Resumo
Glucocorticosteroids (CT) show a powerful anti-inflammatory effect. In view of this, they are extensively used in sports medicine, but their misuse (systemic administration) can lead to severe injuries on athletes and has been restricted by the IOC in recent years. For GC-MS analysis, a derivatization step is necessary in order to convert them to a stable form, thus preventing thermal breakdown with the loss of the ketone side chain. Derivatization of CT is reviewed and a new approach of silylation is presented for screening of CT in urine.
glucocorticosteroids; GC-MS; doping control
glucocorticosteroids; GC-MS; doping control
Artigo
ANÁLISE DE GLICOCORTICOSTERÓIDES POR CG-EM: UMA NOVA ABORDAGEM DE DERIVATIZAÇÃO PARA O CONTROLE DE DOPAGEM NO ESPORTE
Henrique M. G. Pereira*, Marlice A. S. Marques, Jarí N. Cardoso e Francisco R. Aquino Neto
Instituto de Química, Universidade Federal do Rio de Janeiro, Cidade Universitária, 21949-900 Rio de Janeiro ¾ RJ
*e-mail: ladetec@iq.ufrj.br
Recebido em 30/10/01; aceito em 28/3/02
ANALISYS OF GLUCOCORTICOSTEROIDS BY GC-MS: A NEW APPROACH OF DERIVATIZATION STEP FOR DOPING CONTROL IN SPORT. Glucocorticosteroids (CT) show a powerful anti-inflammatory effect. In view of this, they are extensively used in sports medicine, but their misuse (systemic administration) can lead to severe injuries on athletes and has been restricted by the IOC in recent years. For GC-MS analysis, a derivatization step is necessary in order to convert them to a stable form, thus preventing thermal breakdown with the loss of the ketone side chain. Derivatization of CT is reviewed and a new approach of silylation is presented for screening of CT in urine.
Keywords: glucocorticosteroids; GC-MS; doping control.
INTRODUÇÃO
Glicocorticosteróides (CT) são esteróides polifuncionais secretados pela supra-renal que apresentam como funções principais a homeostasia de eletrólitos como o íon sódio (Na+) e potássio (K+), a mediação no metabolismo de carboidratos, proteínas e lipídios e, principalmente, uma potente ação antiinflamatória1. Esta ampla gama de efeitos gerada pela administração de CT endógenos e sintéticos resulta em uma série de aplicações terapêuticas que vão do tratamento da artrite até a terapêutica de neoplasias malignas1,2. A Figura 1 esquematiza a biossíntese dos esteróides endógenos1-3.
A principal utilização de CT em desacordo com a sua destinação médica está relacionada à dopagem esportiva, tanto em humanos como em animais (especialmente cavalos de corrida)4,5. A principal razão para a utilização de CT está relacionada à sua ação antiinflamatória e analgésica6,7. A utilização de CT permite a participação de atletas (e animais) com lesões articulares em provas esportivas reprimindo provisoriamente a dor e a inflamação. Disto resultam conseqüências gravíssimas para o atleta, freqüentemente irreversíveis.
Pelo exposto, o Comitê Olímpico Internacional (COI) restringe a utilização sistêmica de CT. Deste modo, administrações pelas vias oral, retal, intravenosa e intramuscular ficam proibidas. Administrações intra-articulares e tópicas são permitidas, desde que previamente declaradas por um médico responsável8,9.
Ao longo das últimas duas décadas, técnicas analíticas como a cromatografia líquida com detector de ultravioleta (CLAE¾UV)10,11, cromatografia líquida acoplada à espectrometria de massas (CLAE¾EM)4,11-13 e cromatografia gasosa acoplada à espectrometria de massas com ionização negativa (CG¾EM¾IN)5-7,14 e cromatografia gasosa acoplada à espectrometria de massas por ionização positiva (CG-EM)15-27 têm sido empregadas na detecção de esteróides polifuncionais.
A técnica de CLAE-UV apresenta como maiores limitações a baixa sensibilidade e a baixa especificidade devido à falta de informação estrutural sobre o analito de interesse. Tanto a CLAE-EM como a CG¾EM¾IN têm merecido grande destaque por sua alta sensibilidade. Entretanto, o emprego de tais técnicas de triagem em laboratórios de controle de dopagem se torna crítico, uma vez que são exigidos, entre outros requisitos, grande reprodutibilidade e praticidade de manuseio devido ao elevado número de amostras.
Deste modo, a CG-EM tem sido a técnica analítica mais utilizada dentro do cotidiano de laboratórios de controle de dopagem na análise de CT e outros esteróides.
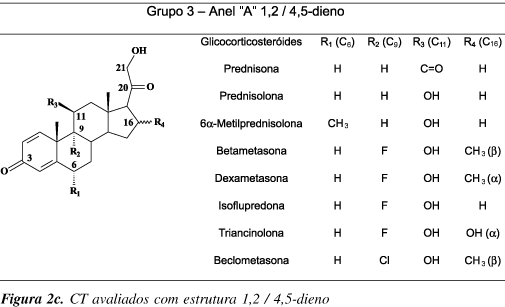
Estruturalmente, CT apresentam grupos polares, hidroxilas ou cetonas conjugadas, na posição 3 (anel A) e uma cadeia lateral hidroxicetônica na posição 17 (anel D). Grupos cetona ou hidroxila são também comuns na posição 11 (anel C), (Figuras 2a e 2b). Alguns CT sintéticos apresentam substituições na posição 16, como a dexamentasona (R = OH) e a triancinolona (R= CH3) (Figura 2c). O metabolismo de CT envolve, normalmente, a hidroxilação da posição 6, como no 6b-hidroxicortisol e a redução dos grupamentos cetona presentes na posição 3, como no tetraidrocortisol e na tetraidrocortisona).
Análise de glicocorticosteróides por CG
O emprego da técnica de cromatografia gasosa (CG) na análise de CT preconiza a adoção de uma etapa de derivatização na preparação da amostra, uma vez que a ramificação hidroxicetônica na posição 17 torna os analitos passíveis de termodegradação16.
Os primeiros estudos de derivatização de esteróides datam do início da década de 196028 (os primeiros estudos de derivatização especificamente sobre CT foram gerados apenas em meados da década seguinte16). A presença de grupamentos hidroxila e cetona sugeriram o emprego de derivados trimetilsilila, inicialmente através de um mecanismo de catálise ácida com trimetilbromosilano17. Entretanto, observou-se diferentes razões de reatividade de acordo com o ambiente químico dos grupos cetonas, com formação de poliderivados e conseqüente queda de rendimento.
Neste contexto, Fales e Luukainen18 utilizaram a clássica reação de formação de metoximas (MO derivados) para a proteção das carbonilas de cetoesteróides (Figura 3).
Os bons resultados observados conduziram Fales18 a recomendar o uso desses derivados na análise estrutural de cetoesteróides por CG-EM. Porém, tal procedimento, como esperado, não protegia os grupamentos OH presentes.
A presença de grupos hidroxila em conjunto com carbonilas cetônicas levaram Horning e colaboradores28 a adotar uma reação em duas etapas. A primeira envolvendo a reação dos CT com hidrocloreto de metoxiamina. A segunda etapa consistindo da reação da metoxima formada com N-N-bis-trimetilsililacetamida (BSA) e trimetilclorossilano (TMCS) à temperatura ambiente por até cinco horas, gerando assim derivados metoximatrimetrilsilila (MO¾TMS). A adoção de duas etapas de derivatização apresentou como vantagem a eliminação da formação de multiderivados através da proteção dos grupos cetonas e posterior silanização dos grupos hidroxila. Além disso, o emprego dos derivados MO¾TMS mostrou-se conveniente devido à sua estabilidade térmica. Entretanto, apesar da derivatização total dos grupos polares no carbono 3, ficou demonstrada a possibilidade de formação de isômeros geométricos sin e anti da metoxima nessa posição, o que para análise quantitativa é considerado uma desvantagem (Figura 4). Não se observa isomerização na formação da metoxima na posição 20.
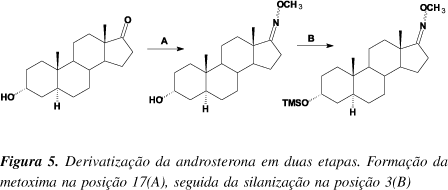
Em 1969, Chambaz e Horning19 desenvolveram estudos sistemáticos de silanização com hidroxicetoesteróides, através de reações não catalisadas e catalisadas com o objetivo de avaliar os graus de impedimento dos grupos hidroxilas, em relação à sua posição na molécula. Na avaliação da androsterona, a cetona em C17 foi previamente protegida pela formação da metoxima, o que possibilitou a formação de apenas um derivado (Figura 5).
Baillie e Brooks16 avaliaram uma série de derivados para CT endógenos como o 11-desoxicortisol (Figura 6), comparando-os com derivados MO¾TMS. Apesar da formação dos produtos propostos, razões como baixos rendimentos e suscetibilidade à hidrólise levaram à não aplicação de tais derivados na análise de CT.
Uma nova abordagem foi introduzida por Chambaz e colaboradores19, quando da proposta de silanização por diferentes reagentes derivatizantes via catálise básica, formando derivados TMS¾enol¾TMS (Figura 7).
Os melhores resultados foram obtidos através do uso de piperidina e acetato (de sódio ou potássio) como catalisador. Nessas condições, observou-se a formação de uma série de derivados de CT com total silanização da cadeia lateral em C17. Grupos hidroxila e cetona na posição 11 não apresentaram reação. CT sintéticos com dupla insaturação no anel A (1,4-dieno) como a triancinolona e a dexametasona não foram investigados. Além disso, os autores discutiram apenas os dados obtidos com acetato de sódio como catalisador.
A silanização de CT também foi alvo da atenção de Simpson20 através da avaliação de CT sintéticos como a triancinolona, betametasona e prednisolona em músculos de rato, utilizando uma mistura de derivatização contendo N-N-bis-trimetilsililacetamida (BSA), N-trimetilsililimidazol (TSIM) e trimetilclorosilano (TMCS) à temperatura ambiente, durante toda a noite.
Observou-se a silanização total dos grupos hidroxila e cetona, inclusive em 1,4-dieno¾CT, ao contrário do observado por Chambaz. Além de demonstrar a possibilidade de formação de um único derivado TMS¾enol¾TMS, sem proteção prévia das carbonilas cetônicas através de metoximas, tal resultado sugere a ocorrência de mecanismos distintos de catálise ácida (Simpson) e básica (Chambaz).
Apesar do resultado de Simpson, derivados MO¾TMS foram exaustivamente utilizados nas décadas de 1970 e 1980. Segundo Pfaffenberger e Horning, derivados MO¾TMS apresentavam os melhores resultados em análises quantitativas e qualitativas de metabólitos de esteróides, uma vez que previnem a enolização de cetonas através da proteção pelo grupo metoxima. As principais vantagens destacadas são a estabilidade térmica, a volatilidade e a impossibilidade de desidratação com perda de simetria do pico cromatográfico. Porém, nada foi mencionado a respeito da formação dos isômeros sin e anti.
Apesar da possibilidade de formação de isômeros geométricos, derivados MO¾TMS foram empregados por Leunissen e Thijsen22 em análises quantitativas e avaliação do perfil urinário de esteróides, por Rodchenkov e colaboradores na determinação de metilprednisonola23, na elucidação do metabolismo de CT sintéticos como a fluorometolona24, betametasona25, dexametasona e triancinolona26 (Figura 8), e na caracterização da prednisona, prednisolona, e metabólitos27.
Entretanto, analitos com substituição na posição 16 apresentam a posição 17 extremamente impedida, o que obriga a realização de reações por até 9 h26. Tal fato não chega a ser um inconveniente significativo em estudos de elucidação de metabolismo de um fármaco. Entretanto, o excessivo tempo de reação impossibilita a utilização de tal derivado como estratégia de triagem no escopo do controle de dopagem. Apesar do exposto, MO¾TMS derivados têm sido reportados na monitoração de CT endógenos como nos trabalhos de Yap e colaboradores29,30.
A falta de um procedimento eficiente na derivatização dos CT tem levado pesquisadores da área a evitar a CG na análise de CT optando, por exemplo, pela CLAE¾EM¾EM com a finalidade de evitar a etapa de derivatização. Porém, em um recente trabalho, Bailloux e colaboradores31 propuseram a adoção de uma etapa de derivatização antes da análise por CLAE¾EM¾EM, visando diminuir os limites de detecção e reduzir alguns problemas inerentes à análise.
Por outro lado, outros esteróides, principalmente aqueles com atividade androgênica, têm sido extensivamente estudados por Schänzer e Donike32. Somente após um trabalho exaustivo de síntese do reagente de silanização metil-N-trimetilsililtrifluoroacetamida (MSTFA, Schänzer e Donike32) e da escolha do catalisador apropriado (NH4I, trimetiliodosilano), tais esteróides foram eficientemente derivatizados. A enolização total de carbonilas cetônicas na posição 3 foi então explorada através do uso do catalisador trimetiliodossilano (TMSI) na presença de MSTFA. Mais tarde, Opfermann e Schänzer33 empregaram uma mistura de silanização que envolve o uso de MSTFA, iodeto de amônio, e eritriol. Posteriormente, o 2-mercaptoetanol substituiu o eritriol por eliminar os problemas de polimerização e conseqüentes interferências observadas na análise. O mecanismo de catálise ácida está baseado na produção de ácido iodídrico in situ34. Até o presente momento, tal mistura não foi avaliada para análise de CT.
Como a maioria dos estudos de derivatização com CT foram conduzidos empregando o reagente N,O-bis-trimetilsililtrifluoroacetamida (o MSTFA só foi sintetizado em 1969), e o iodeto usado por Opfermann e Schänzer33 na derivatização de esteróides androgênicos induz uma via reacional por catálise ácida, o objetivo do presente trabalho foi avaliar o emprego desse procedimento na derivatização de CT. Para tal, comparou-se, para diversos CT endógenos e sintéticos, os resultados obtidos com a mistura MSTFA-NH4I-2-mercaptoetanol (catálise ácida), com os obtidos pelo método descrito por Chambaz (catálise básica)17. Comparou-se ainda o desempenho de derivados TMS¾enol¾TMS com os derivados da metoxima, seguido de silanização (duas etapas).
PARTE EXPERIMENTAL
Reagentes
Todos os padrões de CT foram obtidos da Steraloids (NewPort, EUA). N-metil-N-trimetilsililtrifluoroacetamida (MSTFA), N-trimetilsililimidazol (TSIM), N,O-bis-trimetilsililtrifluoroacetamida (BSTFA), iodeto de amônio, 2-mercaptoetanol, hidrocloreto de metoxiamina e hidrocarbonetos padrões para o cálculo dos índices de retenção foram obtidos da Sigma (St. Louis, EUA). Piperidina foi obtida da Aldrich (Milwaukee, EUA). Piridina foi obtida da Vetec (Rio de Janeiro, BR). Acetato de sódio foi adquirido da Merck (Rio de Janeiro, BR).
Condições de análise no CG-EM
As análises foram realizadas em um sistema cromatográfico Agilent 6890 Series, equipado com um injetor automático 7683 com controle eletrônico de pressão e interfaciado com um detector de massas Agilent 5973. Temperaturas de operação do detector: linha de transferência, 280 oC; fonte iônica, 230 oC; interface, 280 oC; quadrupolo, 150 oC. Detecção por monitoramento seletivo de íons (SIM) com um tempo de varredura de 20 milisegundos. Ionização por impacto de elétrons a 75 eV. Condições de operação do cromatógrafo: injetor, 280 oC; coluna, 180oC (temperatura inicial, 0 min); seguido de um gradiente de 3,0 oC/min até 229 oC/min (0 min) e 40 oC/min até a temperatura final de 310 oC (5,0 min); fluxo total, 18,4 mL/min; pressão, 16,0 psi; velocidade linear média, 38 cm/s; 1 µL de amostra injetada com divisão de fluxo na razão de 1:10. Uma coluna capilar de sílica fundida HP-1 (17,0 m x 0,2 mm x 0,11 µm de espessura de filme) foi utilizada.
Condições de derivatização
A formação dos derivados TMS¾enol¾TMS por catálise ácida foi avaliada utilizando solução de MSTFA¾NH4I¾2¾mercaptoetanol (1000:2:6, v:m:v) a 60 oC por 20 min. Os derivados MO¾TMS foram obtidos pela reação dos CT com uma solução de metoxima¾piridina (8 %) a 60 oC por 30 min. A piridina foi removida sob fluxo de N2 e a silanização foi conduzida com uma mistura MSTFA¾TSIM (2 %) a 70 oC por 15 min. A formação dos derivados TMS-enol-TMS por catálise básica foi avaliada utilizando solução de BSTFA-piperidina (2:1) a 60 oC por 30 min.
Cálculo de índice de Kováts
Os valores do índice de Kováts foram calculados a partir da análise de uma mistura de hidrocarbonetos lineares (n-alcanos entre C8¾ C36), utilizando-se as mesmas condições de análise de CG-EM descritas acima, através da equação 1. Todos ao analitos eluiram durante o gradiente de temperatura.

Equação 1: Cálculo do índice de Kováts. n é o número de carbonos do n-alcano com o tempo de retenção (tRz) imediatamente anterior ao padrão, e tRz+1 o tempo de retenção do n-alcano localizado imediatamente após o padrão avaliado
Cálculo do fator de resposta
No cálculo do fator de resposta dos derivados TMS-enol¾TMS e MO-TMS o 9a-fluoro-17a-metil-4-androsteno-3a,6b,11b,17b-tetrol (Fluoxy ¾ M1) foi utilizado como padrão de referência (Tabelas 1 e 2).
RESULTADOS E DISCUSSÃO
Formação de derivados TMS-enol-TMS via catálise básica
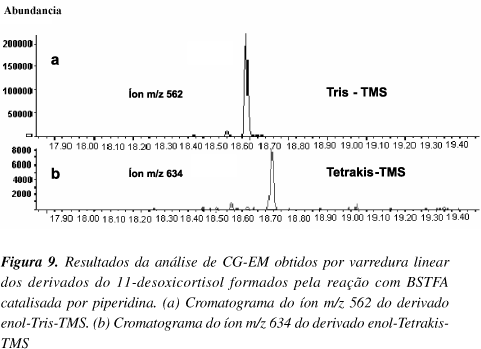
A reprodução do experimento de Chambaz19, realizada pelo LAB DOP utilizando piperidina como catalisador, mostrou que cetonas na posição 3-ceto-4-eno (conjugadas) apresentam enolização parcial, formando dois derivados (Figura 9). A formação de múltiplos derivados é um inconveniente analítico na detecção de CT endógenos como o cortisol, cortisona e 11-desoxicortisol.
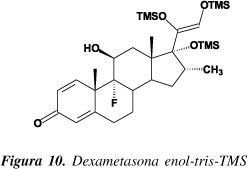
Como esperado 1,4-dieno¾CT não apresentam reação no anel A. Neste estudo, observa-se a formação da dexametasona¾tris¾TMS com total silanização do anel D, inclusive na posição 17, consideravelmente impedida pelo grupo metila na posição 16 (Figura 10). Com relação aos catalisadores utilizados, o autor menciona as vantagens da piperidina sobre os sais de acetato (mesma eficiência, porém maior volatilidade e forma líquida). Foi observado, porém, que a piperidina está sujeita a degradação pela exposição à atmosfera, o que preconiza a adoção de controles de derivatização e cuidados na manipulação, além de purificações periódicas, indesejáveis no contexto de rotina.
Formação de derivados TMS-enol-TMS via catálise ácida
O procedimento de formação de TMS¾enol¾TMS por catálise ácida com NH4I, apresenta maior praticidade se comparado ao uso de metoxiamina, produzindo, com alto rendimento e em um tempo de reação menor, apenas um derivado para cada CT avaliado (Tabela 1). Apesar do grupo hidroxila no carbono 17 não ser silanizado (a perda de água é o processo mais significativo no espectro de massas, Tabela 1 e Figura 11), os picos são simétricos e os resultados reprodutíveis.Nos CT sintéticos e endógenos com grupos cetona no anel A, observou-se enolização total das carbonilas nas posições 3 e 2035. A enolização do anel A em 3-ceto-4-eno conjugados (CT do grupo 2), via catálise ácida com MSTFA, iodeto de amônio, e 2-mercaptoetanol, com formação do 3,5-dienol é esquematizada na Figura 12.
Parâmetros como perfil cromatográfico, fator de resposta e índice de Kováts foram avaliados para vários CT endógenos e sintéticos quanto à formação de derivados TMS-enol-TMS (Tabela 1). Os resultados encontrados para os derivados MO-TMS são relatados com o objetivo de servir como parâmetro de comparação (Tabela 2).
Na análise dos resultados do derivado MO-TMS para a corticosterona, apesar da separação visual dos picos dos isômeros 3-anti/sin, a área referente aos derivados foi integrada como um único pico.
Para cada CT avaliado, os derivados TMS-enol-TMS apresentaram um fator de resposta superior aos derivados MO¾TMS correspondentes, excetuando-se para a tetraidrocortisona e a isoflupredona (Tabelas 1 e 2). Nas condições cromatográficas adotadas, a análise dos índices de Kováts apresentou coeluição entre CT sintéticos e endógenos para alguns derivados MO¾TMS, como observado entre a prednisolona e um dos isômeros geométricos do cortisol (Figura 13), para o 6b-hidroxicortisol (um metabólito do cortisol) e a isoflupredona. Tal fato pode se constituir em uma desvantagem considerável na detecção de CT sintéticos, uma vez que se espera que os CT endógenos sejam excretados em alta concentração. Por outro lado, dentre os derivados TMS¾enol¾TMS, apenas os epímeros dexametasona e betametasona apresentam coeluição. Nenhuma coeluição foi observada entre os derivados TMS¾enol¾TMS sintéticos e endógenos.
CONCLUSÕES
Até o presente momento, não há na literatura um procedimento eficiente para a derivatização dos glicocorticóides (CT). Embora a formação de MO-TMS seja a estratégia mais freqüentemente empregada, tem-se observado a tendência de substituir a CG por técnicas como a CLAE¾EM¾EM com a finalidade de evitar a etapa de derivatização.
Os derivados TMS¾enol¾TMS obtidos através da silanização via catálise ácida com NH4I, por outro lado, mostram comportamento cromatográfico nitidamente superior aos correspondentes derivados metoxima trimetilsilila. Isto se deve, entre outros fatores, ao fato de que a formação de derivados MO¾TMS em compostos que apresentam impedimento estérico no anel D só é possível através de um longo tempo e condições severas de reação. Portanto, tal procedimento torna-se inviável no contexto de análises de triagem. Em adição, para derivados MO¾TMS, alguns CT exógenos mostram coeluição com CT endógenos (excretados em altas quantidades), tornando difícil a avaliação do perfil de endógenos e o diagnóstico do abuso de CT exógenos. Além disto, o processo de derivatização envolvendo a mistura MSTFA¾NH4¾2¾mercaptoetanol mostrou-se eficiente para todos os CT considerados. Estudos posteriores serão realizados em urina para avaliar a viabilidade deste procedimento na etapa de triagem de CT em baixas concentrações no controle de dopagem.
31. Bailloux, I.; De Barros, S.; Lahaussois, A.; Mathurin, J. C.; De Ceaurriz, J.; Ref. 30, p. 203.
34. Van der Kerkhof, D. H.; Zwikker, J. W.; Kaats-Richters, V. E. M.; Bouwman, K.; van Ooijen; de Boer, D.; Maes, R. A. A.; Ref. 30, p. 129.
-
1Haynes Jr., R. C; Murad, F. Em As Bases Farmacológicas da Terapêutica; Goodman, L.S.; Gilman, A.G., eds.; Guanabara Koogan: Rio de Janeiro, 1987, p. 943.
-
2Goth, A.; Shore, P. A.; Farmacologia Médica, Guanabara Koogan: Rio de Janeiro, 1981, p. 393.
-
3Venturelli, E.; Cavarelli, A.; Secreto, G.; J. Chromatogr., B 1995, 671, 363.
-
4Popot, M. A.; Garcia, P.; Bonnaire, Y. Em Recent Advances in Doping Analysis (5); Schänzer, W.; Geyer, H.; Gotzmann, A.; Mareck-Engelke, U., eds.; Sport und Buch Straub: Köln, 1997, p. 185.
-
5Stanley, S. M.; Wilhelmi, B. S.; Rodgers, J. P.; J. Chromatogr. 1993, 614, 77.
-
6Kim, Y.; Kim, T.; Lee, W.; Rapid Commun. Mass Spectrom 1997, 11, 863.
-
7Her, G. R.; Watson, J. T.; Anal. Biochem 1985, 151, 292.
-
8The OMAC 1999. Olympic Movement Anti-doping Code Appendix A. 4.
-
9Catlin, D. H.; Murray, T. H.; J. Am. Med. Assoc. 1996, 276, 231.
-
10Jusko, W. J.; Pyszczynski, N. A.; Bushway, M. S.; D'Ambrosio, R.; Mis, S. M.; J. Chromatogr., B 1994, 658, 47.
-
11Barbarulo, M. V.; Buiarelli, F.; Ciardi, M.; Giarrusso, A.; Rosati, F. Em 10th Cologne Workshop on Dope Analysis; Proccedings.; Donike, M.; Geyer, H.; Gotzmann, A.; Mareck-Engelke, U.; Rauth, S., eds.; Sport and Buch Straub: Cologne, 1992, p. 249.
-
12Creaser, C. S.; Feely, S. J.; Houghton, E.; Seymour, M.; J. Chromatogr., A 1998, 794, 37.
-
13Savu, S. R.; Silvestro, L.; Haag, A.; Sörgel, F.; J. Mass Spectrom. 1996, 31, 1351.
-
14Delahaut, Ph.; Jacquemin, P.; Colemonts, Y.; Dubois, M.; De Graeve, J.; Deluyker, H.; Chromatography 1997, 696, 203.
-
15Ishibashi, M.; Takayama, H.; Nakagawa, Y.; Noriaki, H.; Chem. Pharm. Bull. 1988, 36, 845.
-
16Baillie, T. A.; Brooks, C. J. W.; Anal. Chem. 1972, 44, 30.
-
17Chambaz, E. M.; Defaye, G.; Madani, C.; Anal. Chem 1973, 45, 1090.
-
18Fales, H. M.; Luukkainen, T.; Anal. Chem. 1965, 37, 955.
-
19Chambaz, E. M.; Horning, E. C.; Anal. Biochem 1969, 30, 7.
-
20Simpson, P. M.; J. Chromatogr. 1973, 77, 161.
-
21Pfaffenberger, C. D.; Horning, E. C.; High Resol. Biomed. Gas Chromatog 1975, 112, 581.
-
22Leunissen, W. J.; Thijsen, J. H. H.; J. Chromatogr. 1978, 146, 365.
-
23Rodchenkov, G. M.; Uralets, V. P.; Semenov, V.; J. Chromatogr. 1987, 423, 15.
-
24Rodchenkov, G. M.; Uralets, V. P.; Semenov, V. A.; J. Chromatogr. 1988, 426, 399.
-
25Rodchenkov, G. M.; Uralets, V. P.; Semenov, V. A.; J. Chromatogr. 1988, 432, 283.
-
26Rodchenkov, G. M.; Uralets, V. P.; Semenov, V. A.; Leclercq, P. A.; J. High Resolut. Chromatogr. Chromatogr. Commun. 1988, 11, 283.
-
27Rodchenkov, G. M.; Vedenin, A. N.; Uralets, V. P.; Semenov, V. A.; J. Chromatogr. 1991, 565, 45.
-
28Horning, M. G.; Moss, A. M.; Horning, E. C.; Anal. Biochem 1968, 22, 284.
-
29Yap, B. K.; Johnston, G. A.; Kazlauskas, R.; J. Chromatogr. 1992, 573, 183.
-
30Yap, B.; Kazlauskas, R. Em Recent Advances in Doping Analysis (7); Schänzer, W.; Geyer, H.; Gotzmann, A.; Mareck-Engelke, U., eds.; Sport and Buch Straub: Cologne, 1999, p. 85.
- 32. Schänzer, W.; Donike, M.; Anal. Chim. Acta, 1993, 275, 23.
- 33. Opfermann, G.; Schänzer, W. Em Recent Advances in Doping Analysis (4); Schänzer, W.; Geyer, W.; Gotzmann, A.; Mareck-Engelke, U., eds.; Sport and Buch Straub: Cologne, 1996, p. 247.
- 35. Pereira, H.M.G.; Marques, M.A,S.; Cardoso, J.N.; de Aquino Neto, F. R. Em Recent Advances in Doping Analysis (9); Schänzer, W.; Geyer, H.; Gotzmann, A.; Mareck-Engelke, U., eds.; Sport and Buch Straub: Cologne, 2001, p. 203.
Datas de Publicação
-
Publicação nesta coleção
12 Dez 2002 -
Data do Fascículo
Dez 2002
Histórico
-
Aceito
28 Mar 2002 -
Recebido
30 Out 2001