Resumo
Although electrospray ionization mass spectrometry (ES-MS) has been extensively applied to study organic and biochemical species, it is also specially suitable to study inorganic and organo-metallic species. Such species, even those that are low-volatile or thermo-unstable, can be easily ionized or simply transferred from liquid to gas phase by electrospray. However, chemical transformations frequently occur during the process. This paper discusses the fundamental aspects of electrospray ionization as well as its analytical applicability to inorganic and organo-metallic species in order to spread the technique and make its characteristics more familiar to potential users.
mass spectrometry; electrospray; metal ions
mass spectrometry; electrospray; metal ions
DIVULGAÇÃO
Espectrometria de massas com ionização por "electrospray" aplicada ao estudo de espécies inorgânicas e organometálicas
Electrospray ionization mass spectrometry applied to study inorganic and organo-metallic species
Maria Carolina B. MoraesI, # * e-mail: claudemi@iq.usp.br # endereço atual: Embrapa Cernagen PqEB, W/5 Norte (final), 70770-900 Brasília - DF ; Claudimir Lucio do LagoII, * * e-mail: claudemi@iq.usp.br # endereço atual: Embrapa Cernagen PqEB, W/5 Norte (final), 70770-900 Brasília - DF
ICentro de Energia Nuclear na Agricultura, Universidade de São Paulo, CP 96, 13400-970 Piracicaba - SP
IIDepartamento de Química Fundamental, Instituto de Química, Universidade de São Paulo, Av. Prof. Lineu Prestes, 748, 05508-900 São Paulo - SP
ABSTRACT
Although electrospray ionization mass spectrometry (ES-MS) has been extensively applied to study organic and biochemical species, it is also specially suitable to study inorganic and organo-metallic species. Such species, even those that are low-volatile or thermo-unstable, can be easily ionized or simply transferred from liquid to gas phase by electrospray. However, chemical transformations frequently occur during the process. This paper discusses the fundamental aspects of electrospray ionization as well as its analytical applicability to inorganic and organo-metallic species in order to spread the technique and make its characteristics more familiar to potential users.
Keywords: mass spectrometry; electrospray; metal ions.
INTRODUÇÃO
Um espectrômetro de massas pode ser entendido, via de regra, como um instrumento contendo uma fonte de íons, um separador ou filtro de massas na realidade, massa/carga (m/z) e um detector. Embora, existam várias estratégias para separação e detecção, a etapa de ionização é aquela com o maior número de diferentes estratégias. Isso se deve à grande variedade de tipos de amostras e espécies de interesse. Amostras sólidas, líquidas ou gasosas contendo espécies voláteis ou não e com interesses voltados desde a análise elementar até a composição de proteínas requerem diferentes processos de ionização. Uma vez gerados os íons, os processos de separação e detecção podem ser escolhidos de acordo com características mais ou menos comuns como sensibilidade, resolução e precisão de m/z e custo. Neste contexto, surgiu a ionização por "electrospray" como uma alternativa para geração de íons a partir de espécies pouco voláteis presentes em fase líquida.
Um levantamento na literatura mostra que, de 1980 a 2000, o número aproximado de artigos publicados usando "electrospray" passou de 100 para 7800, sendo que 80% destes estão relacionados com análise de biomoléculas e de compostos orgânicos, 10% são relativos à instrumentação e estudos sobre os aspectos fundamentais da formação do "electrospray" e 10% das publicações estão associadas a espécies organometálicas e inorgânicas nas mais diferentes formas. Apesar deste fato, vários pesquisadores ainda desconhecem a potencialidade desta técnica, o que motivou este artigo de divulgação.
ASPECTOS FUNDAMENTAIS DA IONIZAÇÃO POR "ELECTROSPRAY"
Embora seja normalmente considerada como uma fonte de ionização, o "electrospray" é, na realidade, um processo de transferência de íons pré-existentes em solução para a fase gasosa. Pode-se dizer que a efetiva ionização (transformação de uma espécie neutra em um íon) é um efeito secundário. De qualquer forma, é fácil entender porque uma técnica que permite a transferência de íons de uma solução para a fase gasosa para análise por espectrometria de massas, em tão pouco tempo, teve um impulso tão grande: isto se deve ao fato da maioria dos processos químicos e bioquímicos ocorrerem em fase líquida, envolvendo muitas vezes espécies pouco voláteis.
O "electrospray" foi sugerido como um possível modo de ionização para espectrometria de massas por Dole em 19681. No entanto, seus experimentos não foram convincentes pois estes visavam a análise de espécies poliméricas, como poliestireno, que não estão ionizados em solução. Foi somente em 1984 que Yamashita e Fenn demonstraram a aplicabilidade da fonte de "electrospray" como um método de ionização branda2.
Métodos de ionização anteriores ao "electrospray", no qual íons são transferidos da fase líquida para a fase gasosa, tal como bombardeamento de átomos rápido (FAB) ou dessorção por plasma, provocam não só a dessolvatação dos íons, mas, muitas vezes, sua fragmentação e formação de íons a partir de moléculas neutras. Isto se deve ao fato de que, nestas técnicas, uma grande quantidade de energia é fornecida de forma focalizada e em um curto período de tempo. A principal vantagem do "electrospray" sobre estas outras técnicas é que a dessolvatação ocorre gradualmente em temperaturas relativamente baixas (tipicamente, de temperatura ambiente até 80 °C), de forma a não gerar fragmentos nem moléculas ionizadas. Assim, muitos dos íons gerados na fase gasosa mantêm exatamente a mesma estrutura e carga das espécies em solução, o que é perfeito para análise de espécies não voláteis e para estudos de especiação. No entanto, isto não ocorre para todas as espécies. Muitas vezes, uma espécie com carga maior ou igual a dois está estabilizada devido à camada de solvatação. À medida que a espécie é dessolvatada, tende a se envolver em processos que levem à redução de sua carga. Além disto, espécies neutras podem ser ionizadas por processos eletrolíticos na fase líquida, formação de agregados iônicos durante o processo de "electrospray" e por dissociação induzida por colisão (CID) já na fase gasosa. A preservação ou não dos íons originais está intimamente ligada ao processo de ionização por "electrospray"3.
A ionização por "electrospray" envolve a formação de um "spray" eletrostático, a partir do qual são geradas pequenas gotas carregadas e destas são liberados os íons. A implementação de uma fonte de "electrospray" é relativamente simples, comparando-se com outras fontes para espectrometria de massas3. É necessária uma fonte de alta tensão (1000 a 7000 V) que esteja em contato com a solução contendo eletrólitos. Tipicamente, esta solução é bombeada através de um capilar (d.i. 50 a 100 µm) com uma vazão inferior a 10 µL/min. No caso de fluxos menores que 1 µL/min, o processo é chamado de "nanoelectrospray".
A Figura 1 mostra uma representação esquemática da fonte de "electrospray" de um espectrômetro Platform II (Micromass, Inglaterra). Toda a região da fonte está à pressão atmosférica. Quando um potencial positivo, por exemplo, é aplicado na solução, os íons positivos tendem a se afastar para uma região menos positiva, isto é, em direção ao contra-eletrodo. Assim, a gota sendo formada na ponta do capilar estará enriquecida em íons positivos. Este tipo de separação de carga é chamado de processo eletroforético. Conforme a densidade de carga aumenta na gota, o campo elétrico formado entre o capilar e o contra eletrodo aumenta provocando a deformação da gota. A gota ganha a forma de um cone que é denominado de cone de Taylor.
Esta gota na forma de cone permanece "presa" ao capilar até o momento em que a densidade de carga na superfície da gota e o aumento da repulsão entre os íons vençam a tensão superficial do líquido, ocorrendo então a liberação de pequenas gotas com alta densidade de carga. A freqüência deste processo depende da magnitude do campo elétrico, da tensão superficial do solvente e da condutividade da solução.
A FONTE DE "ELECTROSPRAY" COMO UMA CÉLULA ELETROQUÍMICA
Como foi relatado anteriormente, a fonte de "electrospray" pode gerar espécies eletroliticamente. Isto porque esta pode ser comparada a uma célula eletrolítica de dois eletrodos. O capilar metálico ou um fio metálico em contato com a solução é um dos eletrodos da célula. Apesar de não estar diretamente em contato com a solução, o contra-eletrodo completa o circuito. Devido ao potencial aplicado e à proximidade entre a ponta do capilar e o contra-eletrodo, há um alto campo elétrico nesta região que provoca a formação do cone de Taylor. O sistema de "electrospray" funciona como um tipo especial de célula eletrolítica. Especial porque o transporte de íons não ocorre ininterruptamente pela solução, como nas células eletrolíticas convencionais. Parte do transporte dos íons ocorre na fase gasosa. Uma reação de oxidação normal ocorre no eletrodo positivo, isto é, na interface solução/fio metálico. Esta reação fornece íons positivos à solução, convertendo átomos do metal em íons metálicos positivos e liberando elétrons (Equação 1). Outra alternativa é remover íons negativos presentes na solução através da oxidação (Equação 2) ou oxidação do solvente ou outra espécie neutra (Equação 3). A redução das espécies ocorre no contraeletrodo (Equação 4). Estes processos são válidos se considerarmos o "electrospray" como fonte de íons positivos. Obviamente, com a reversão dos potenciais sobre os eletrodos, os processos de oxidação e redução seriam invertidos.

Como nas células eletrolíticas convencionais, predominará a reação com menor potencial de oxidação. Isto vai depender da composição do eletrodo, do material na solução e da natureza do solvente. Prova da ocorrência de oxidação eletroquímica no capilar metálico foi fornecida por Blades e colaboradores4,5. Estes usaram um capilar de sílica recoberta com um depósito de zinco como ponta do capilar. Quando o potencial foi aplicado, íons Zn2+ foram detectados. Resultados similares foram obtidos usando capilares de ácido inoxidável, quando íons Fe2+ foram detectados.
O processo eletroquímico poderá ter uma influência desprezível no espectro de massas da espécie em estudo. Por exemplo, no caso da maior parte dos estudos conduzidos por "electrospray" estudos de biomoléculas como peptídeos, proteínas e oligonucleotídoes , as alterações eletroquímicas sobre estas espécies são desprezíveis porque normalmente outras espécies comuns no sistema como eletrólitos, o metal do capilar e o próprio solvente sofrem muito mais facilmente o processo redox6-8. O mesmo também ocorre se, ao invés de aplicar o potencial diretamente no capilar, for utilizado um eletrodo imerso no reservatório da solução. Neste caso, devido ao grande volume de solução comparado com a pequena extensão das transformações eletródicas, há uma diluição das espécies transformadas e pequena ou nenhuma alteração no espectro de massas acaba sendo observada.
DESSOLVATAÇÃO DAS GOTAS
Depois da liberação das gotas com alta densidade de carga do cone de Taylor, estas passam pela região entre a ponta do capilar e o contra eletrodo e vão sofrendo dessolvatação. A evaporação do solvente é favorecida pela energia térmica do ambiente e pelo auxílio de um gás secante, normalmente nitrogênio. Conforme a gota perde solvente, a densidade de carga aumenta até um ponto em que as forças de repulsão vencem a tensão superficial e gotículas são liberadas pela fissão da gota inicial.
A carga máxima Q que uma gota de raio R pode manter antes de se fragmentar é fornecida pela Equação 5.

onde e0 é a permissividade livre no espaço e g é a tensão superficial da gota. Esta dedução foi proposta por Lord Rayleigh (1882) e é comumente conhecida como o limite de Rayleigh9.
Este processo de dessolvatação e fissão das gotas vem sendo estudado por vários grupos. Taflin e colaboradores7 observaram que as gotas para sofrerem fissão não precisam alcançar o limite de Rayleigh. A ruptura ocorre em torno de 80% deste limite e resulta na expulsão de aproximadamente 20 gotas descendentes somando de 2 a 3% da massa e 10 a 18% da carga da gota ascendente. Assim, as gotas descendentes são bem menores que as gotas ascendentes e possuem uma razão massa/carga menor. A fissura precoce da gota deve estar relacionada a pelo menos dois fatores: (1) normalmente, as gotas vibram, o que estimula sua ruptura e (2) a gota que possui um desequilíbrio de cargas sofre deformação por estar imersa em um intenso campo elétrico. Esta deformação pode ser entendida considerando-se o que ocorre originalmente com a solução na ponta do capilar.
A Figura 2 mostra uma representação esquemática do processo de formação das gotas descendentes. As fissões subseqüentes também são mostradas. A Figura 3 mostra esquematicamente o processo de deformação da gota quando esta é inserida na região com um alto campo elétrico. Estas ocorrem em tempos, progressivamente, mais curtos. O tempo de evolução total e produção dos íons na fase gasosa é em torno de microssegundos10.
FORMAÇÃO DO ÍON NA FASE GASOSA
Todo este processo de dessolvatação da gota até atingir o raio de aproximadamente 30 nm já está muito bem entendido. No entanto, ainda não se sabe exatamente como o íon é gerado na fase gasosa. Há dúvidas se o íon é expelido da gota ou se o processo de fissões sucessivas ocorre até restar um único íon. Existem dois diferentes mecanismos que foram propostos para a formação dos íons na fase gasosa.
O primeiro depende da formação de gotas extremamente pequenas, R » 1 nm, e que contenham somente um íon. A evaporação do solvente da gota converterá esta em um íon na fase gasosa. Esse mecanismo foi proposto por Dole em 1968 e foi denominado modelo da carga residual (CRM)1.
O outro mecanismo foi proposto por Thomson e Iribarne modelo da evaporação do íon (IEM)11,12 e sugere que a emissão dos íons ocorre diretamente de gotas muito pequenas e altamente carregadas. Para começar a ocorrer este processo, a gota precisa ter um raio de 8 nm e 70 cargas aproximadamente. Nestas condições, a gota não sofre ruptura mas pode emitir íons para a fase gasosa. Assim, a gota não precisaria atingir tamanhos tão pequenos como 1 nm e podem existir outros solutos.
O IEM foi proposto porque, em espectros de soluções 1 mmol/L de NaCl em metanol:água, o único íon detectado foi Na+ e espécies solvatadas. Íons como Nan(NaCl)n+m, que deveriam ser formados caso o modelo CRM ocorresse, não foram observados em seus experimentos. Somente espécies solvatadas, protonadas e íons livres foram detectados. No entanto, os experimentos de Wang e Cole mostraram a existência de íons como Cs+ e Cs(CsCl)+ em uma solução 50:50 água:metanol13. É ainda provável que ambos os processos estejam ocorrendo simultaneamente.
DISSOCIAÇÃO INDUZIDA POR COLISÃO
Depois de formado, o íon alcança a região do capilar ou cone de amostragem que tem um orifício bem pequeno (da ordem de poucos micrometros). A partir daí, o sistema passa para a região de baixa pressão. Na entrada, há um conjunto de lentes que conduzem os íons ao analisador de massas (Figura 1). O sistema de bombas e ótica varia conforme o produtor do espectrômetro. A região entre o capilar ou cone de amostragem, o "skimmer" e as lentes é muito importante, pois nesta região pode ocorrer dissociação induzida por colisão (CID).
Esta região contém íons formados no "electrospray" e o gás secante (nitrogênio). Como há um campo elétrico, os íons são acelerados provocando a colisão entre estes e as moléculas do gás secante. Assim, por exemplo, se um íon solvatado entrar nesta região ele pode sofrer colisão e perder moléculas do solvente ou até mesmo sofrer processos de rearranjo intramolecular ou fragmentação.
O processo de CID pode ser interessante em alguns estudos, mas pode dificultar a visualização das espécies na forma como elas se encontravam em solução. O fenômeno de CID pode ser minimizado controlando a queda de potencial sentida pelo íon na região do cone de amostragem e o "skimmer".
INTENSIDADE DO PICO VERSUS CONCENTRAÇÃO
Um dos grandes problemas no "electrospray" é a falta de linearidade da intensidade com a concentração. A vazão da amostra, do gás nebulizante, do gás secante e a distância da ponta do capilar ao contra eletrodo são parâmetros que afetam a estabilidade do "electrospray" e, portanto, a formação das espécies na fase gasosa. No entanto, estes são facilmente controlados para se obter um fluxo de amostra constante. Já a presença de outros eletrólitos na solução não é tão fácil de ser controlada e compromete a linearidade de resposta da intensidade de um pico em função da concentração. A seção linear do "electrospray" trabalha numa faixa estreita de concentração (de 10-6 a 10-3 mol/L). Todavia, esta linearidade pode ser prejudicada se outros eletrólitos estiverem presentes na solução.
No solvente utilizado sempre há impurezas. Por exemplo, no metanol as impurezas são principalmente sais de amônio e sódio numa concentração 10-5 mol/L3. Muitas vezes, essas impurezas são necessárias, porque não há formação do "electrospray" se a condutividade for menor do que 10-7 ohm-1 cm-1. Por exemplo, considerando um analito A+ e uma impureza B+, quando A+ estiver em baixas concentrações, B+ predominará na gota e será transportado em maior número para a fase gasosa. Conforme a concentração de A+ aumenta, este começa a dominar. Normalmente, para uma concentração do analito acima de 10-5 mol/L, a intensidade de B+ (IB) começa a diminuir. Os íons A+ competem com os íons B+ no processo de conversão para a fase gasosa. Baseando-se em tais considerações, Kebarle e Tang propuseram a Equação 6. Esta foi elaborada para um sistema de dois eletrólitos, mas pode ser estendida para n eletrólitos10.

onde IA é a intensidade do íon A+ que foi detectada no espectrômetro de massas, [A+] e [B+] são as concentrações dos eletrólitos na solução, ƒ é fração de cargas na gota que são convertidas para a fase gasosa e p é a fração de íons detectados em relação aos íons gerados, kA e kB são constantes que expressam a taxa de transferência de íons da gota carregada para a fase gasosa. A Equação 6 expressa a competição entre os íons em solução pela transferência para a fase gasosa. Assim sendo, a intensidade de uma espécie na fase gasosa, IA, não dependerá somente da sua concentração na solução e sim da razão entre kA / kB .
Através da Equação 6 pode-se perceber que, aumentando a concentração do analito A+, há um aumento da corrente, mas como esta também está no denominador, existe um limite para este aumento. O aumento da concentração não altera a corrente devido a efeitos interiônicos, o que diminui a mobilidade iônica e provoca saturação da espécie na gota10.
No geral, em concentrações acima de 1 mmol/L, a intensidade da corrente mantém-se constante e algumas vezes pode até diminuir. O raio das primeiras gotas formadas por "electrospray" deve ser da ordem de 0,5 a 1 µm para sofrerem todo o processo de dessolvatação e gerarem os íons na fase gasosa (Figura 2). Para tanto, deve-se utilizar capilares com diâmetro interno menor que 100 µm e fluxo de amostra menor que 10 µL/min. Gotas grandes não sofrem dessolvatação eficiente para gerar os íons na fase gasosa. Diminuição do fluxo de amostra e aumento da condutividade são dois parâmetros que permitem obter gotas menores, aumentando a eficiência da dessolvatação.
A supressão do íon devido à competição entre os ligantes pelo átomo central pode ser observada na Figura 4. Esta figura mostra o estudo14 de supressão usando soluções com concentração constante, 100 mmol/L, de Zn(NO3)2 e diferentes concentrações de iodeto, que variaram de 10 µmol/L a 10 mmol/L. Quando o iodeto está numa concentração inferior a 100 µmol/L, a espécie favorecida é o complexo Zn(NO3)3- , com o aumento da concentração de iodeto há um favorecimento da espécie ZnI3-.
DESCARGA CORONA
Um dos principais problemas enfrentados para se obter uma corrente constante de íons é a ocorrência de descarga corona, principalmente no modo de operação para íons negativos. O efeito de descarga corona ocorre quando elétrons livres presentes no gás são acelerados a uma velocidade suficiente para ionizar moléculas de gases presentes na câmara de ionização, provocando a ionização destas e gerando íons como CH3O- a partir do metanol e O2- do ar. A descarga corona diminui a intensidade dos íons do analito e os picos correspondentes às espécies geradas pela descarga aparecem com uma intensidade muito alta. A presença de espécies que capturam elétrons, como os gases O2, SF6 e os hidrocarbonetos policlorados, inibem a descarga corona1. A Figura 5 ilustra este problema. O íon de m/z 32 (Figura 5a) indica a ocorrência de descarga corona. Este espectro foi obtido de uma solução 100 µmol/L de NaNO3 e NaCl em metanol:água 50:50 (v/v) e com uma tensão de 4 kV aplicada no capilar. A Figura 5b mostra o espectro da mesma solução obtida com a tensão no capilar em 2 kV, onde não se observa a formação de espécies características da descarga corona. Além disto, pode-se observar na Figura 5a que há uma supressão do sinal do NO3- e do Cl- comparativamente ao espectro da Figura 5b.
TRANSFORMAÇÕES QUÍMICAS DOS ÍONS PRESENTES NA GOTA CARREGADA DURANTE O PROCESSO DE "ELECTROSPRAY"
À primeira vista, o "electrospray" deveria ser a técnica ideal para se fazer especiação. Bastaria controlar os eletrólitos presentes na solução e facilmente poder-se-ia identificar qualquer espécie iônica. Todavia, quando a gota começa a sofrer o processo de dessolvatação, a concentração das espécies e o pH da gota podem ser significativamente alterados15. Além disso, as mudanças posteriores na fase gasosa normalmente conduzem à redução de carga das espécies e à dissociação induzida por colisão.
A concentração do soluto, que é menos volátil do que o solvente, aumenta porque o volume da gota diminui com a evaporação do solvente. Mudanças na concentração do soluto levam a mudanças do pH e, portanto, do espectro de massas, uma vez que o grau de ionização das espécies depende do pH.
É possível estimar a concentração final do analito na gota através dos raios das gotas formadas conforme estas evaporam. Solutos não voláteis como o NaCl e outros eletrólitos podem aumentar a sua concentração em até 50 vezes na gota final.
O processo de redução da carga da espécie é bastante complexo e pode ocorrer por CID, através da dessolvatação ou por influência do solvente. A redução da carga do complexo devido à evaporação da água é mostrada nas Equações 7 e 8.

Conforme as moléculas do solvente são removidas, o complexo íon-solvente não consegue mais estabilizar a carga devido à diminuição da esfera de solvatação do íon. Embora o estado de oxidação do metal permaneça o mesmo, tais reações podem conduzir à formação de espécies diferentes daquelas encontradas na fase aquosa. Assim, é difícil observar na fase gasosa íons metálicos livres com cargas +3 ou +2, a não ser que o íon esteja complexado.
Além disto, a observação de íons com múltiplas cargas dependerá também do tipo de solvente utilizado. Lantanídeos com carga +3 só podem ser observados com solventes como dimetilsulfóxido (DMSO) e dimetilformamida3-5. Cu(II) tende a sofrer redução na presença de soluções preparadas com acetonitrila como solvente.
ALGUMAS APLICAÇÕES DE ES-MS PARA ESPÉCIES INORGÂNICAS E ORGANOMETÁLICAS
Existem diferentes tipos de analisadores de massas interfaceados com o modo de ionização por "electrospray". O mais comum, devido à facilidade de manuseio e ao menor custo, é o analisador quadrupolar. O "electrospray" com filtro quadrupolar está sendo amplamente utilizado para estudos do comportamento dos íons na fase gasosa, especiação de complexos metálicos e estudos sobre os processos fundamentais de ionização. Outros tipos de analisadores de massa como tempo de vôo, setor magnético, "ion-trap" e ressonância ciclotrônica também já existem comercialmente9.
A técnica de "electrospray" permite a análise de compostos com peso molecular de até 200.000 Da em equipamentos com pequeno alcance de massas (30 a 3000 u). Isto porque, no "electrospray", moléculas com alto peso molecular podem ter múltiplas cargas. Estas cargas podem ser adquiridas pela ionização da macromolécula em diversos pontos da cadeia ou, mais comumente, pela incorporação de espécies iônicas pequenas que estejam presentes na solução. Este é o caso, por exemplo, da incorporação de NH4+ ou Na+ no modo íons positivos ou de HCOO- ou OH- no modo íons negativos. Obviamente, com a incorporação destas espécies, além do aumento de carga, há também o aumento de massa.
As principais aplicações da ionização por "electrospray" têm sido os estudos na área de bioquímica e química orgânica. No entanto, vem crescendo o interesse na técnica para aplicação em estudos fundamentais, de organometálicos e complexos inorgânicos16-26. ES-MS vem sendo utilizado nos mais diferentes estudos como na investigação da acidez de dióis27, estabilidade dos íons na fase gasosa28, identificação de íons organometálicos e íons inorgânicos16-29 e especiação28,33. No Brasil, os estudos para aplicações bioquímicas e orgânicas têm avançado bastante nos últimos três anos, mas tem sido muito modesta a aplicação da técnica para estudos eletroquímicos e de complexos inorgânicos, entre outros.
Pesquisadores como Cole3, Fenn2 e Kebarle30 vêm estudando intensamente o comportamento de íons relativamente simples como metais alcalinos e alcalinos terrosos a fim de tentar esclarecer o processo de transferência dos íons da fase líquida para a fase gasosa31,4,5. Os estudos de Kerbale e Tang 30,31 e Ikomonou4,5 para o entendimento da formação da gota carregada e produção dos íons na fase gasosa foram realizados com íons inorgânicos simples como cloretos e iodetos de sódio, potássio e césio, contribuindo muito para o entendimento sobre o comportamento destes íons em ES-MS.
Uma das questões a serem respondidas é se os complexos observados no espectro de massas são formados na fase gasosa, durante o processo de "electrospray", ou se já estão presentes na fase líquida. Existe uma série de trabalhos que mostra que quando o íon gerado não está no estado de oxidação esperado, este resultou da formação na fase gasosa. Moraes e colaboradores mostraram que espécies como NiI2-, com níquel no estado de oxidação um, são formadas na fase gasosa32.
Os estudos de Horlick e Agnes16-19 mostraram a possibilidade de se utilizar o "electrospray" para a especiação de metais e semimetais. Eles estudaram a dependência da intensidade do sinal com a concentração do analito e de outros eletrólitos presentes na solução e a influência dos parâmetros de formação do "spray" na intensidade do sinal e na composição dos íons gerados. Os experimentos de Horlick e Agnes concordam com os experimentos de Kebarle e Tang e com a teoria por eles desenvolvida (Equação 6). Estes mostraram que a formação de íons por "electrospray" só é gerada dentro de um mínimo de condutividade. Isto é, é necessária uma quantidade mínima de eletrólito na solução para a formação de um "electrospray" estável. No entanto, isto pode suprimir a ionização da espécie de interesse. Por exemplo, entre as espécies I¯ e Cl¯ , a primeira apresenta uma sensibilidade muito maior. A quantificação e especiação das espécies por "electrospray" estão diretamente relacionadas com a supressão do sinal e respostas relativas das intensidades das espécies presentes na solução. As diferentes espécies presentes na solução, o analito, o solvente e as impurezas, podem vir a ter um comportamento completamente diferente do esperado, durante a formação do "spray", como sofrer processos de oxi-redução, agregação, fragmentação e supressão.
Recentemente, diferentes trabalhos vêm sendo publicados onde os autores demonstram a possibilidade de fazer especiação utilizando condições brandas no "electrospray"16-18,20. No entanto, têm-se observado que há algumas dificuldades. Obter íons estáveis com múltiplas cargas no "electrospray" não é uma tarefa fácil. Mesmo íons como [Fe(CN)6]4- e [Fe(CN)6]3- que em fase líquida permitem a estabilização do ferro nos estados de oxidação II e III, apresentaram, em fase gasosa, predominantemente espécies com uma carga com o ferro em diferentes estados de oxidação: I, II, III e IV32. A especiação por "electrospray" é favorecida no caso de complexos volumosos e com múltiplas insaturações, o que ajuda a acomodar cargas maiores. A ionização por "electrospray" de complexos pequenos e monoatômicos, ao contrário dos complexos volumosos, sofre grande influência dos processos eletroquímicos.
Raymond e colaboradores33 demonstraram a possibilidade de utilizar o "electrospray" para identificação de espécies de poliselenetos em soluções aquosas. Orians estudou a especiação de complexos de diferentes metais com ligantes orgânicos. Por exemplo, no caso do complexo de cobre com a 8hidroxiquinolina, estes mostraram que em soluções metanólicas a espécie predominante é o complexo com Cu(II). No entanto, quando o solvente usado foi acetonitrila, a espécie predominante é o complexo com Cu(I)20.
O processo eletroquímico poderá ter uma influência desprezível no espectro de massas se os íons produzidos, consumidos, ou alterados pelo processo eletroquímico não são convertidos para a fase gasosa. No entanto, como discutido por Van Berkel34, Fernandez de La Mora6 e outros22,23, o processo eletroquímico pode ter importância crucial em alguns estudos. Como na especiação de soluções de complexos metálicos que sofrem facilmente processos de oxi-redução.
Van Berkel34 e Moraes e colaboradores14 observaram que, durante o processo de "electrospray" usando-se um capilar de aço inoxidável, no modo íons negativos, pode haver redução do metal no capilar metálico. Isto tem duas conseqüências negativas: (1) alteração do estado de oxidação da espécie podendo inibir ou até impedir a sua detecção e (2) efeito de memória. Como o metal é reduzido e depositado no capilar, ao mudar a solução é necessário um longo tempo passando-se o solvente, com a polaridade positiva, para a limpeza do mesmo. O uso de um simples capilar de sílica fundida ao invés do capilar de aço inoxidável minimiza de forma significativa o processo de redução e o efeito de memória14,35,36 .
Além deste fenômeno, outros processos durante o "electrospray" podem provocar alterações eletroquímicas na solução. Dependendo da tensão aplicada no cone de amostragem, alguns estados de oxidação são observados14.
Um exemplo onde o estado de oxidação do metal não foi alterado devido à formação do "electrospray" pode ser ilustrado pelo estudo para identificação de ftalocianinas sulfonadas de zinco, lutécio e disprósio no modo íons negativos37. Os espectros obtidos para as três ftalocianinas apresentaram clusters do tipo [MPcs2H]3-, [MPcs2NaH2On]3-, [MPcs2Na2H2On ]3-. A Tabela 1 mostra os principais íons identificados através do espectro obtido para uma solução 1 mmol/L de ftalocianina de disprósio em meio metanol:acetonitrila no modo íons negativos. Acredita-se que as estruturas das ftalocianinas consigam estabilizar a carga do complexo e manter suas características mesmo na fase gasosa.
Um exemplo contrário é o estudo realizado por Prada e colaboradores38 para a determinação da composição e especiação de complexos do agente quelante di-2-piridilcetonabenzoilidrazona (DPKBH) com íons de Cu(II), Fe(II) e Fe(III). Estes estudos mostraram que a composição do complexo Cu(II)/DPKBH é 1:2 metal/ligante. Os íons de m/z 667 e 669 indicam a presença do complexo com Cu(II) protonado, [Cu(DPKBH)2H]+. Além disto, foi possível observar os íons de m/z 666 e 668 que indicam a formação do complexo com Cu(III), [Cu(DPKBH)2]+. A oxidação do Cu(II) a Cu(III) pode estar ocorrendo durante a dessolvatação das gotas no processo de ionização. O mesmo comportamento foi observado para o espectro com Fe(II), com a formação de [FeII(DPKBH)2H]+ e [FeII(DPKBH)2]+ (Figura 6). Para o complexo com Fe(III), somente o cluster de picos respectivos a [FeIII(DPKBH)2]+ foi observado. Estas espécies indicam a labilidade do complexo, uma vez que os espectros foram obtidos a baixas tensões do cone, quando não há favorecimento de dissociação por CID38. Apesar das estruturas destes compostos serem volumosas e apresentarem um número de sítios que poderiam estabilizar a carga do complexo, o metal sofreu alteração do seu estado de oxidação. Na fase aquosa, seriam esperados somente os complexos com Cu(II).
Importantes estudos vêm sendo conduzidos para a identificação e quantificação de metalotioneínas em fluidos biológicos. Os primeiros estudos de complexos envolvendo metalotioneínas foram conduzidos por Fenselau e colaboradores15, mostrando a possibilidade de se utilizar o "electrospray" para estudos envolvendo as metalotioneínas nos organismos vivos. Eles monitoraram o fluxo de entrada e saída de cádmio e zinco das metalotioneínas por ES-MS.
A compreensão da importância dos metais nos mecanismos moleculares e sua toxicidade e essencialidade nos seres vivos e nos ciclos biogeoquímicos depende de técnicas analíticas capazes de fornecerem informação da forma química na qual os elementos estão presentes nos organismos e na natureza. Muitos estudos relacionados à biometilação de metais (principalmente mercúrio, chumbo e cádmio) e semimetais (arsênio, selênio e telúrio) têm sido feitos, uma vez que estes elementos podem apresentar um alto nível de toxicidade, dependendo do estado de oxidação em que se encontram.
O acoplamento de eletroforese capilar à ES-MS, ou CE-ES-MS, é uma poderosa ferramenta na especiação, pois associa as vantagens da separação eficiente das espécies em pequenos volumes por eletroforese capilar com a alta sensibilidade da espectrometria de massas.
A proposta de uma interface para o acoplamento do sistema de CEES-MS tem sido objeto de estudo de várias pesquisas em química analítica. Entretanto, certas considerações com respeito ao acoplamento CE-ES-MS devem ser avaliadas tais como a diferença de intensidade de corrente operando no sistema. O circuito CE opera com uma corrente de dezenas de micro amperes, enquanto o "electrospray" opera com uma corrente em torno de centenas de nano amperes. Isto pode provocar uma mudança na tensão na ponta do capilar, o que altera a formação do "spray". Hau e Roberts39 propuseram um sistema bastante simples para o acoplamento do sistema CE-ES-MS, onde este efeito é minimizado aterrando-se o capilar do eletrólito auxiliar e usando um capilar para este fluxo com um diâmetro bem maior do que o da eletroforese. Assim, há uma diminuição da resistência e pode-se garantir que a tensão na ponta do capilar é mantida constante.
Estudos para especiação usando CE-ES-MS ainda não são muitos. Corr e Anacleto40 fizeram um estudo inicial para avaliar a interface para especiação de metais, o grau de formação de cluster e fragmentação. Os estudos foram realizados com soluções padrão de 29 cátions e 15 ânions inorgânicos. Os espectros foram obtidos tanto no modo íons negativos como positivos. Schramel e colaboradores22 realizaram estudos com espécies metálicas usando um sistema comercial CE-ES-MS. No entanto, os autores relataram que o sistema comercial não produz "electrospray" estável durante um período longo de aquisição. Assim, estes mudaram o capilar de amostragem do sistema comercial por um capilar de eletroforese de 50 µm de diâmetro interno. Inicialmente os autores estudaram os complexos CuEDTA, CuCl2, cloreto de trimetrilantimônio (TMSbCl2) e espécies como selenometionina e selenocisteína sem o acoplamento. O sistema hifenado foi avaliado para a especiação de selenometionina, selenocistina e selenocistamina. Lintschinger e colaboradores41 investigaram a forma química do dicloreto de trimetilantimônio (TMSbCl2) em soluções aquosas. Zheng e colaboradores42 mostraram a viabilidade do ES-MS sem acoplamento a técnicas de separação para a especiação dos compostos TMSbCl2 e TMSb(OH)2, tanto no modo íons positivos como negativos e compostos inorgânicos de Sb(II) e Sb(V) no modo íons negativos.
Um outro aspecto bastante relevante é que o "electrospray" pode ser uma ferramenta alternativa às fontes de plasma acoplado indutivamente (ICP) e ionização térmica para estudos de medidas isotópicas. Ketterrer e Guzowisk43, em 1997, foram os primeiros a propor o uso da técnica para medidas de razão isotópica. No entanto, estes propuseram o uso do "electrospray" como uma técnica para geração de espécies monoatômicas. Usando condições drásticas de ionização, é possível gerar espécies monoatômicas, como em ICP-MS. Estes mostraram a aplicabilidade da técnica para prata, chumbo e tálio. No caso da prata e tálio não houve problema. No entanto, para o chumbo, foi detectada a espécie PbH+ que foi comprovada usando solvente deuterado, sendo detectada a espécie PbD+. Assim, não é possível usar o espectro de massas para obter diretamente o padrão isotópico do chumbo. Uma alternativa seria determinar o padrão isotópico do "cluster" do PbH+ e subtraí-lo do padrão isotópico do Pb+. Moraes e colaboradores desenvolveram um procedimento matemático para eliminação de interferentes isobáricos gerados pela perda de um ou mais átomos de hidrogênio em conjuntos de picos45. Recentemente, Brito e Lago45 publicaram um outro procedimento matemático bastante genérico para calcular o padrão isotópico e eliminar interferentes isobáricos em um determinado "cluster".
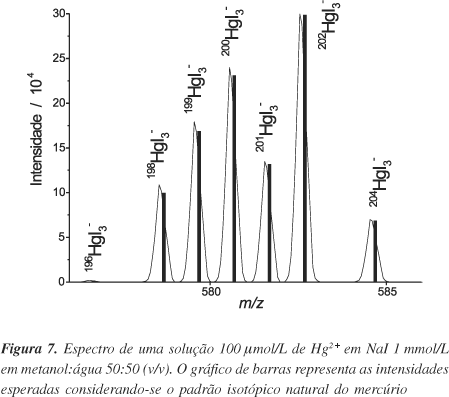
A técnica de ES-MS assim como a espectrometria de massas com ionização térmica (TIMS) permite a utilização de espécies poliatômicas, aumentando as possibilidades de se trabalhar em regiões de m/z livres de interferências isobáricas comuns. Moraes e colaboradores32 estudaram a viabilidade de se utilizar ES-MS para determinações de padrões isotópicos usando complexos de Zn, Cd, Ni, Fe, Hg, Cu, Tl e B47 usando ligantes monoatômicos, como I- e F-, que não interferem no padrão isotópico. A Figura 7 apresenta o espectro de uma solução 100 µmol/L de Hg2+ e 1 mmol/L de NaI no modo íons negativos e o padrão isotópico do mercúrio, mostrando que, além da boa concordância, o espectro está livre de interferentes como observado por Ketterrer e Guzowisk43.
CONCLUSÕES
ES-MS é uma técnica bastante conveniente para estudos de compostos pouco voláteis. Embora, em geral, os íons pré-existentes em fase líquida sejam observados através dos espectros, outros podem ser produzidos durante o "electrospray". O surgimento destas novas espécies é dependente da composição da solução solvente, pH, presença de espécies ligantes e supressores e das condições da fonte de "electrospray" tensões no capilar, contra-eletrodo e lentes e vazões dos gases. Estes fatores devem ser cuidadosamente considerados já que estas alterações costumam levar a uma interpretação equivocada da composição da solução original.
A técnica de ionização por "electrospray" apresenta uma enorme versatilidade e é uma importante ferramenta nos mais variados estudos desde a identificação e quantificação de espécies inorgânicas e organometálicos até medidas isotópicas e especiação. A técnica acoplada a métodos de separação apresenta uma alternativa aos métodos espectroscópicos normalmente utilizados para detecção de metais, como ICP-MS. A técnica é bastante promissora mas ainda há muitos estudos a serem realizados e otimizados principalmente nos campos do acoplamento CE-ES-MS, das medidas isotópicas e do desenvolvimento da instrumentação.
Uma das grandes vantagens da técnica é a relativa simplicidade da fonte de ionização comparada a outras fontes utilizadas na espectrometria de massas, o que permite uma versatilidade grande de configurações, dependendo da criatividade do pesquisador.
9. Gatlin, C. L.; Turecek, F. Em ref. 3, p. 527.
Recebido em 27/5/02
Aceito em 20/11/02
- 1. Dole, M.; Mack, L. L.; Hines, R. L.; Mobley, R. C.; Fergunson, L. D.; Alice, M. B.; J. Chem. Phys. 1968, 49, 2240.
- 2. Yamashita, M.; Fenn, J. B.; J. Phys. Chem. 1984, 88, 4671.
- 3. Cole, R. B. Em Electrospray Ionization Mass Spectrometry: Fundamentals, Instrumentation, and Applications; Cole, R.B., ed.; John Wiley & Sons: New York, 1997, p. 12.
- 4. Blades, A. T.; Jayaweera, P.; Ikonomou, M. G.; Kebarle, P.; Int. J. Mass Spectrom. 1990, 101, 325.
- 5. Blades, A. T.; Jayaweera, P.; Ikonomou, M. G.; Kebarle, P.; Int. J. Mass Spectrom. 1990, 102, 251.
- 6. De La Mora, J. F.; Van Berkel, G. J.; Enke, C. G.; Cole, R. B.; Sanchez, M. M.; Fenn, J. B; J. Mass Spectrom. 2000, 35, 939.
- 7. Taflin, D. C.; Ward, T. L.; Davis, E. J.; Langmuir 1989, 5, 376.
- 8. Bond, A. M.; Colton, R.; D'Agostino, A.; Downard, A. J.; Traeger, J. C.; Anal.Chem. 1995, 67, 1691.
- 10. Tang, L.; Kebarle, P.; Anal. Chem 1993, 65, 3654.
- 11. Thomson, B. A.; Iribarne, J. V.; J. Chem. Phys 1979, 71, 4451.
- 12. Iribarne, J. V.; Thomson, B. A.; J. Chem. Phys 1976, 64, 2287.
- 13. Wang, G.; Cole, R. B.; Anal. Chem. 1994, 66, 3702.
- 14. Moraes, M. C. B.; Tese de Doutorado, Universidade de São Paulo, Brasil, 2001.
- 15. Kelly, M. A.; Vestling, M. M.; Fenselau, C. C.; Smith, P. B.; Org. Mass Spectrom. 1992, 27, 1143.
- 16. Agnes, G. R.; Horlick, G.; Appl. Spectrosc 1992, 46, 401.
- 17. Agnes, G. R.; Horlick, G.; Appl. Spectrosc 1994, 48, 649.
- 18. Agnes, G. R.; Horlick, G.; Appl. Spectrosc 1994, 48, 655.
- 19. Agnes, G. R.; Horlick, G.; Appl. Spectrosc 1995, 49, 324.
- 20. Ross, A. R. S.; Ikonomou, M. G.; Thompson, A. J.; Orians, K. J.; Anal. Chem 1998, 70, 2225.
- 21. Mcsheedy, S.; Pohl, P.; Szpunar, J.; Potin-Guatier, M.; Lóbinsk, R.; J. Anal. At. Spectrom 2001, 16, 6873.
- 22. Schramel, O.; Michalke, B.; Kettrup, A.; J. Chromatogr., A 1998, 819, 231.
- 23. Schramel, O.; Michalke, B.; Kettrup, A.; Fresenius' J. Anal. Chem. 1999, 363, 459.
- 24. Madren, A. D.; Goessler, W.; Pederson, S. N.; Francesconi, K.V.; J. Anal. At. Spectrom. 2000, 15, 657.
- 25. Stewart, I. I.; Horlick, G.; Trends Anal.Chem 1996, 15, 80.
- 26. Stewart, I. I.; Spectrochim. Acta, Part B 1999, 54, 1649.
- 27. Crowder, C. A.; Batmess, J. E.; J. Am. Chem. Soc. Mass Spectrom. 1993, 4, 723.
- 28. Blades, A. T.; Kebarle, P.; J. Am. Chem. Soc 1994, 16, 10761.
- 29. Colton, R.; Traeger, J. C.; Inorg. Chim. Acta 1992, 202, 153.
- 30. Kebarle, P.; Tang, L.; Anal Chem 1993, 65, 972A.
- 31. Gómez, A.; Tang, K.; Phys. Fluids 1994, 6, 404.
- 32. Moraes, M. C. B.; Brito-Neto, J. G. A.; Lago, C. L.; Int. J. Mass Spectrom. 2000, 198, 121.
- 33. Raymond, C. C.; Dick, D. C.; Dorhout, P. K.; Inorg Chem 1997, 36, 2678.
- 34. Van Berkel, G. J.; J. Mass Spectrom 2000, 35, 773.
- 35. Jackson, G. S.; Enke, C. G.; Anal. Chem. 1998, 13, 603.
- 36. Kertesz, V.; Van Berkel, G. J.; J. Mass Spectrom. 2001, 36, 204.
- 37. Santos, M. R.; Tese de Doutorado; Universidade de São Paulo, Brasil, 2000.
- 38. Prado, S. M.; Moraes, M. C. B.; Suarez-Iha, M. E.; Lago, C. L.; 23º Encontro Nacional de Química Analítica, Santa Maria, Brasil, 2000.
- 39. Hau, J.; Roberts, M.; Anal. Chem 1999, 71, 3977.
- 40. Corr, J. J.; Anaceleto, J. F.; Anal. Chem 1996, 68, 2155.
- 41. Scramel, O.; Michalke, B.; Kettrup, A.; J. Chromatogr., A 1998, 819, 231.
- 42. Lintschinger, J.; Schramel, O.; Kettrup, A.; Fresenius' J. Anal. Chem 1998, 361, 96.
- 43. Zheng, J.; Takeda, A.; Furuta, N.; J Anal. At. Spectrom 2001, 16, 62.
- 44. Ketterer, M. E; Guzowisk, J.; Anal. Chem 1996, 68, 883.
- 45. Moraes, M. C. B.; Brito, J. G. A.; Lago, C. L.; Int. J. Mass Spectrom 1998, 178, 129.
- 46. Brito-Neto, J. G. A.; Lago, C. L.; Int. J. Mass Spectrom. 2002, 216, 95.
- 47. Moraes, M. C. B.; Brito, J. G. A.; Lago, C. L.; J. Anal. At. Spectrom. 2001, 16, 1259.
Datas de Publicação
-
Publicação nesta coleção
04 Ago 2003 -
Data do Fascículo
Ago 2003
Histórico
-
Aceito
20 Nov 2002 -
Recebido
27 Maio 2002