys. Acta 1984, 795, 100.
- 302. Frankel, E. N.; Neff, W. E.; Selke, E.; Lipids 1983, 18, 353.
- 303. Frankel, E. N.; Neff, W. E.; Brooks, D. D.; Fujimoto, K.; Biochim. Biophys. Acta 1987, 919, 239.
- 304. Hasegawa, K.; Fujimoto, K.; Kaneda, T.; Frankel, E. N.; Biochim. Biophys. Acta 1988, 962, 371.
- 305. Bao, M. L.; Barbieri, K.; Burrini, D.; Griffini, O.; Pantani, F.; Water Res. 1997, 31, 1719.
- 306. Milo, C.; Grosch, W.; J. Agric. Food Chem. 1996, 44, 2366.
- 307. Yoshiwa, T.; Morimoto, K.; Sakamoto, K.; Ishikawa, Y.; Tokita, M.; Morita, M.; Nippon Suisan Gakkaishi 1997, 63, 222.
- 308. Cha, Y. J.; Cadwallader, K. R.; J. Agric. Food Chem. 1998, 46, 1123.
- 309. Brewer, M. S.; Vega, J. D.; J. Food Sci. 1995, 60, 592.
- 310. Konopka, U. C.; Guth, H.; Grosch, W.; Zeitschrift fur Lebensmittel-Untersuchung Und-Forschung 1995, 201, 339.
- 311. Farkas, P.; Sadecka, J.; Kovac, M.; Siegmund, B.; Leitner, E.; Pfannhauser, W.; Food Chem. 1997, 60, 617.
- 312. Kerler, J.; Grosch, W.; Zeitschrift fur Lebensmittel-Untersuchung Und-Forschung A-Food Research and Technology 1997, 205, 232.
- 314. Moshonas, M. G.; Shaw, P. E.; J. Agric. Food Chem. 1979, 27, 210.
- 315. Boosfeld, J.; Vitzthum, O. G.; J. Food Sci. 1995, 60, 1092.
- 316. Boeve, J. L.; Gfeller, H.; Schlunegger, U. P.; Francke, W.; Biochem. Syst. Ecol. 1997, 25, 195.
- 317. Almosnino, A. M.; Belin, J. M.; Biotechnol. Lett. 1991, 13, 893.
- 318. Andrianarison, R. H.; Rabinovitch-Chable, H.; Beneytout, J. L.; Biochem. Biophys. Res. Commun. 1991, 180, 1002.
- 319. Kaneko, T.; Honda, S.; Nakano, S.; Matsuo, M.; Chem. Biol. Interact. 1987, 63, 127.
- 320. Kaneko, T.; Kaji, K.; Matsuo, M.; Chem. Biol. Interact. 1988, 67, 295.
- 321. Nappez, C.; Battu, S.; Beneytout, J. L.; Cancer Lett. 1996, 99, 115.
- 322. Carvalho, V. M.; Di Mascio, P.; Arruda-Campos, I. P.; Douki, T.; Cadet, J.; Medeiros, M. H. G.; Chem. Res. Toxicol. 1998, 11, 1042.
- 323. Cadet, J.; Carvalho, V. M.; Onuki, J.; Douki, T.; Medeiros, M. H. G.; Di Mascio, P.; IARC Sci. Publ. 1999, 103.
- 324. Carvalho, V. M.; Asahara, F.; Di Mascio, P.; Arruda-Campos, I. P.; Cadet, J.; Medeiros, M. H. G.; Chem. Res. Toxicol. 2000, 13, 397.
- 325. Loureiro, A. P. M.; Di Mascio, P.; Gomes, O. F.; Medeiros, M. H. G.; Chem. Res. Toxicol. 2000, 13, 601.
- 326. Rindgen, D.; Nakajima, M.; Wehrli, S.; Xu, K.; Blair, I. A.; Chem. Res. Toxicol. 1999, 12, 1195.
- 327. Lee, S. H.; Rindgen, D.; Bible, R. H., Jr.; Hajdu, E.; Blair, I. A.; Chem. Res. Toxicol. 2000, 13, 565.
- 328. Hecht, S. S.; Young-Sciame, R.; Chung, F. L.; Chem. Res. Toxicol. 1992, 5, 706.
- 329. Liu, Z.; Young-Sciame, R.; Hecht, S. S.; Chem. Res. Toxicol. 1996, 9, 774.
- 330. Xu, G.; Sayre, L. M.; Chem. Res. Toxicol. 1998, 11, 247.
- 331. Pryor, W. A.; Porter, N. A.; Free Radical Biol. Med. 1990, 8, 541.
- 332. Grosch, W.; Trends Food Sci. Technol. 1993, 4, 68.
- 333. Gardner, H. W.; Selke, E.; Lipids 1984, 19, 375.
- 334. Frankel, E. N.; Neff, W. E.; Selke, E.; Lipids 1981, 16, 279.
- 335. Hidalgo, F. J.; Zamora, R.; J. Biol. Chem. 1993, 268, 16190.
Datas de Publicação
-
Publicação nesta coleção
13 Nov 2002 -
Data do Fascículo
Set 2002
Histórico
-
Recebido
05 Jul 2001 -
Aceito
28 Nov 2001