Resumo
This paper provides a survey of general aspects involved in the coordination chemistry of low-valent (mainly +III,+II), low-spin (d p5,d p6) ruthenium ions with ethylenediamine-N,N,N',N'-tetraacetate (edta) and their substituted derivatives. The topics covered herein include structure, reactivity, kinetics, thermodynamics, electrochemistry and spectroscopy. The contributions from either our research group or the literature over the last three decades are focused in this review.
ruthenium; ethylenedinitrilotetraacetate; edta
ruthenium; ethylenedinitrilotetraacetate; edta
REVISÃO
Química de complexos de (etilenodiaminatetraacetato)rutenato(III/II)
Ruthenium complexes containing ethylenediaminetetraacetate
Francisca N. Rein; Reginaldo C. Rocha; Henrique E. Toma* * e-mail: henetoma@iq.usp.br
Instituto de Química, Universidade de São Paulo, CP 26077, 05513-970 São Paulo - SP
ABSTRACT
This paper provides a survey of general aspects involved in the coordination chemistry of low-valent (mainly +III,+II), low-spin (dp5,dp6) ruthenium ions with ethylenediamine-N,N,N',N'-tetraacetate (edta) and their substituted derivatives. The topics covered herein include structure, reactivity, kinetics, thermodynamics, electrochemistry and spectroscopy. The contributions from either our research group or the literature over the last three decades are focused in this review.
Keywords: ruthenium; ethylenedinitrilotetraacetate; edta.
INTRODUÇÃO
A química dos complexos derivados de etilenodiaminotetraacetatorutenato(III/II), [RuIII/II(edta)]-,2-, desenvolveu-se principalmente ao longo das últimas três décadas1,2. Inicialmente, a maior parte dos trabalhos foi direcionada ao estudo cinético das reações de substituição desses complexos em solução aquosa, onde se descobriu que, diferentemente da maioria das séries de complexos de rutênio e ferro com configuração eletrônica d5/d6 de spin baixo então conhecidas (tais como rutênio-poliaminas, rutênio-polipiridinas ou cianoferratos), a espécie de partida [Ru(edta)(H2O)]n apresenta alta reatividade no estado de oxidação formal +III. Além disso, verificou-se que os complexos substituídos do tipo [RuIII,II(edta)(L)]n são bastante estáveis em ambos os estados redox. Essa característica especial de tais sistemas é atribuída, entre outros fatores, à natureza química mista do ligante edta, que possui a habilidade de estabilizar tanto o estado reduzido +II (devido à presença dos grupos amina) como o estado oxidado +III (devido à presença dos grupos carboxilatos). Mais tarde, complexos mono-, bi- e polinucleares contendo os mais variados tipos de ligantes (i.e., mono-, bi- ou polidentados e multifuncionais), também foram sintetizados e investigados tanto em solução, como no estado sólido. A importância biológica e a habilidade catalítica desses compostos em várias transformações orgânicas também têm sido recentemente exploradas.
De fato, a importância dos complexos de rutênio-edta deve-se, em parte, à versatilidade de sua química de coordenação, no sentido de que o ligante edta (que também é chamado de "co-ligante" no contexto da maioria dos trabalhos de literatura) possui a capacidade de se coordenar através de modos hexa-, penta-, tetra- e até mesmo tri- ou bidentados, permitindo assim a formação de espécies mono-, bi- ou polissubstituídas em reações de substituição do complexo de partida, [RuIII,II(edta)]-,2-, tanto com ligantes p-receptores (ácidos) como com ligantes s,p-doadores (básicos). Os complexos mono- e dissubstituídos do tipo [Ru(edta)(L)]n e [Ru(edta)(LL)]n, respectivamente, são mais comumente encontrados e por isso são particularmente enfocados no presente trabalho.
Apesar da relevância dos derivados de rutênio-edta no contexto da química inorgânica de coordenação, nenhum trabalho de revisão havia sido voltado especificamente para tais sistemas até a presente data (exceto por um artigo acerca de rutênio-poliaminocarboxilatos em geral3). Tomando isso como principal motivação, esta revisão tem como objetivo proporcionar uma visão abrangente de diversos aspectos envolvidos na química de coordenação dos complexos de íons rutênio (em particular, RuIII e RuII) com etilenodiaminotetraacetato (Figura 1). Os tópicos aqui descritos incluem estrutura, reatividade, cinética, termodinâmica, eletroquímica e espectroscopia.
Observação: Embora a abordagem das seções seguintes seja essencialmente descritiva, seu foco principal incide sobre as idéias e avanços relacionados ao campo, sem qualquer compromisso em oferecer detalhes ou mesmo resultados numéricos acerca de cada um dos estudos a serem mencionados. Portanto, algumas informações mais específicas, tais como dados cinéticos e termodinâmicos, dados eletroquímicos e dados espectroscópicos foram coletadas no final do artigo como tabelas (Apêndice B). Os métodos de síntese do material de partida e as abreviaturas empregadas ao longo do texto também encontram-se disponíveis nos Apêndices A e C, respectivamente.
REATIVIDADE E ESTRUTURA
Apesar de os complexos de Ru-edta4 serem conhecidos desde a década de 605-7, os aspectos relacionados à sua reatividade só foram cuidadosamente estudados no final da década de 70 por Matsubara e Creutz8,9, os quais descreveram uma série extensa de complexos do tipo [RuIII,II(edta)(L)]-,2-, onde L varia de água a ligantes heterocíclicos nitrogenados de diferentes naturezas químicas. Nesses estudos, foi demonstrado que o aquacomplexo de partida, RuIII(Hedta)(H2O), pode sofrer dois processos de desprotonação, cujas constantes de equilíbrio ácido-base correspondem a pKa(1) = 2,37 e pKa(2) = 7,60:9


A espécie [RuIII(edta)(H2O)]- , gerada em solução aquosa pela dissociação ácida do complexo RuIII(Hedta)(H2O) em pH > 3, exibe uma labilidade incomum e elevada reatividade com muitos tipos de ligantes (Equação 3), cuja cinética apresenta um pH ótimo ao redor de 5 (que coincide com o pH médio entre os dois pKa do aquacomplexo, onde a presença da espécie [RuIII(edta)(H2O)]- se dá em maior quantidade).

As elevadas constantes de velocidade observadas para as reações de substituição em [RuIII(edta)(H2O)]- , segundo um mecanismo cinético associativo, foram atribuídas a fatores estruturais, onde a formação de uma ponte entre o hidrogênio do ligante água e o oxigênio do grupo carboxilato livre (não-coordenado) do edta provocaria uma distorção geométrica e o aparecimento de uma região molecular estericamente desimpedida (vacância) para ataques nucleofílicos do tipo SN18-10.
A existência de uma ponte de hidrogênio tal como descrita acima foi confirmada por estudos de Oyama e Anson11, os quais estudaram o comportamento eletroquímico do aquacomplexo depositado sobre a superfície de um eletrodo de grafite por meio da condensação do grupo carboxilato livre (não-coordenado) do Ru-edta com grupos amina introduzidos na superfície do eletrodo, verificando que há substituição da molécula de água em [Ru(edta)(H2O)]n por ligantes como piridina, isonicotinamida e nicotinamida somente quando o metal está na forma reduzida +II (n = -2). Resultados similares foram obtidos posteriormente pelos mesmos autores12, desta vez por meio da coordenação de Ru(III)-edta ao ligante 4-metilaminopiridina, previamente depositado na superfície de um eletrodo de grafite. Além disso, estudos cinéticos efetuados com aquacomplexos derivados de edta modificado (substituição de um dos acetatos por um grupo alquila ou hidroxialquila)13 indicaram que o par acetato/água é aquele que confere maior reatividade ao complexo. A troca de um acetato por uma alquila (ou hidroxialquila) provoca uma diminuição de 1-4 ordens de grandeza na constante de velocidade. Assumindo-se que a estabilidade da ligação e o mecanismo de reação nos complexos de edta e seus derivados devem ser semelhantes, algum tipo de interação entre a água (coordenada) e o grupo carboxilato livre (não-coordenado) deve ser considerado para explicar uma variação dessa magnitude na constante cinética.
Outro mecanismo envolvendo a coordenação transiente do grupo pendente, o qual auxiliaria na eliminação da molécula de água no complexo [RuIII(edta)(H2O)]- , também aparece na literatura14. Posteriormente, evidências contra essa proposta foram observadas no estudo da substituição de [(NH3)5RuIII(edta)RuIII(H 2O)]2+ com tiouréia15.
As estruturas molecular e cristalina do complexo NH4[RuIII(Hedta)(Cl)] foram caracterizadas por Jolley et al.16. Tais estudos confirmaram que, nesse complexo, o Hedta (monoprotonado) atua como um co-ligante pentadentado, coordenando-se ao metal através de dois átomos de nitrogênio terciários e três oxigênios de grupos carboxilatos. O ligante cloreto completa a geometria octaédrica ao redor do íon metálico.
Resultados de modelagem molecular obtidos por Rein1 e Rocha2 através de técnicas de mecânica molecular e cálculos quânticos semi-empíricos para as diversas conformações geométricas possíveis do complexo [RuIII(edta)(H2O)]- concordam com a proposição de formação da ponte de hidrogênio, conforme pode-se depreender da estrutura molecular otimizada representada na Figura 2.
Conforme ilustrado, a esfera de coordenação do aquacomplexo é constituída pelo co-ligante edta pentadentado (via dois sítios N-amina e três O-carboxilato) mais o ligante água ocupando a sexta posição (que é lábil e pode ser substituído por um ligante L qualquer). No caso de complexos dissubstituídos do tipo [Ru(edta)(L)2]n (ou [Ru(edta)(LL)]n), estes são constituídos do ligante edta tetradentado (via dois átomos N-amina e dois O-carboxilato) mais dois ligantes monodentados (2×L) ou um ligante bidentado (LL) ocupando a quinta e sexta posições vicinais de coordenação. Em ambos os casos o(s) átomo(s) coordenante(s) do(s) ligante(s) substituinte(s) e os dois nitrogênios dos grupos amina oriundos do edta e ligados ao íon rutênio são coplanares (i.e., tanto mono- como dissubstituição ocorrem com deslocamento de um ou dois grupo(s) carboxilato(s) do plano equatorial incluindo o fragmento etilenodiamina, gerando assim uma geometria local cis no caso do complexo com edta tetradentado).
Estudos por cristalografia de raios-X17 também revelaram que, quando o complexo se encontra protonado através do grupo carboxílico pendente, i.e. Ru(Hedta)(H2O), então o grupo -COOH (não-coordenado) do Hedta se encontra afastado da molécula de água coordenada. Naquele estado sólido cristalino, contudo, foi observada uma aproximação intermolecular entre a carbonila do grupo carboxílico livre e um átomo de hidrogênio de uma molécula de água de solvatação/hidratação, perfazendo uma distância típica de pontes de hidrogênio. Vale mencionar ainda o fato de que, neste caso, os resultados de cálculos moleculares18 também estão de acordo com aqueles obtidos experimentalmente por meio de cristalografia de raios-X para o complexo protonado, Ru(Hedta)(H2O).
FORMAÇÃO DA ESPÉCIE RUIV-EDTA
No começo da década de 80, Ikeda et al.19 verificaram que [RuIII(edta)(H2O)]- na presença de um agente oxidante, tal como clorato de sódio ou peróxido de hidrogênio, fornece um produto com intensa coloração verde. Um comportamento similar já havia sido observado por Ezerskaya e Solovykh6, que atribuíram a cor à existência de um dímero contendo um ligante-ponte µ-oxo ou µ-peroxo, mas não apresentaram nenhuma caracterização do dímero. As propriedades espectroscópicas e eletroquímicas reportadas no trabalho de Ikeda et al.19 são coerentes com a formação do dímero [(edta)RuIV-O-RuIII(edta)]3-, no qual as cargas encontram-se deslocalizadas e os íons rutênio apresentam, na verdade, um estado de oxidação médio igual a III½. Segundo os autores, esse dímero é razoavelmente estável, podendo ser reduzido eletroquimicamente a [(edta)RuIII-O-RuIII(edta)]4-, o qual se dissocia rapidamente, regenerando o aquacomplexo [RuIII(edta)(H2O)]- .
As oxidações química e eletroquímica do complexo [RuIII(edta)(H2O)]- foram descritas mais tarde por Baar e Anson20, que determinaram os potenciais formais de redução do dímero [RuIII½(edta)]2 e também verificaram que sua oxidação gera a espécie [RuIV(edta)]2. Segundo esse estudo, em solução aquosa ocorre a redução espontânea de [RuIV(edta)]2 a [RuIII½(edta)]2, às custas da oxidação de água a oxigênio. Assim, os autores sugerem que o composto [RuIV(edta)]2 possa ser usado como catalisador na oxidação da água e que os íons metálicos constituintes do dímero encontram-se ligados por meio de uma ponte do tipo µ-oxo ou µ-hidroxo, concordando com os resultados obtidos por Ikeda et al.19. Outro estudo mostrou que o complexo µ-peroxo-rutênio(IV)-dioxigênio age como um relé eletrônico em um sistema de geração fotoquímica de hidrogênio e oxigênio21. Posteriormente, a cinética de dissociação do dímero [RuIII(edta)]2 foi investigada utilizando técnicas polarográficas de pulso22. Nesse caso, o dímero foi gerado através da redução de [RuIII½(edta)]2, o qual se decompõe nos compostos de partida em meio ácido.
A interação de O2 e H2O2 com Ru-edta foi estudada por Khan et al.23 através de técnicas potenciométricas, espectrofotométricas, eletroquímicas e de absorção gasosa. Em pH 7-9,5, [RuIII(edta)(OH)]2- reage com O2 formando a espécie [(edta)(OH)RuIV-(O22- )-RuIV(OH)(edta)]4-. Em pH acima de 9,5, o dímero [(edta)(OH)2RuIV-(O2 )-RuIV(OH)2(edta)]4- também é formado e se decompõe irreversivelmente formando [RuIII(edta)(OH)2]3- com liberação de O2. Quando H2O2 é adicionado a Ru(Hedta)(H2O) na proporção molar 1:0,5, o dímero (Hedta)RuIII-(H2O2 )-RuIII(Hedta) é formado em meio ácido. A mesma reação, na proporção 1:1, resulta na formação do complexo (Hedta)RuIV-(O22-) -RuIV(Hedta).
Novos estudos com o dímero de Ru-edta foram divulgados mais tarde por Zhou et al.24, com enfoque na possível catálise da oxidação da água a oxigênio. Através de espectroscopia Raman, polarografia, espectroscopia de massa e cromatografia gasosa, o dímero foi caracterizado como sendo do tipo [(edta)RuIV-O-RuIV(edta)]2-, onde verificou-se que o tal complexo não é capaz de oxidar água a oxigênio mesmo quando em meio alcalino, contrariando os resultados obtidos por Baar e Anson20.
REAÇÕES DE SUBSTITUIÇÃO
Enquanto a maioria dos estudos descritos acima envolve aspectos estruturais e redox relevantes à reatividade do rutênio-edta e seus mecanismos cinéticos, outros trabalhos exploram a síntese e caracterização de seus derivados substituídos contendo os mais variados tipos de ligantes.
Conforme mencionado acima, os estudos de Matsubara e Creutz8,9 foram pioneiros na investigação sistemática da cinética e reatividade de complexos de rutênio-edta. Além de propor o tipo de mecanismo envolvido na formação dessas espécies e discutir a importância estrutural (da esfera interna de coordenação, ditada pelo co-ligante edta) na alta reatividade de [RuIII(edta)(H2O)]- , esses trabalhos também estabeleceram uma ampla avaliação comparativa das propriedades espectrais e eletroquímicas desses complexos. Do ponto de vista da química redox, em particular, os resultados obtidos para tais sistemas seguem as mesmas tendências qualitativas observadas anteriormente para outras séries envolvendo [RuIII,II(NH3)5(L)]n e [FeIII,II(CN)5(L)]n . Uma observação relevante acerca desses estudos é a constatação de que na série do tipo [RuIII,II(edta)(L)]n a diferença de afinidade entre as formas oxidada e reduzida do metal por ligantes p-receptores é menor que a observada para a série análoga com pentaaminas de rutênio.
No começo da década de 80, Diamantis e Dubrawski25,26 também reportaram as propriedades eletroquímicas e espectroscópicas de uma ampla série de novos complexos contendo edta penta- ou tetradentado, do tipo [Ru(edta)(L)]n ou [Ru(edta)(LL)]n (onde L e LL são ligantes mono- e bidentados, respectivamente). Os estudos mostraram que o ligante L ou LL assume uma posição equatorial nesses complexos, e que a resistência à oxidação, tanto por eletroquímica (RuII®RuIII) como induzida opticamente por transferência de carga (RuIIdp® Lp*/LLp*), depende da natureza química do ligante substituinte e segue a ordem: NO, CO > RCN > bpy, phen > py > SCN- > acac > dmg. Nota-se, portanto, que os ligantes doadores de elétrons (s,p-básicos) favorecem a estabilização do íon oxidado Ru(III), enquanto os receptores (p-ácidos) tendem a estabilizar o íon reduzido Ru(II), principalmente por meio de interações de retrodoação-p.
Os trabalhos acima marcaram as pesquisas sobre Ru-edta pelo fato de envolverem séries de ligantes com diferentes naturezas químicas. Muitos outros estudos, contudo, contribuíram com o desenvolvimento da química dos complexos de Ru-edta através de investigações sistemáticas de tipos específicos de ligantes em derivados do tipo [RuIII,II(edta)(L)]n. Nas seções seguintes as propriedades gerais desses sistemas são coletadas através de uma abordagem descritiva, baseando-se em uma distinção seletiva dos ligantes substituintes. Dessa forma, os sistemas encontram-se agrupados de acordo com o tipo de átomo coordenante (N, O, P e S) ou característica principal de cada série (ênfases em isomeria, nuclearidade do sistema, transferência de elétrons, interesse biológico e aplicações em catálise ou polímeros).
Ligantes nitrogenados
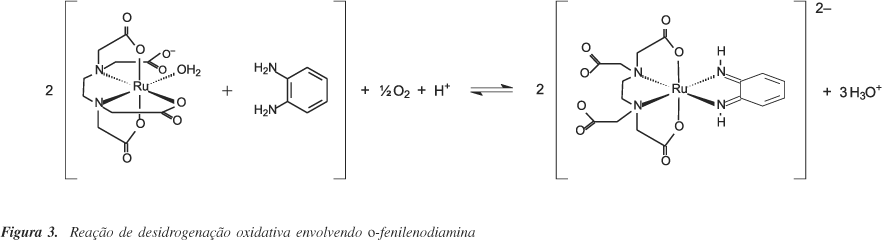
O primeiro trabalho envolvendo uma reação de desidrogenação oxidativa em derivados de Ru(III)-edta foi descrito por Yoshino et al.27, os quais estudaram o produto diamagnético formado pela interação entre [RuIII(edta)(H2O)]- e o-fenilenodiamina, em condições aeróbicas. Essa reação envolve um processo de transferência eletrônica intramolecular, onde o Ru(III) é reduzido a Ru(II) e o ligante é oxidado a o-benzoquinona diimina (bqdi), gerando [RuII(edta)(bqdi)]2- (Figura 3).
Posteriormente, Rein et al.28 descreveram em detalhes a química redox daquele mesmo sistema, em solução aquosa, com base em estudos eletroquímicos e espectroeletroquímicos. Enquanto o comportamento eletroquímico do ligante livre o-fenilenodiamina é um tanto complicado e envolve reações químicas secundárias subseqüentes à oxidação química irreversível envolvendo 2 prótons/2 elétrons, seu complexo correspondente é eletroquimicamente bem comportado com dois processos redox quimicamente reversíveis: um par monoeletrônico associado ao metal (RuIII/RuII) e uma outra etapa bieletrônica centrada no ligante coordenado (bqdi/opda). Os espectros eletrônicos na região do UV-Vis obtidos por eletrólise, in situ, do complexo em todos os seus estados redox acessíveis nas condições de voltametria cíclica mostraram que essas espécies são bastante estáveis e totalmente interconversíveis.
Outro exemplo de desidrogenação oxidativa em complexos de Ru-edta foi estudado por Toma e Tsurumaki29, os quais verificaram que o mecanismo da reação entre 2-aminometilpiridina e [RuIII(edta)(H2O)]- é consistente com a desprotonação da amina seguida por uma etapa de transferência monoeletrônica reversível induzida pelo metal, onde o Ru(III) é reduzido a Ru(II) e o ligante é oxidado à sua forma imínica (impy), gerando o complexo [RuII(edta)(impy)]2- (Figura 4).
A interação de Ru-edta com o cátion N-metilpirazínio foi investigada por Araki et al.30, onde observou-se que a constante de equilíbrio para o complexo oxidado é muito menor que a do complexo reduzido, em virtude da diferença de afinidade entre o ligante catiônico e o íon metálico nesses dois estados de oxidação. Posteriormente, Stadler et al.31 mostraram que a reação de [RuII(Hedta)(H2O)]- com o ligante tridentado diacetil-monooxima-b-piridil(2)metilimina (dmp) resulta as espécies [RuII(Hedta)(dpm)]- e [RuII(dpm)2)]2+. As constantes totais de formação foram obtidas por métodos espectrofotométrico e voltamétrico para o complexo reduzido e por método potenciométrico para o complexo oxidado. Os resultados foram racionalizados com base na interação entre o complexo de rutênio e o ligante imínico.
Mais recentemente, interessados no estudo de processos redox dependentes do pH, Rocha et al.32 estudaram complexos contendo ligantes ionizáveis/desprotonáveis do tipo [RuIII,II(edta)(LH)]-,2- (LH = benzotriazol ou benzoimidazol), os quais foram caracterizados em soluções aquosas por meio de métodos eletroquímicos e espectroeletroquímicos. Os valores de pKa e dos potenciais formais de redução para as espécies protonada e desprotonada em ambos os estados redox foram obtidos através de diagramas de Pourbaix, os quais apresentam um comportamento tipicamente Nernstiano (Figura 5). A região ativa na qual existe dependência do par redox MIII/MII com o pH foi usada na avaliação desses sistemas como modelos simples de transferência eletrônica acoplada a próton. Os resultados foram interpretados em termos do caráter eletrônico doador/receptor dos ligantes e interações-s,p metal-ligante em ambos os estados redox do íon metálico.
Ligantes oxigenados
Complexos metal-catecol podem exibir efeitos eletrônicos interessantes associados à distribuição de carga variável entre o metal e o ligante, em função do delicado balanço de energia entre os orbitais de fronteira do dioxoleno e do metal. Os trabalhos envolvendo Ru-edta e quinonóides derivados de catecolatos foram todos desenvolvidos em nosso grupo33,34. O comportamento eletroquímico exibido pelo catecol (1,2-diidroxibenzeno) e seus derivados (tiron, ácido 3,4-diidróxido benzóico, dopamina e L-dopa) em solução aquosa é um tanto complicado, mas pode ser convenientemente controlado através da coordenação desses ligantes a Ru-edta (Figura 6). Em suas formas livres, os catecóis sofrem um processo primário de oxidação eletroquímica na faixa de 0,1-0,6 V vs EPH, gerando produtos derivados de quinona, os quais são susceptíveis a um grande número de reações químicas secundárias dependentes do pH (Figura 7a). Contudo, quando coordenados a Ru(III)-edta, o comportamento eletroquímico e espectroeletroquímico dessas espécies é alterado drasticamente (Figura 7b), aproximando-se daquele observado para outros complexos metálicos com dioxolenos. Nesse caso, a redução do íon metálico Ru(III), acima de pH 9, ocorre reversivelmente na região de -0,5 V a -0,7 V e o processo de oxidação centrado nos ligantes catecolatos torna-se reversível, levando à formação exclusiva de espécies semiquinona, sem evidência de complicações geradas por reações secundárias. Essas mudanças no comportamento eletroquímico dos neurotransmissores dopamina e L-dopa tornam suas ondas voltamétricas de redução/oxidação mais definidas, favorecendo uma análise quantitativa mais precisa. Com base nos resultados empíricos de eletroquímica e espectroscopia eletrônica, um diagrama qualitativo de energia para os níveis orbitais de fronteira para uma série de catecolatos de rutênio(III)-edta foi proposto34. Uma considerável diferença entre o comportamento redox dos complexos contendo dioxolenos33,34 e diiminas28 foi revelado através da comparação de tais sistemas, a qual foi racionalizada em termos da natureza eletrônica p-receptora eficiente do ligante o-benzoquinona diimina28.
Ligantes fosforados e arsenados
Os estudos sobre a reatividade e a estrutura de complexos de Ru-edta com fosfinas e arsinas foram realizados por Khan et al.35 na década de 90. A substituição de RuIII(Hedta)(H2O) ou RuII(H2edta)(H2O) com bis(difenilfosfino)metano (dppm), 1,2-bis(difenilfosfino)etano, NPr(i)(PPh2)2 e 1,2-bis(difenilarsino)etano em uma mistura de água-etanol produz os complexos mononucleares de fórmula geral RuIII(Hedta)(LL) ou RuII(H2edta)(LL), que apresentam uma combinação de átomos doadores duros e moles em suas esferas de coordenação. A redução dos complexos RuIII(Hedta)(LL) com hidrogênio molecular em dimetilsulfóxido gera os produtos [RuII(H2edta)(LL)]×dmso× H2O. Estudos por cristalografia de raios-X da espécie RuII(H2edta)(dppm)×dmso× H2O, em particular, indicaram que os sítios de coordenação do octaedro distorcido de Ru(II) encontram-se ocupados pelo co-ligante edta tetradentado e pelo ligante substituinte bis(difenilfosfino)metano, que se coordena de modo bidentado formando um anel quelato de quatro membros, onde cada um dos átomos de fósforo ocupa uma posição trans em relação aos átomos de nitrogênio (do edta) no plano equatorial. Os átomos de hidrogênio dos dois grupos -COOH livres do edta e da molécula de água participam de uma extensiva rede de pontes de hidrogênio.
Ligantes sulfurados
A reatividade e as propriedades dos complexos de Ru(III/II)-edta com dimetilsulfóxido (dmso) foram descritas por Toma e Araki36. Verificou-se que a espécie [RuII(edta)(kS{dmso})]2- é extraordinariamente estável (K = 7,7x109 M-1), enquanto sua correspondente forma oxidada [RuIII(edta)(kS{dmso})]- apresenta-se lábil (K = 1,8 M-1). Esse sistema difere do análogo [RuIII(NH3)5(dmso)]3+ devido a uma maior estabilidade da espécie Ru(III)-kS com respeito ao seu isômero Ru(III)-kO, ainda que ambas espécies apresentem pronunciada labilidade no estado oxidado.
Interessados no comportamento de sistemas onde o ligante se coordena ao Ru-edta de forma bidentada, Araki et al.37 descreveram o comportamento cinético e espectroeletroquímico em solução aquosa do complexo [RuIII(edta)(dedtc)]- (dedtc = ion dietilditiocarbamato) , propondo a formação de um precursor monodentado, o qual se converte rapidamente em um produto quelato estável. A constante de equilíbrio desse complexo é maior que 107 M-1, indicando que o composto é bastante estável e que dietilditiocarbamato poderia ser usado na determinação quantitativa de Ru-edta.
A reação entre [RuIII(edta)(H2O)]- e sulfito foi investigada por Chatterjee et al.38, os quais notaram que a formação do composto diamagnético [RuII(edta)(SO3)]4- ocorre com a geração de uma espécie intermediária, [(edta)RuIII-O-RuIII(edta)]4-, em uma etapa rápida e independente da concentração de sulfito. Ao contrário do sistema contendo ferro (no qual a espécie [FeIII(edta)(SO3)]3- inicia o processo catalítico), no caso com rutênio o complexo oxidado [RuIII(edta)(SO3)]3- é extremamente instável e sofre um rápido processo de hidrólise, gerando a espécie aqua/hidroxo correspondente. Essa diferença de comportamento deve ser causada pela estabilização preferencial de sistemas 3d5 por óxidos de enxofre p-receptores. A espécie reduzida [RuII(edta)(SO3)]4- , entretanto, é completamente inerte e insensível a oxigênio molecular.
Um extensivo estudo cinético envolvendo uma série de complexos do tipo [RuIII(edta)(SR)]n (SR- = cisteína desprotonada, N-acetilcisteína, 2-mercaptoetanol, glutationa ou penicilamina) foi descrito por Povse e Olabe39. Todos os complexos exibem intensas bandas LMCT na região do visível40, associadas à coloração vermelha típica de espécies sulfuradas. As constantes cinéticas de formação são de primeira ordem em relação tanto ao ligante, quanto ao metal. Em pH~8,0, um significativo aumento na velocidade de reação foi observado para a cisteína e para a penicilamina devido à desprotonação dos reagentes envolvendo o grupo tiol, o qual já havido sido previamente observado por Chatterjee e Bajaj41. As elevadas constantes de equilíbrio (da ordem de 105 M-1) indicam a alta estabilidade dos complexos formados.
ISOMERIA DE LIGAÇÃO
O primeiro trabalho sobre isomeria de ligação42 em complexos de Ru-edta foi reportado na década de 80 por Toma et al.43. A interação entre [RuIII(edta)(H2O)]- e 2-mercaptopiridina, em meio ácido, gera um complexo vermelho (lmáx = 550 nm), no qual o ligante encontra-se coordenado de modo monodentado através do átomo de enxofre. A desprotonação da 2-mercaptopiridina promove a formação de um produto verde (lmáx = 630 nm), onde a coordenação ocorre de forma bidentada através dos átomos de nitrogênio e enxofre (Figura 8). O valor do pKa associado ao equilíbrio entre as espécies vermelha e verde é igual a 4,35 e a constante cinética de conversão entre as duas espécies é 0,31 s-1. Posteriormente, um comportamento análogo foi observado para a reação entre [RuIII(edta)(H2O)]- e 4,6-dimetil-2-mercaptopirimidina44.
A caracterização dos complexos envolvendo Ru(III)-edta e isonicotinamida (isonic) foi descrita por Chatterjee e Bajaj45, os quais verificaram que, em meio ácido, o ligante se coordena através do átomo de nitrogênio da piridina, formando a espécie [RuIII(edta)(kNpy{isonic})]-. A adição de base (pH ~ 9,6) promove uma isomerização de ligação intramolecular gerando [RuIII(edta)(kNam{isonic})]-, onde a coordenação ocorre através do átomo de nitrogênio do grupo amida. Os dados cinéticos sugerem que a reação diretamente em meio básico ocorre em duas etapas: inicialmente ocorre a formação de [RuIII(edta)(kNpy{isonic})]- (etapa rápida), a qual é convertida no isômero [RuIII(edta)(kNam{isonic})]- (etapa lenta). Em seguida, uma série de estudos cinéticos e termodinâmicos para a reação entre [RuIII(edta)(H2O)]- e 4-sulfanilpiridina46 mostrou que a formação do produto coordenado através do átomo de enxofre é favorecida termodinamicamente, enquanto que o isômero N-substituído é favorecido cineticamente.
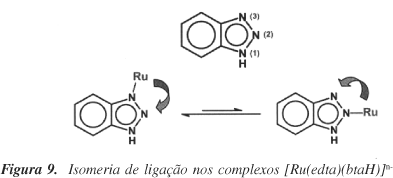
No final da década de 90, Rocha et al.47 reportaram o estudo do complexo [RuIII(edta)(btaH)]- (btaH = benzotriazol) em água (pH 4,7) por meio de técnicas de cinética rápida, eletroquímica e espectroeletroquímica. A reação de substituição entre o aquacomplexo (no estado oxidado; RuIII) e benzotriazol conduz essencialmente à formação do isômero coordenado através do átomo de nitrogênio N(3) do anel triazol. No caso da espécie de partida reduzida (RuII), os produtos apresentam um equilíbrio de isomerização intramolecular envolvendo N(3) e o sítio intermediário N(2), com favorecimento da espécie isômera Ru-kN(3) (Figura 9). As constantes de velocidade e de equilíbrio para o processo de isomerização foram estimadas por meio de desconvolução dos voltamogramas cíclicos com velocidade e sentido de varredura variáveis (Figura 10), nos quais o potencial redox para a espécie Ru-kN(2) aparece cerca de 0,1 V mais positivo em relação ao seu isômero Ru-kN(3). Os dados cinéticos e termodinâmicos são coerentes com os resultados de cálculos moleculares teóricos2,47, refletindo a importância das interações de retrodoação-p nas espécies reduzidas.
Mais recentemente, outro exemplo de isomeria de ligação envolvendo ligantes N-heterocíclicos foi descrito por Rein et al.48. Em pH 5, a reação entre Ru(III)-edta e 3-hidroxipicolinato (Hhpic) gera o complexo [RuIII(edta)(kN,kO{Hhpic})]2-. A desprotonação do grupo fenólico do ligante 3-hidroxipicolinato (em pH ³ 9) promove uma isomerização de ligação intramolecular formando a espécie [RuIII(edta)(kO,kO{hpic})]3- (Figura 11). Ambos os isômeros podem ser reduzidos eletroquimicamente convergindo ao mesmo complexo, [RuII(edta)(kN,kO{Hhpic})]3-, que é altamente estabilizado por interações de transferência de carga do tipo dp®pp* (Ru®piridinacarboxilato). As diferentes propriedades de ligação observadas em função dos estados de oxidação e do pH foram racionalizadas com base em cálculos teóricos semi-empíricos para os complexos.
COMPLEXOS DINUCLEARES
O primeiro trabalho envolvendo complexos dinucleares de Ru-edta foi descrito no final da década de 70 por Creutz et al.49, os quais estudaram as propriedades espectrais e eletroquímicas dos complexos [(edta)Ru(pz)Ru(edta)]3- e [(NH3)5Ru(pz)Ru(edta)]+ (pz = pirazina), e efetuaram a medida direta da velocidade de transferência eletrônica intramolecular para tais sistemas no estado de valência mista. Com base nos resultados obtidos, sugeriu-se um modelo para processos de transferência eletrônica em sistemas nos quais as barreiras de ativação originam-se principalmente da reorganização do solvente.
Interessados na química de complexos contendo carbonila, que são potenciais catalisadores em reações de hidroformilação e carbonilação, Khan et al.50 descreveram que K[RuIII(edta)(CO)(H2O)] e K3[(edta)RuIII-µ(CO)-µ(OH)-RuIII(edta)] são os produtos da reação entre Ru(III)-edta e CO em meio básico. Dados de EPR do dímero indicaram que a interação metal-metal é do tipo anti-ferromagnética.
Na década de 90, vários grupos realizaram estudos envolvendo complexos dinucleares assimétricos do tipo [(NC)5M(CN)Ru(edta)]n (M = Fe51-53, Ru52,54 ou Os52), os quais apresentam banda de intervalência intensa na região de 600-1000 nm e acoplamento eletrônico moderado entre os centros metálicos. As constantes de equilíbrio são similares para os três íons metálicos estudados. O complexo K5[(NC)5MII(CN)RuIII(edta)] é a espécie predominante na presença de excesso de M(CN)6, mas espécies tri- e polinucleares também são obtidas quando se aumenta a concentração de Ru-edta.
Estudos com complexos dinucleares do tipo [(edta)RuIII-L-M]n (M = RuII(edta)55 ou FeII(CN)556; L = pirazina, 4,4'-bipiridina, trans-1,2-bis(4-piridil)etileno, 3,3'-dimetil-4,4'-bipiridina ou 1,3-bis(4-piridil)propano) também foram reportados por Das et al.57. Tais sistemas apresentam acoplamento metal-metal fraco, cuja intensidade é dependente da distância e da coplanaridade do ligante-ponte entre os centros redox. A formação do complexo dinuclear assimétrico contendo Ru-edta e trans-1-(4-piridil)etilenoferroceno também foi descrita57.
Rein e Toma58 investigaram o complexo dinuclear formado pela interação de Ru-edta com ácido 2-mercaptonicotínico, que é um ligante bastante interessante, devido à presença de três átomos potencialmente coordenantes vicinais (N, S, O). Os dados espectroscópicos e eletroquímicos sugerem que o ligante-ponte se coordena a uma das unidades de Ru-edta de modo bidentado através dos átomos de nitrogênio e enxofre (N,S)43 e a outra unidade, através dos átomos de enxofre e oxigênio (S,O)59 (Figura 12). A correspondente espécie de valência mista apresenta uma banda de intervalência intensa em 1250 nm, envolvendo uma energia de acoplamento eletrônico equivalente a 990 cm-1.
Em continuidade aos trabalhos com o ligante benzotriazol, Rocha et al.60,61 estudaram a química de valência mista62 de complexos simétricos e assimétricos do tipo [RuIII(edta)-L-M]n (M = RuII(edta) ou RuII(NH3)5; L = benzotriazolato ou benzoimidazolato). Propriedades de transferência de intervalência contrastantes foram observadas (Figura 13): enquanto o derivado de benzoimidazol apresenta localização de cargas e se comporta como um sistema de classe II (no contexto da classificação de Robin-Day63), o análogo envolvendo benzotriazol exibe significativa deslocalização eletrônica e é melhor descrito como um sistema de classe intermediária II/III. Nesse caso, a forte interação eletrônica entre os centros metálicos é promovida por um mecanismo de mobilidade fluxional sobre a coordenação do ligante-ponte benzotriazolato. Cálculos moleculares semi-empíricos indicaram que tais efeitos estruturais dinâmicos podem ser muito importantes na capacidade do ligante-ponte intensificar o acoplamento intermetálico, adicionalmente ao mecanismo usual onde a comunicação eletrônica se propaga através da conjugação-p entre as extremidades terminais do ligante-ponte64. Com o intuito de se explorar os limites da teoria de Hush65 para sistemas na fronteira dos regimes localizado/deslocalizado, medidas eletroquímicas e espectroeletroquímicas do complexo (NH3)5RuII(bta)RuIII(edta) 61 foram conduzidas em vários solventes orgânicos, confirmando a natureza limítrofe (classe II/III) desses sistemas.
Mais recentemente, Rocha e Toma66 reportaram um exemplo interessante de interruptor molecular do tipo "ON/OFF", induzido por próton, baseado na comunicação intermetálica dos dímeros de valência mista [(edta)RuIII-Lb-RuII (edta)]n- (Lb = bta- ou btaH; n = 4 ou 3, respectivamente). Assim, dependendo do pH, o ligante-ponte pode se encontrar nos estados protonado ou desprotonado e os graus de deslocalização de cargas e acoplamento eletrônico metal-metal podem ser modulados de bastante elevados (no caso de Lb = bta-; classe II/III) a apenas moderados (no caso de Lb = btaH; classe II) (Figuras 14 e 15).
Interessados no estudo de agentes captadores de NO baseados em complexos de rutênio, Cameron et al.67 descreveram a formação de um dímero de rutênio contendo bis-tiolato como ponte, por meio da interação de Ru(III)-edta com tiofenol. Dados de raios-X indicaram a presença de uma ligação simples Ru-Ru com uma distância de 2,866 Å. Apesar de os resultados de espectroscopia de massa apontarem para a existência de íons metálicos no estado de oxidação +III, os dados de 1H RMN mostraram uma natureza diamagnética para esse complexo dinuclear, revelando a existência de um forte acoplamento eletrônico e magnético entre os dois sítios redox de Ru(III). Esse parece ser o primeiro exemplo de um complexo dimérico Ru(III)-Ru(III) contendo tiolato como ligante-ponte e que não possui grupos ciclopentadienilas em sua esfera de coordenação.
COMPLEXOS POLINUCLEARES
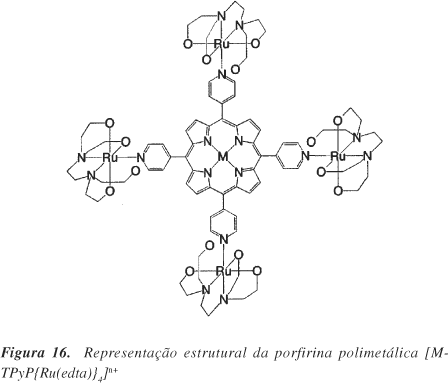
Os trabalhos envolvendo complexos polinucleares de Ru-edta foram desenvolvidos em nosso grupo, usando entidades supramoleculares como ponte entre duas ou mais unidades de Ru-edta. O primeiro desses sistemas foi descrito no início da década de 9068,69, onde investigou-se a porfirina polimetalada Na4[{FeIII(TPyP)Cl}{RuIII (edta)}4] (Figura 16) resultante da reação entre meso-tetra(4-piridil)porfirina ferro(III) e [RuIII(edta)(H2O)]- em proporções estequiométricas. Essa espécie supramolecular em sua forma totalmente reduzida (FeIIRuII), em altas concentrações, é capaz de reduzir oxigênio molecular a água com pouca influência dos grupos Ru(II)-edta periféricos. Contudo, conforme a concentração da porfirina diminui, observa-se um efeito cooperativo envolvendo ferroporfirina e Ru(II)-edta. Nesse caso, a redução do oxigênio é iniciada no centro da porfirina às custas da oxidação do íon Fe(II), o qual, por sua vez, é regenerado através de transferência de elétrons com grupos periféricos Ru(II)-edta. Observa-se, portanto, um processo de transferência eletrônica de várias etapas, o qual converte oxigênio em água de maneira similar ao citocromo-c oxidase. Araki e Toma70,71 estudaram ainda outra espécie supramolecular similar, onde o íon ferro central foi substituído por cobalto. Essa porfirina tetrametalada de cobalto apresentou um aumento na atividade eletrocatalítica envolvida na redução do oxigênio molecular, o que também parece estar associado a um efeito cooperativo entre o centro de Co(III) e as unidades Ru(II)-edta.
Toma et al.72,73 também descreveram a caracterização e a determinação das constantes de equilíbrio das espécies polinucleares formadas pela reação entre [RuIII(edta)(H2O)]- e [RuII(bpz)3]2+ (Figura 17). A química desses compostos, em solução aquosa, é caracterizada por equilíbrios dinâmicos complexos e por interações fracas envolvendo os grupos Ru-edta periféricos. A estabilidade dos complexos aumenta bastante no estado totalmente reduzido, como conseqüência de interações de retrodoação dp-pp* entre os íons Ru(II) e o ligante bipirazina.
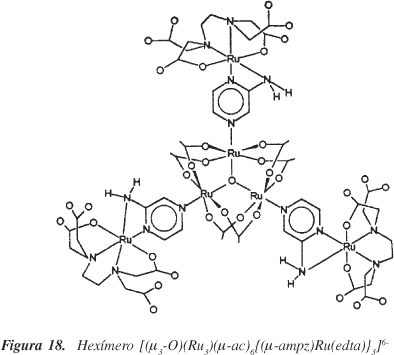
Outro sistema interessante é o "cluster" supramolecular polimetalado formado pela coordenação de Ru(II)-edta ao ligante periférico aminopirazina do "cluster" trinuclear [(µ3-O)(RuIIIRuIII RuII)(µ-ac)6(ampz)3 ]74. A reação ocorre rapidamente em solução aquosa e o produto [(µ3-O)(Ru3)(µ-ac)6 {(µ-ampz)Ru(edta)}3]6- (Figura 18) exibe uma intensa banda MLCT ao redor de 500 nm e constantes de estabilidade da ordem de 104 M-1. As constantes cinéticas e termodinâmicas do sistema supramolecular são bastante parecidas àquelas observadas na associação de Ru-edta ao ligante aminopirazina.
Estudos eletroquímicos e espectroeletroquímicos de complexos bi- e trinucleares (Figura 19) formados pela associação de cis- ou trans-[Ru(NH3)4(pz)2] 3+/2+ com Ru(III,II)-edta foram descritos por Toma e Sernaglia75. Com base nos valores das constantes termodinâmicas e nos potenciais redox do sistema, uma intrincada rede de equilíbrios envolvendo as espécies de valência mista foi elucidada.
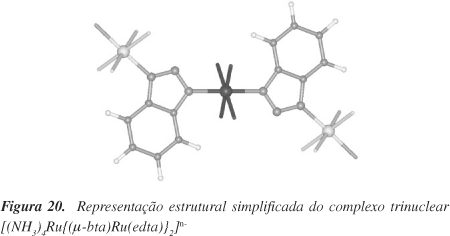
Recentemente, Rocha e Toma76 reportaram as propriedades espectroscópicas e eletroquímicas de uma nova série de complexos mono-, di- e trinucleares contendo benzotriazolato (trans-[Ru(NH3)4(bta)2]x+, trans-[(NH3)4(bta)Ru(bta)Ru(edta)]y-, e trans-[(NH3)4Ru{(bta)Ru(edta)}2]z-; onde x = 0 ou 1, y = 0, 1 ou 2, z = 1, 2 ou 4). As propriedades de valência mista das espécies dinuclear (RuIIRuIII) e trinuclear (RuIIIRuIIRuIII) indicaram um forte acoplamento eletrônico entre os centros metálicos com estados de oxidação localizados. Análise dos resultados no contexto da teoria perturbacional de Mulliken-Hush65 revelou que a interação eletrônica metal-metal aumenta ligeiramente com a nuclearidade do sistema, sugerindo que o complexo trinuclear simétrico não-linear (Figura 20) age como modelo/precursor de cadeias moleculares estendidas do tipo "fios moleculares". Comparação dos resultados com uma série de complexos análogos reforça o caráter condutor e a aplicabilidade de triazolato-derivados como espaçadores ricos em elétrons no planejamento de sistemas supramoleculares.
TRANSFERÊNCIA DE ELÉTRONS
Uma série de complexos dinucleares de fórmula geral [(edta)RuIII(L)CoIII(NH3 )5]2+ (L = pirazina, 4,4'-bipiridina, 3,3'-dimetil-4,4'-bipiridina, trans-1,2-bis(4-piridil)etileno e 1,4-bis(4-piridil)butadieno) foi investigada no início da década de 90 por de Oliveira et al.77. Demonstrou-se que a reação desses complexos com ácido ascórbico ou ditionita promove a redução preferencial de Ru(III) a Ru(II), produzindo [(edta)RuII(L)CoIII(NH3 )5]+, no qual ocorre, então, uma transferência de elétrons intramolecular de Ru(II) para Co(III). Verificou-se ainda que as constantes de velocidade diminuem com o aumento da extensão do ligante"-ponte, devido ao aumento da energia de reorganização do solvente com o aumento da distância entre os centros metálicos.
Estudos cinéticos da oxidação de complexos dinucleares por peroxidissulfato têm sido reportados por Ram e Haim78,79. No caso particular de (edta)RuII(pz)RuII(NH3 )5, o produto primário da oxidação é o produto instável [(edta)RuII(pz)RuIII(NH3 )5]+, o qual sofre uma rápida transferência eletrônica intramolecular formando o isômero eletrônico estável [(edta)RuIII(pz)RuII(NH3 )5]+. A oxidação com peroxidissulfato da mistura dos isômeros em equilíbrio se processa através do isômero mais estável. A reação de oxidação do complexo (edta)RuII(pz)RuII(NH3 )5 representa um exemplo de ativação de um centro metálico através de transferência de elétrons.
Com o objetivo de investigar reações com transferência de elétrons, Chatterjee estudou a cinética e o mecanismo de redução do complexo [RuIII(edta)(pz)]- com ácido ascórbico80, catecol80 e sulfito81, em soluções aquosas. Embora [RuIII(edta)(pz)]- não apresente bandas de absorção na região do visível, a redução do complexo a Ru(II) promove o surgimento de uma banda MLCT dependente do pH, a qual foi usada na caracterização dos produtos de reação. Os dados cinéticos e os parâmetros de ativação são consistentes com um mecanismo de transferência eletrônica de esfera externa. Em continuidade a seus trabalhos sobre cinética de transferência de elétrons, Chatterjee82 também descreveu a redução do complexo [(edta)RuIII(NC)FeIII(CN)5]4- com ácido ascórbico e a oxidação de [(edta)RuIII(NC)FeII(CN)5]5- com peroxidissulfato, para os quais a velocidade de redução/oxidação é de primeira ordem tanto em relação ao complexo, quanto ao agente químico redutor/oxidante. Um mecanismo de esfera externa centrado no íon Fe(II/III) foi proposto em ambos os casos.
Mais recentemente, Ward et al.83 mostraram que a reação entre [RuIII(edta)(H2O)]- e [FeIII(CN)6]3-, em solução aquosa, resulta na rápida formação do complexo dinuclear [(edta)RuIII(NC)FeIII(CN)5]4-, que sofre um processo lento de transferência eletrônica intramolecular espontânea gerando o complexo de valência mista [(edta)RuIV(NC)FeII(CN)5 ]4-.
BIOINORGÂNICA
Uma série de estudos espectroscópicos e eletroquímicos envolvendo complexos de Ru-edta com pirimidinas84 e purinas85 foi realizada por Khan e Annapoorna. Na série das pirimidinas84, verificou-se que uracila se coordena ao íon metálico de forma monodentada, enquanto que citosina, 2-tiocitosina e 2-aminopirimidina atuam como ligantes bidentados. O ligante 5-aminouracila forma um complexo organometálico interessante, no qual o seu átomo de carbono C(6) forma uma ligação covalente com Ru(III) através da coordenação simultânea do nitrogênio do grupo amino a um segundo íon rutênio, resultando um complexo bimetálico contendo duas moléculas de ligante como ponte. Na série das purinas85, notou-se que adenina, 2,6-diaminopurina e 2-tioxantina coordenam-se ao metal de modo bidentado e também formam complexos bimetálicos contendo dois ligantes-ponte. Guanina e hipoxantina formam complexos 1:1 e se coordenam de modo monodentado, através do átomo N(7). Os mesmos autores também investigaram outra série de complexos de Ru(III)-edta com nucleosídeos86, onde propôs-se que os nucleosídeos derivados de purina (adenosina, guanosina, xantosina e inosina) se coordenam ao íon metálico através do átomo de nitrogênio N(7) e os derivados de pirimidina (uridina) se ligam através do átomo N(3). Contudo, no caso específico da citidina ocorre a formação de um quelato através dos sítios C(2)O e N(3).
Estudos de Ru-edta com bases nucleicas (adenina, adenosina, citosina, citidina ou timina) e DNA, em meio aquoso, foram realizados por Chatterjee et al.87. Os espectros eletrônicos dos complexos, em pH 5,0, são caracterizados por uma banda LMCT intensa na região do UV. Os potenciais de redução centrados no metal foram observados na região de -0,28 a -0,15 V vs ECS. A reatividade dos ligantes frente à substituição da água em [RuIII(edta)(H2O)]- obedece à seqüência adenina > adenosina > citosina > citidina. Com base nos resultados espectroscópicos, eletroquímicos e cinéticos, propôs-se que a interação do centro metálico com DNA ocorre através da adenina.
A investigação cinética da reação entre Ru-edta e cisteína41, em função do pH e da temperatura, mostrou que o processo é rápido, de primeira ordem em relação aos dois reagentes, e a máxima constante de velocidade é obtida em pH 7,8-8,0. Sugeriu-se que o complexo poderia ser eficiente no tratamento de doenças renais, considerando sua solubilidade e estabilidade no pH fisiológico.
Estudos cinéticos, eletroquímicos e espectroscópicos do produto da reação entre Ru(II/III)-edta e guanosina-5'-monofosfato88 revelaram a presença de dois isômeros: kN(3) e kN(7). Quando o produto é gerado a partir do metal na forma oxidada (RuIII), apenas a espécie kN(7) encontra-se presente em grande quantidade. Posteriormente, novos resultados cinéticos envolvendo adenina, adenosina e os correspondentes 5'-nucleotídeos (adenosina-5'-monofosfato, adenosina-5'-difosfato e adenosina-5'-trifosfato)89 indicaram que a ligação dos nucleotídeos se dá em uma etapa rápida e dependente da concentração do nucleófilo, seguida por uma reação de fechamento do anel quelato independente da concentração.
Complexos do tipo Na6[(edta)RuIII-(µ-L)2 (µ-O)-RuIII(edta)] contendo alanina, fenilalanina ou valina como ligante-ponte também foram caracterizados90. Essas espécies são estáveis sob atmosfera inerte e sofrem desaminação oxidativa em presença de oxigênio, conforme mostrado pela formação de um a-cetoácido e liberação de amônia em uma reação catalítica.
Em outro trabalho interessante, demonstrou-se que o produto da reação de tricloreto de rutênio e ácido etilenodiaminotetraacético, em HCl, é o complexo antitumor [Ru(H3edta)(Cl)2]×4H2O 91, cuja estrutura obtida por raios-X sugere o primeiro exemplo de uma ligação Ru-O=COHR (glicina) estável, a qual foi confirmada através de dados de espectroscopias fotoeletrônica de raios-X e infravermelho.
Recentemente, Marmion et al.92 propuseram um modelo simples para a síntese fisiológica do NO, o qual seria conseguido através da interação de Ru(III)-edta e H2O2, na presença de arginina, para dar um complexo do tipo RuII-NO+ e citrulina. Em seguida, estudos sobre a termodinâmica e a cinética da reação direta entre [RuIII(edta)(H2O)]- e NO, em soluções aquosas, foram reportados por Davies et al.93 e por Wanat et al.94, que concluíram que a coordenação de óxido nítrico a Ru-edta é extremamente rápida (para os limites de detecção de técnicas cinéticas convencionais) e forma um nitrosil-complexo bastante estável. Tentativas de se determinar a velocidade de formação do complexo utilizando técnicas de fotólise rápida por laser foram prejudicadas devido à ocorrência de reações secundárias. Com base nas elevadas constantes de equilíbrio para os complexos em ambos os estados de oxidação, sugeriu-se que Ru-edta é um capturador ideal e versátil para NO.
CATÁLISE
A grande maioria dos trabalhos relacionados a aplicações de Ru-edta em catálise foi desenvolvida por Khan e colaboradores, os quais demonstraram que esse complexo pode catalisar a oxidação de vários substratos orgânicos95 (fosfinas96, aminas97, sulfeto98, hidrocarbonetos (alcanos e alcenos)99, álcoois100,101, etc) com oxigênio molecular ou [O=RuV(edta)]-. Contudo, a velocidade de oxidação é muito baixa para ser utilizada com sucesso em aplicações comerciais. Rutênio(III)-edta pode ainda catalisar reações de redução102, epoxidação103,104, carbonilação105, hidratação106, hidrogenação107, hidroformilação108 e transferência de átomo de enxofre109 em substratos orgânicos.
Khan et al. também investigaram um análogo de rutênio(III) do sistema de Udenfriend110 (i.e., [RuIII(edta)(H2O)]- /ácido ascórbico/H2O2 ou O2) em reações de transferência de átomo de oxigênio em compostos orgânicos saturados e insaturados101,104,111, onde a formação de um intermediário do tipo RuIV=O foi proposta. Estudos semelhantes foram realizados por Shukla, posteriormente112.
A importância de espécies intermediárias oxo-metálicas de alta valência na oxigenação catalítica de alguns substratos orgânicos (XO) como NaClO113, KHSO5114, PhIO115 também tem sido demonstrada. A formação do complexo metal-oxo consiste na etapa-chave que governa a eficiência do processo catalítico. A interação de Ru-edta com XO produz a espécie [RuV(edta)(O)]-, a qual foi isolada e caracterizada por métodos físico-químicos116.
Na década de 90, vários trabalhos sobre a redução fotocatalítica de nitrogênio a amônia catalisada por [RuIII(edta)(H2O)]- ou [RuII(edta)(N2)]2- (na presença de um sistema particulado semicondutor em solução aquosa e em condições ambientes) mostraram que a reação é de primeira ordem em relação ao catalisador e que a formação do complexo de rutênio(II) dinitrogenado é uma etapa essencial no processo de redução. A fotoxidação da água a oxigênio foi confirmada através de estudos com 18OH2117. Chatterjee reportou um estudo similar envolvendo a redução de hidrazina a amônia, confirmando que a formação do complexo com hidrazina é essencial no processo de redução118. Khan et al.119 também realizaram a fotorredução de CO2 e N2 em uma superfície semicondutora de TiO2 modificada pela adsorção de K[RuIII(edta)(bpy)] ou K[RuIII(edta)(phen)]. A fotorredução de CO2 foi conseguida em uma energia menor que a energia de separação de bandas do dióxido de titânio.
Através do estudo eletroquímico de RuII(Hedta)(NO+) em função do pH, Rhodes et al.120,121 verificaram que esse complexo é um eletrocatalisador efetivo na redução de NO2- e HNO2, gerando como produtos N2O, N2, NH3OH+ ou NH4+. Notou-se que a seleção do produto depende da escolha apropriada do pH e do potencial aplicado, e que a habilidade catalítica do complexo é restrita a soluções ácidas. O mecanismo de redução de nitrito é similar aos encontrados para derivados de polipiridinas de rutênio e ósmio, bem como para porfirinas de ferro solúveis em água. Ramachandraiah et al.122-124 realizaram estudos semelhantes com os complexos RuIII(edta)(N2H5) e RuIII(edta)(PhN2H5 ).
Posteriormente, Khan et al.125 também observaram que o complexo RuIII(edta)(NO+) é um excelente agente catalisador nas reações de hexeno-1 formando hexanona-2 e cicloexeno formando epóxido, não havendo necessidade de um co-catalisador e retendo sua atividade durante muitos ciclos de reação.
A síntese e a caracterização de um polímero de Ru-edta com atividade catalítica foram descritas mais recentemente126. O complexo coordenado ao polímero foi usado no estudo da hidrogenação de hexeno-1 a n-hexano sob condições brandas, para a qual sugeriu-se a expressão de velocidade e explorou-se a eficiência do catalisador.
POLÍMEROS E SUPERFÍCIES
No começo da década de 90, Kobayashi e Anson127 reportaram estudos sobre a interação de Ru(III)-edta com um polissiloxano solúvel contendo 4,4'-bipiridina como grupo pendente. As respostas eletroquímicas foram utilizadas na determinação de coeficientes de difusão e constantes de equilíbrio, as quais também foram medidas através de diálise de equilíbrio. Os resultados indicaram que o equilíbrio de ligação dessas espécies é mais estático que dinâmico na escala de tempo da eletroquímica.
A coordenação de Ru(III/II)-edta a poli(4-vinilpiridina) também foi investigada em seguida por Jiang e Anson128, os quais observaram que a velocidade de difusão do complexo RuIII(edta)(PVP) é aumentada substancialmente por transferência eletrônica homogênea entre os centros de Ru(III)-edta ligados a PVP e [RuII(edta)(H2O)]2- gerado na superfície do eletrodo. O coeficiente de difusão do complexo RuII(edta)(PVP) (igual a 1,4×10-7 cm2 s-1) forneceu uma estimativa do coeficiente de difusão das próprias cadeias de PVP protonadas.
Mais tarde, Sabatani e Anson129 estudaram as cinéticas de transferência de elétrons entre [RuIII(edta)(H2O)]- e complexos catiônicos de rutênio ([Ru(NH3)6]2+ ou [Ru(NH3)5(py)]2+) confinados em membranas de Nafion® depositadas em eletrodos, as quais foram analisadas de acordo com um modelo no qual as espécies imobilizadas exibem uma distribuição Gaussiana de potencial. Demonstrou-se que a correlação linear entre as constantes de velocidade e a termodinâmica das reações de transferência de elétrons cruzadas preditas pela teoria de Marcus não é afetada quando um dos co-reagentes apresenta um arranjo distribuído de potenciais formais ao invés de um único valor.
Lindall et al.130 mostraram que a coordenação de [RuIII(edta)(H2O)]- a sítios de piridina em poli(vinilpiridina-co-estireno) (90% PVP; 10% poliestireno) é induzida pela interação de uma solução diluída do complexo com um eletrodo de carbono vítreo revestido com PVPS. Esse polímero é capaz de incorporar [MoIV(CN)8]4- e a quantidade de ânion incorporado é aumentada se o complexo de rutênio for mantido no estado reduzido (RuII). Estudos semelhantes foram realizados com um polímero contendo {[RuIII(edta)(H2O)(py)]2 [RuII(NH3)5(py)]}, o qual pode ser variado em seis estados redox.
A preparação de polímeros metálicos mistos [Os(bpy)2(PVPS)nRu(edta)Cl] (n = 10, 15, 20), pela reação de [RuIII(edta)(H2O)]+ e [Os(bpy)2(PVPS)nCl]Cl depositado em eletrodo, foi descrita em seguida131. O polímero resultante é razoavelmente estável, apesar de sua redução em -0,4 V promover a perda gradual de centros de rutênio. Estudos envolvendo troca de íons mostraram que metilviologênio induz o decaimento na eletroatividade tanto nas unidades de rutênio coordenadas, como nos sítios de ósmio. Nenhuma evidência foi encontrada de que a redução de Ru(III)-edta induz a incorporação de MV2+. A oxidação de Os(II), por outro lado, induz a incorporação de [Fe(bathophen)3]4-. A possível incorporação de MV2+ e [Fe(bathophen)3]4-, simultaneamente, gera um polímero contendo seis diferentes estados redox. Experimentos de microbalança de quartzo demonstraram a perda rápida de Ru-edta pelo polímero. Observou-se que a estabilidade do polímero se torna muito maior em meio orgânico, no qual o potencial do par RuIII/RuII é mais negativo que em água, indicando uma forte afinidade do polímero por Ru(III).
Interessados no estudo de géis poliméricos com transições de fase associadas a parâmetros químicos e físicos, Iwaku et al.132 estudaram a eletroquímica de Ru-edta coordenado ao gel NIPA-VP, formado por N-isopropilacrilamida copolimerizada com 4-vinilpiridina e N,N'-metileno(bis)acrilamida. As espécies Ru(III)-edta e Ru(II)-edta coordenam-se a um e dois ligantes piridina, respectivamente. O último se dissocia gradualmente, formando Ru-edta monossubstituído durante a varredura de potencial. O coeficiente de difusão aparente para a propagação de carga no gel inchado, tendo Ru(II/III)-edta coordenado a uma piridina, foi estimado como sendo 20 vezes maior que o do gel encolhido.
Também no contexto de pesquisas envolvendo géis, Gushikem et al.133 verificaram que o complexo [RuIII(edta)(H2O)]- é fortemente adsorvido em uma superfície de sílica-gel revestida com óxido de zircônio(IV). A substituição da molécula de água no complexo de rutênio promove um deslocamento dos potenciais associados ao metal (água: -0,29 V; tiocianato: -0,20 V; piridina: -0,18 V; 4-cianopiridina: -0,08 V; pirazina: -0,05 V vs SCE).
Mais recentemente, Lazarin e Sernaglia134 descreveram um estudo sobre a funcionalização e a caracterização de sílica-gel com a espécie [3-(2-aminoetil)aminopropil]trimetoxisilano por meio de análises químicas e espectroscopia na região do infravermelho. Neste trabalho, enfatizou-se a capacidade da sílica modificada adsorver o complexo Ru(III)-edta presente em soluções etanólicas. Verificou-se também que o coeficiente de seletividade do complexo formado no suporte é da ordem de 104 M-1 e o número médio de ligantes coordenados a um íon metálico no suporte é ~ 1.
ALGUMAS PERSPECTIVAS
Inicialmente, o enfoque dos estudos envolvendo complexos de Ru-edta esteve voltado ao entendimento da cinética e do mecanismo das reações de substituição envolvendo [RuIII(edta)(H2O)]- , na busca de um modelo que justificasse a alta reatividade dessa espécie no estado de oxidação +III. Isso porque em sistemas similares (p. ex. poliaminas de rutênio) a forma reduzida (+II) do metal é a mais reativa. Contudo, à medida que os trabalhos confirmavam o modelo estrutural proposto por Matsubara e Creutz9, as pesquisas começaram a caminhar para novas áreas.
Os poliaminocarboxilatos de rutênio(III) têm uma potencial aplicação como metalofármacos135, pois são solúveis em água (permitindo rápida liberação in vivo), baixa toxicidade e apresentam um sítio disponível para coordenação de moléculas simples (p. ex. NO93). O óxido nítrico é um componente essencial em muitos processos fisiológicos, e sua liberação leva a uma diminuição da pressão sanguínea. A disfunção no seu metabolismo, como a liberação excessiva de NO, está associada a um grande número de doenças. Uma alternativa para se remover o excesso de NO seria o uso de um capturador específico, de alta afinidade e atóxico. O complexo Ru-edta é um candidato promissor à aplicação biológica como capturador de NO, pois, além de apresentar alta afinidade química por essa entidade, possui elevada estabilidade e insensibilidade a O294. Na verdade, tal nitrosil-complexo tem recebido bastante atenção desde o final da década de 80, quando foi publicado o primeiro trabalho sobre RuII(Hedta)(NO+)120 . A cinética e a termodinâmica da reação entre [RuIII(edta)(H2O)]- e NO93,94 parecem ser especialmente interessantes do ponto de vista catalítico120,121,125 e médico136.
Diversos trabalhos demonstraram a habilidade catalítica de derivados de Ru-edta em vários tipos de transformações orgânicas, tais como oxidação95, redução102, epoxidação103,104, carbonilação105, etc. Portanto, aplicações no campo de catálise também parecem promissoras. Novos estudos usando complexos de Ru-edta na modificação de superfícies sólidas (p. ex. matrizes poliméricas) devem promover uma maior seletividade catalítica em relação às reações de catálise homogênea observadas em fase líquida.
Ainda na direção de sistemas estruturados e interfaces, vários estudos mostraram que unidades de Ru-edta podem ser ancoradas ou incorporadas a polímeros e superfícies (de eletrodos, por exemplo) através de interações envolvendo tanto o íon metálico como o(s) grupo(s) carboxilato(s) livre(s) do edta. Tais pesquisas são de especial interesse, visto que superfícies/eletrodos modificados têm potencial emprego em catálise, células de conversão de energia, dispositivos mostradores de informação, sensores, etc137.
Por fim, algumas características particulares [RuIII(edta)(H2O)]- , tais como elevada solubilidade em água, labilidade do aquacomplexo de partida e estabilidade de seus derivados substituídos em ambos os estados redox (+III e +II), são importantes no direcionamento desses sistemas em diversas áreas de fronteira da química, cujas aplicações podem variar de detecção (bio)analítica de substâncias (p. ex. neurotransmissores34) a preparação de novos materiais ou planejamento de dispositivos funcionais em escala molecular66.
AGRADECIMENTOS
À Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) e ao Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) pelo apoio financeiro.
REFERÊNCIAS E NOTAS
1. Rein, F. N.; Tese de Doutorado, Universidade de São Paulo, Brasil, 1999.
2. Rocha, R. C.; Tese de Doutorado, Universidade de São Paulo, Brasil, 2000.
3. Chatterjee, D.; Coord. Chem. Rev. 1998, 168, 273.
4. Embora a existência do complexo com edta hexacoordenado (i.e., [RuIII/II(edta)]-,2-) seja reconhecida, nenhum dos estudos coletados nesta revisão é dirigido especificamente a tal espécie. Sendo assim, a terminologia genérica do tipo "Ru-edta" empregada neste artigo pode se referir tanto ao material de partida, [Ru(edta)(H2O)]-, como ao fragmento {Ru(edta)} penta- ou tetracoordenado de complexos mono- ou dissubstituídos, dependendo do contexto onde ela aparece.
5. Mukaida, M.; Okuno, H.; Ishimori, T.; Nippon Kagaku Zasshi 1965, 86, 589.
6. Ezerskaya, N. A.; Solovykh, T. P.; Russ. J. Inorg. Chem. 1966, 11, 991.
7. Scherzer, J.; Clapp, L. B.; J. Inorg. Nucl. Chem. 1968, 30, 1107; Shimizu, K.; Matsubara, T.; Sato, G. P.; Bull. Chem. Soc. Jpn. 1974, 47, 1651.
8. Matsubara, T.; Creutz, C.; J. Am. Chem. Soc. 1978, 100, 6255.
9. Matsubara, T.; Creutz, C.; Inorg. Chem. 1979, 18, 1956.
10. Bajaj, H. C.; van Eldik, R.; Inorg. Chem. 1988, 27, 4052.
11. Oyama, N.; Anson, F. C.; J. Electroanal. Chem. 1978, 88, 289.
12. Oyama, N.; Anson, F. C.; J. Am. Chem. Soc. 1979, 101, 1634.
13. Ogino, H.; Shimura, M.; Adv. Inorg. Bioinorg. Mechanisms 1986, 4, 107; Bajaj, H. C.; van Eldik, R.; Inorg. Chem. 1989, 28, 1980; Bajaj, H. C.; van Eldik, R.; Inorg. Chem. 1990, 29, 2855.
14. Ogino, H.; Katsuyama, T.; Ito, S.; Bull. Chem. Soc. Jpn. 1990, 63, 1370.
15. Chatterjee, D.; Bajaj, H. C.; J. Chem. Soc., Dalton Trans. 1993, 1065.
16. Jolley, J.; Campbell, C. J.; Castineiras, A.; Yanovsky, A. I.; Nolan, K. B.; Polyhedron 1998, 18, 49.
17. Okamoto, K.; Hidaka, J.; Iida, I.; Higashino, K.; Kanamori, K.; Acta Crystallogr. Section C: 1990, 46, 2327.
18. Detalhes computacionais são fornecidos nas refs. 1 e 2.
19. Ikeda, M.; Shimizu, K.; Sato, G. P.; Bull. Chem. Soc. Jpn. 1982, 55, 797.
20. Baar, R. B.; Anson, F. C.; J. Electroanal. Chem. 1985, 187, 265.
21. Khan, M. M. T.; Bhardwaj, R. C.; Jadhav, C. M.; J. Chem. Soc., Chem. Commun. 1985, 1690.
22. Ni, C. L.; Anson, F. C.; Osteryoung, J. G.; J. Electroanal. Chem. 1986, 202, 101.
23. Khan, M. M. T.; Hussain, A.; Ramachandraiah, G.; Moiz, M. A.; Inorg. Chem. 1986, 25, 3023.
24. Zhou, J. H.; Xi, W.; Hurst, J. K.; Inorg. Chem. 1990, 29, 160.
25. Diamantis, A. A.; Dubrawski, J. V.; Inorg. Chem. 1981, 20, 1142.
26. Diamantis, A. A.; Dubrawski, J. V.; Inorg. Chem. 1983, 22, 1934.
27. Yoshino, Y.; Kasahara, M.; Saito, M.; Polyhedron 1985, 4, 1019.
28. Rein, F. N.; Rocha, R. C.; Toma, H. E.; Electrochem. Commun. 2002, 4, 436.
29. Toma, H. E.; Tsurumaki, M.; J. Braz. Chem. Soc. 1990, 1, 17.
30. Araki, K.; Shu, C. F.; Anson, F. C.; Inorg. Chem. 1991, 30, 3043.
31. Stadler, C. C.; Mangrich, A. S.; Szpoganicz, B.; Stadler, E.; J. Braz. Chem. Soc. 1995, 6, 93.
32. Rocha, R. C.; Rein, F. N.; Toma, H. E.; J. Braz. Chem. Soc. 2001, 12, 234.
33. Rein, F. N.; Rocha, R. C.; Toma, H. E.; J. Electroanal. Chem. 2000, 494, 21.
34. Rein, F. N.; Rocha, R. C.; Toma, H. E.; J. Inorg. Biochem. 2001, 85, 155.
35. Khan, M. M. T.; Venkatasubramanian, K.; Shirin, Z.; Bhadbhade, M. M.; J. Chem. Soc., Dalton Trans. 1992, 885.
36. Toma, H. E.; Araki, K.; J. Coord. Chem. 1991, 24, 1.
37. Araki, K.; Rein, F. N.; Camera, S. G.; Toma, H. E.; Transition Met. Chem. 1992, 17, 535.
38. Chatterjee, D.; Bajaj, H. C.; Hussain, A.; J. Coord. Chem. 1994, 31, 329.
39. Povse, V. G.; Olabe, J. A.; Transition Met. Chem. 1998, 23, 657.
40. Yoshino, Y.; Uehiro, T.; Saito, M.; Bull. Chem. Soc. Jpn. 1979, 52, 1060.
41. Chatterjee, D.; Bajaj, H. C.; J. Coord. Chem. 1996, 39, 117.
42. Toma, H. E.; Rocha, R. C.; Croat. Chem. Acta 2001, 74, 499.
43. Toma, H. E.; Santos, P. S.; Mattioli, M. P. D.; de Oliveira, L. A. A.; Polyhedron 1987, 6, 603.
44. Khan, M. M. T.; Chatterjee, D.; Hussain, A.; Moiz, M. A.; Polyhedron 1990, 9, 2681.
45. Chatterjee, D.; Bajaj, H. C.; J. Chem. Soc., Dalton Trans. 1995, 3415.
46. Bajaj, H. C.; Das, A.; van Eldik, R.; J. Chem. Soc., Dalton Trans. 1998, 1563.
47. Rocha, R. C.; Araki, K.; Toma, H. E.; Transition Met. Chem. 1998, 23, 13.
48. Rein, F. N.; Rocha, R. C.; Toma, H. E.; J. Coord. Chem. 2001, 53, 99.
49. Creutz, C.; Kroger, P.; Matsubara, T.; Netzel, T. L.; Sutin, N.; J. Am. Chem. Soc. 1979, 101, 5442.
50. Khan, M. M. T.; Hussain, A.; Moiz, M. A.; Polyhedron 1992, 11, 687.
51. Chatterjee, D.; Bajaj, H. C.; Das, A.; Inorg. Chem. 1993, 32, 4049.
52. Forlano, P.; Cukiernik, F. D.; Poizat, O.; Olabe, J. A.; J. Chem. Soc., Dalton Trans. 1997, 1595.
53. Khoshtariya, D. E.; Bajaj, H. C.; Tregloan, P. A.; van Eldik, R.; J. Phys. Chem. A 2000, 104, 5535.
54. Bajaj, H. C.; Das, A.; Polyhedron 1997, 16, 3851.
55. Das, A.; Bajaj, H. C.; Chatterjee, D.; Polyhedron 1995, 14, 3585.
56. Das, A.; Bajaj, H. C.; Polyhedron 1997, 16, 1023.
57. Das, A.; Bajaj, H. C.; Bhadbhade, M. M.; J. Organomet. Chem. 1997, 544, 55.
58. Rein, F. N.; Toma, H. E.; Polyhedron 1998, 17, 1439.
59. Aikawa, M.; Yoshino, Y.; Inorg. Chim. Acta 1998, 282, 38.
60. Rocha, R. C.; Araki, K.; Toma, H. E.; Inorg. Chim. Acta 1999, 285, 197.
61. Rocha, R. C.; Toma, H. E.; Inorg. Chim. Acta 2000, 310, 65.
62. Rocha, R. C.; Toma, H. E.; Quim. Nova 2002, 25, 624.
63. Robin, M. B.; Day, P.; Adv. Inorg. Chem. Radiochem. 1967, 10, 247.
64. Creutz, C.; Prog. Inorg. Chem. 1983, 30, 1.
65. Hush, N. S.; Prog. Inorg. Chem. 1967, 8, 391.
66. Rocha, R. C.; Toma, H. E.; Inorg. Chem. Commun. 2001, 4, 230.
67. Cameron, B. R.; Bridger, G. J.; Maresca, K. P.; Zubieta, J.; Inorg. Chem. 2000, 39, 3928.
68. Toma, H. E.; Araki, K.; J. Chem. Res., Synop. 1990, 82.
69. Araki, K.; Santos, P. S.; Toma, H. E.; Spectrosc. Lett. 1993, 26, 1417.
70. Araki, K.; Toma, H. E.; Inorg. Chim. Acta 1991, 179, 293.
71. Toma, H. E.; Araki, K.; Coord. Chem. Rev. 2000, 196, 307.
72. Toma, H. E.; Sernaglia, R. L.; Talanta 1993, 40, 515.
73. Toma, H. E.; Santos, P. S.; Camera, S. G.; Sernaglia, R. L.; Spectrosc. Lett. 1997, 30, 507.
74. Toma, H. E.; Olive, M. A. L.; Polyhedron 1994, 13, 2647.
75. Toma, H. E.; Sernaglia, R. L.; Talanta 1995, 42, 1867.
76. Rocha, R. C.; Toma, H. E.; Polyhedron 2002, 21, 2089.
77. de Oliveira, L. A. A.; Dellaciana, L.; Haim, A.; Inorg. Chim. Acta 1994, 225, 129.
78. Ram, M. S.; Haim, A.; Inorg. Chem. 1991, 30, 1319.
79. Haim, A.; Adv. Chem. Ser. 1997, 253, 239.
80. Chatterjee, D.; J. Chem. Soc., Dalton Trans. 1996, 4389.
81. Chatterjee, D.; Transition Met. Chem. 2000, 25, 227.
82. Chatterjee, D.; Polyhedron 1999, 18, 1767.
83. Ward, M. S.; Chatterjee, D.; Shepherd, R. E.; Polyhedron 2000, 19, 1339.
84. Khan, B. T.; Annapoorna, K.; Inorg. Chim. Acta 1990, 171, 157.
85. Khan, B. T.; Annapoorna, K.; Inorg. Chim. Acta 1990, 176, 241.
86. Khan, B. T.; Annapoorna, K.; Polyhedron 1991, 10, 2465.
87. Chatterjee, D.; Bajaj, H. C.; Das, A.; J. Chem. Soc., Dalton Trans. 1995, 2497.
88. Chatterjee, D.; Ward, M. S.; Shepherd, R. E.; Inorg. Chim. Acta 1999, 285, 170.
89. Chatterjee, D.; Mitra, A.; Hamza, M. S. A.; van Eldik, R.; J. Chem. Soc., Dalton Trans. 2002, 962.
90. Khan, M. M. T.; Hussain, A.; Moiz, M. A.; Chatterjee, D.; Thorat, R. B.; Polyhedron 1993, 12, 1437.
91. Vilaplanaserrano, R.; Basallote, M. G.; Ruizvalero, C.; Gutierrezpuebla, E.; Gonzalezvilchez, F.; J. Chem. Soc., Chem. Commun. 1991, 100.
92. Marmion, C. J.; Murphy, T.; Nolan, K. B.; Chem. Commun. 2001, 1870.
93. Davies, N. A.; Wilson, M. T.; Slade, E.; Fricker, S. P.; Murrer, B. A.; Powell, N. A.; Henderson, G. R.; Chem. Commun. 1997, 47.
94. Wanat, A.; Schneppensieper, T.; Karocki, A.; Stochel, G.; van Eldik, R.; J. Chem. Soc., Dalton Trans. 2002, 941.
95. Khan, M. M. T.; Rao, A. P.; Bhatt, S. D.; J. Mol. Catal. 1992, 75, 41.
96. Khan, M. M. T.; Chatterjee, D.; Siddiqui, M. R. H.; Bhatt, S. D.; Bajaj, H. C.; Venkatasubramanian, K.; Moiz, M. A.; Polyhedron 1993, 12, 1443.
97. Khan, M. M. T.; Mirza, S. A.; Bajaj, H. C.; J. Mol. Catal. 1986, 37, 253; Khan, M. M. T.; Mirza, S. A.; Bajaj, H. C.; J. Mol. Catal. 1987, 39, 323; Khan, M. M. T.; Chatterjee, D.; Kumar, S.; Merchant, R. R.; Bhatt, K. N.; J. Mol. Catal. 1991, 67, 317; Khan, N. N. T.; Chatterjee, D.; Bhatt, S. D.; Bajaj, H. C.; Mirza, S. A.; React. Kinet. Catal. Lett. 1991, 44, 505.
98. Khan, M. M. T.; Bajaj, H. C.; Chatterjee, D.; J. Mol. Catal. 1992, 71, 177; Chatterjee, D.; J. Mol. Catal., A 1997, 127, 57; Chatterjee, D.; J. Mol. Catal. A: Chem. 1999, 150, 49.
99. Khan, M. M. T.; Mirza, S. A.; Bajaj, H. C.; J. Mol. Catal. 1987, 42, 323; Khan, M. M. T.; Shukla, R. S.; J. Mol. Catal. 1988, 44, 85; Khan, M. M. T.; Bajaj, H. C.; Shukla, R. S.; Mirza, S. A.; J. Mol. Catal. 1988, 45, 51.
100. Bajaj, H. C.; Khan, M. M. T.; React. Kinet. Catal. Lett. 1985, 28, 339; Khan, M. M. T.; Chatterjee, D.; Merchant, R. R.; Bhatt, A.; Kumar, S.; J. Mol. Catal. 1991, 66, 289; Khan, M. M. T.; Merchant, R. R.; Chatterjee, D.; Bhatt, K. N.; J. Mol. Catal. 1991, 67, 309.
101. Khan, M. M. T.; Rao, A. P.; Shukla, R. S.; J. Mol. Catal. 1989, 50, 45.
102. Khan, M. M. T.; Rao, N. N.; Chatterjee, D.; J. Photochem. Photobiol., A 1991, 60, 311.
103. Khan, M. M. T.; Rao, A. P.; J. Mol. Catal. 1987, 39, 331; Khan, M. M. T.; Chatterjee, D.; Bhatt, S. D.; Rao, A. P.; J. Mol. Catal. 1992, 77, 23.
104. Khan, M. M. T.; Shukla, R. S.; J. Mol. Catal. 1991, 70, 129.
105. Khan, M. M. T.; Halligudi, S. B.; Abdi, S. H. R.; J. Mol. Catal. 1988, 44, 179; Khan, M. M. T.; Halligudi, S. B.; Shukla, S.; J. Mol. Catal. 1989, 49, L69; Khan, M. M. T.; Halligudi, S. B.; Shukla, S.; Abdi, S. H. R.; J. Mol. Catal. 1989, 51, 129; Khan, M. M. T.; Hussain, A.; Moiz, M. A.; Naik, R. M.; Polyhedron 1989, 8, 2199.
106. Khan, M. M. T.; Halligudi, S. B.; Shukla, S.; J. Mol. Catal. 1990, 58, 299.
107. Khan, M. M. T.; Rao, N. N.; J. Mol. Catal. 1990, 58, 323.
108. Khan, M. M. T.; Halligudi, S. B.; Abdi, S. H. R.; J. Mol. Catal. 1988, 45, 215; Khan, M. M. T.; Halligudi, S. B.; Abdi, S. H. R.; J. Mol. Catal. 1988, 48, 7.
109. Khan, M. M. T.; Siddiqui, M. R. H.; Inorg. Chem. 1991, 30, 1157.
110. Udenfriend, S.; Clark, C. T.; Axelrod, J.; Brodie, B. B.; J. Biol. Chem. 1954, 208, 731; Udenfriend, S.; Clark, C. T.; Axelrod, J.; Brodie, B. B.; J. Biol. Chem. 1954, 208, 741.
111. Khan, M. M. T.; Shukla, R. S.; J. Mol. Catal. 1987, 39, 139; Khan, M. M. T.; Shukla, R. S.; J. Mol. Catal. 1988, 44, 73; Khan, M. M. T.; Shukla, R. S.; J. Mol. Catal. 1988, 45, 25; Khan, M. M. T.; Shukla, R. S.; Rao, A. P.; Inorg. Chem. 1989, 28, 452; Khan, M. M. T.; Shukla, R. S.; J. Mol. Catal. 1989, 51, 171; Khan, M. M. T.; Shukla, R. S.; J. Mol. Catal. 1990, 58, 405; Khan, M. M. T.; Chatterjee, D.; Shukla, R. S.; J. Mol. Catal. 1991, 69, 33; Khan, M. M. T.; Shukla, R. S.; J. Mol. Catal. 1992, 71, 157; Khan, M. M. T.; Shukla, R. S.; J. Mol. Catal. 1992, 72, 361.
112. Shukla, R. S.; Stud. Surf. Sci. Catal. 1998, 113, 897.
113. Khan, M. M. T.; Chatterjee, D.; Samad, S. A.; Merchant, R. R.; J. Mol. Catal. 1990, 61, 55.
114. Khan, M. M. T.; Chatterjee, D.; Merchant, R. R.; Bhatt, A.; J. Mol. Catal. 1990, 63, 147.
115. Khan, M. M. T.; Moiz, M. A.; Bhatt, S. D.; Merchant, R. R.; Chatterjee, D.; J. Mol. Catal. 1991, 67, 1.
116. Khan, M. M. T.; Chatterjee, D.; Merchant, R. R.; Paul, P.; Abdi, S. H. R.; Srinivas, D.; Siddiqui, M. R. H.; Moiz, M. A.; Bhadbhade, M. M.; Venkatasubramanian, K.; Inorg. Chem. 1992, 31, 2711.
117. Khan, M. M. T.; Rao, N. N.; J. Mol. Catal. 1989, 52, L5; Khan, M. M. T.; Rao, N. N.; J. Photochem. Photobiol. A 1991, 56, 101; Khan, M. M. T.; Chatterjee, D.; Krishnaratnam, M.; Bala, M.; J. Mol. Catal. 1992, 72, 13; Khan, M. M. T.; Bhardwaj, R. C.; Bhardwaj, C.; Rao, N. N.; J. Photochem. Photobiol., A 1992, 68, 137; Rao, N. N.; J. Mol. Catal. 1994, 93, 23.
118. Chatterjee, D.; J. Mol. Catal. A: Chem. 2000, 154, 1.
119. Khan, M. M. T.; Chatterjee, D.; Bala, M.; J. Photochem. Photobiol., A 1992, 67, 349; Khan, M. M. T.; Chatterjee, D.; Hussain, A.; Moiz, M. A.; J. Photochem. Photobiol., A 1993, 76, 97.
120. Rhodes, M. R.; Meyer, T. J.; Inorg. Chem. 1988, 27, 4772.
121. Rhodes, M. R.; Barley, M. H.; Meyer, T. J.; Inorg. Chem. 1991, 30, 629.
122. Ramachandraiah, G.; J. Am. Chem. Soc. 1994, 116, 6733.
123. Ramachandraiah, G.; Tyagi, B.; Prakash, R.; Bull. Electrochem. 1997, 13, 294.
124. Prakash, R.; Tyagi, B.; Chatterjee, D.; Ramachandraiah, G.; Polyhedron 1997, 16, 1235.
125. Khan, M. M. T.; Venkatasubramanian, K.; Shirin, Z.; Bhadbhade, M. M.; J. Chem. Soc., Dalton Trans. 1992, 1031.
126. Dalal, M. K.; Ram, R. N.; Bull. Mater. Sci. 2001, 24, 237.
127. Kobayashi, J.; Anson, F. C.; J. Phys. Chem. 1991, 95, 2595.
128. Jiang, R. Z.; Anson, F. C.; J. Phys. Chem. 1992, 96, 452.
129. Sabatani, E.; Anson, F. C.; J. Electroanal. Chem. 1995, 386, 111.
130. Lindall, C. M.; Crayston, J. A.; Colehamilton, D. J.; J. Mater. Chem. 1995, 5, 955.
131. Lindall, C. M.; Crayston, J. A.; ColeHamilton, D. J.; Glidle, A.; Peacock, R. D.; J. Mater. Chem. 1996, 6, 1259.
132. Iwaku, M.; Haseba, T.; Tatsuma, T.; Oyama, N.; J. Electroanal. Chem. 1998, 442, 27.
133. Gushikem, Y.; Peixoto, C. R. M.; Filho, U. P. R.; Kubota, L. T.; Stadler, E.; J. Colloid Interface Sci. 1996, 184, 236.
134. Lazarin, A. M.; Sernaglia, R. L.; Quim. Nova 1999, 22, 342.
135. Clarke, M. J.; Gaul, J. B.; Struct. Bond. 1993, 81, 147.
136. Fricker, S. P.; Platinum Met. Rev. 1995, 39, 150.
137. Merz, A.; Top. Curr. Chem. 1990, 152, 51.
138. Kane-Maguire, L. A. P.; Sheridan, P. S.; Basolo, F.; Pearson, R. G.; J. Am. Chem. Soc. 1970, 92, 5865.
139. A parametrização eletroquímica dos complexos de Ru-edta também foi realizada recentemente por Rocha et al.140, a partir da qual uma lista de parâmetros eletroquímicos (EL) de novos ligantes foi compilada.
140. Rocha, R. C.; Rein, F. N.; Toma, H. E.; Inorg. Chem. Commun. 2002, 5, 891.
Recebido em 15/10/02; aceito em 15/5/03
APÊNDICES
Apêndice A: Síntese do material de partida
Diferentemente da maioria dos complexos de partida na síntese de derivados de rutênio (p. ex., RuCl2(bpy)2 e [RuCl(NH3)5]Cl2), as espécies Ru(Hedta)(H2O) e K[Ru(Hedta)Cl] não são disponíveis comercialmente. Porém, esses precursores podem ser sintetizados de acordo com procedimentos descritos por Yoshino et al.40 (método A; uma modificação do método de Mukaida et al.5) e Diamantis e Dubrawski25 (método B; uma modificação do método de Ezerskaya e Solovykh6). Embora nenhum dos procedimentos apresente vantagens aparentes em termos de simplicidade nas etapas sintéticas, o método A dá um rendimento ligeiramente superior.
Apêndice B: Miscelânea de dados empíricos
Apêndice C: Abreviaturas

- 1. Rein, F. N.; Tese de Doutorado, Universidade de São Paulo, Brasil, 1999.
- 2. Rocha, R. C.; Tese de Doutorado, Universidade de São Paulo, Brasil, 2000.
- 3. Chatterjee, D.; Coord. Chem. Rev. 1998, 168, 273.
- 5. Mukaida, M.; Okuno, H.; Ishimori, T.; Nippon Kagaku Zasshi 1965, 86, 589.
- 6. Ezerskaya, N. A.; Solovykh, T. P.; Russ. J. Inorg. Chem. 1966, 11, 991.
- 7. Scherzer, J.; Clapp, L. B.; J. Inorg. Nucl. Chem. 1968, 30, 1107;
- Shimizu, K.; Matsubara, T.; Sato, G. P.; Bull. Chem. Soc. Jpn. 1974, 47, 1651.
- 8. Matsubara, T.; Creutz, C.; J. Am. Chem. Soc. 1978, 100, 6255.
- 9. Matsubara, T.; Creutz, C.; Inorg. Chem. 1979, 18, 1956.
- 10. Bajaj, H. C.; van Eldik, R.; Inorg. Chem. 1988, 27, 4052.
- 11. Oyama, N.; Anson, F. C.; J. Electroanal. Chem. 1978, 88, 289.
- 12. Oyama, N.; Anson, F. C.; J. Am. Chem. Soc. 1979, 101, 1634.
- 13. Ogino, H.; Shimura, M.; Adv. Inorg. Bioinorg. Mechanisms 1986, 4, 107;
- Bajaj, H. C.; van Eldik, R.; Inorg. Chem. 1989, 28, 1980;
- Bajaj, H. C.; van Eldik, R.; Inorg. Chem. 1990, 29, 2855.
- 14. Ogino, H.; Katsuyama, T.; Ito, S.; Bull. Chem. Soc. Jpn. 1990, 63, 1370.
- 15. Chatterjee, D.; Bajaj, H. C.; J. Chem. Soc., Dalton Trans. 1993, 1065.
- 16. Jolley, J.; Campbell, C. J.; Castineiras, A.; Yanovsky, A. I.; Nolan, K. B.; Polyhedron 1998, 18, 49.
- 17. Okamoto, K.; Hidaka, J.; Iida, I.; Higashino, K.; Kanamori, K.; Acta Crystallogr. Section C: 1990, 46, 2327.
- 19. Ikeda, M.; Shimizu, K.; Sato, G. P.; Bull. Chem. Soc. Jpn. 1982, 55, 797.
- 20. Baar, R. B.; Anson, F. C.; J. Electroanal. Chem. 1985, 187, 265.
- 21. Khan, M. M. T.; Bhardwaj, R. C.; Jadhav, C. M.; J. Chem. Soc., Chem. Commun. 1985, 1690.
- 22. Ni, C. L.; Anson, F. C.; Osteryoung, J. G.; J. Electroanal. Chem. 1986, 202, 101.
- 23. Khan, M. M. T.; Hussain, A.; Ramachandraiah, G.; Moiz, M. A.; Inorg. Chem. 1986, 25, 3023.
- 24. Zhou, J. H.; Xi, W.; Hurst, J. K.; Inorg. Chem. 1990, 29, 160.
- 25. Diamantis, A. A.; Dubrawski, J. V.; Inorg. Chem. 1981, 20, 1142.
- 26. Diamantis, A. A.; Dubrawski, J. V.; Inorg. Chem. 1983, 22, 1934.
- 27. Yoshino, Y.; Kasahara, M.; Saito, M.; Polyhedron 1985, 4, 1019.
- 28. Rein, F. N.; Rocha, R. C.; Toma, H. E.; Electrochem. Commun. 2002, 4, 436.
- 29. Toma, H. E.; Tsurumaki, M.; J. Braz. Chem. Soc. 1990, 1, 17.
- 30. Araki, K.; Shu, C. F.; Anson, F. C.; Inorg. Chem. 1991, 30, 3043.
- 31. Stadler, C. C.; Mangrich, A. S.; Szpoganicz, B.; Stadler, E.; J. Braz. Chem. Soc. 1995, 6, 93.
- 32. Rocha, R. C.; Rein, F. N.; Toma, H. E.; J. Braz. Chem. Soc. 2001, 12, 234.
- 33. Rein, F. N.; Rocha, R. C.; Toma, H. E.; J. Electroanal. Chem. 2000, 494, 21.
- 34. Rein, F. N.; Rocha, R. C.; Toma, H. E.; J. Inorg. Biochem. 2001, 85, 155.
- 35. Khan, M. M. T.; Venkatasubramanian, K.; Shirin, Z.; Bhadbhade, M. M.; J. Chem. Soc., Dalton Trans. 1992, 885.
- 36. Toma, H. E.; Araki, K.; J. Coord. Chem. 1991, 24, 1.
- 37. Araki, K.; Rein, F. N.; Camera, S. G.; Toma, H. E.; Transition Met. Chem. 1992, 17, 535.
- 38. Chatterjee, D.; Bajaj, H. C.; Hussain, A.; J. Coord. Chem. 1994, 31, 329.
- 39. Povse, V. G.; Olabe, J. A.; Transition Met. Chem. 1998, 23, 657.
- 40. Yoshino, Y.; Uehiro, T.; Saito, M.; Bull. Chem. Soc. Jpn. 1979, 52, 1060.
- 41. Chatterjee, D.; Bajaj, H. C.; J. Coord. Chem. 1996, 39, 117.
- 42. Toma, H. E.; Rocha, R. C.; Croat. Chem. Acta 2001, 74, 499.
- 43. Toma, H. E.; Santos, P. S.; Mattioli, M. P. D.; de Oliveira, L. A. A.; Polyhedron 1987, 6, 603.
- 44. Khan, M. M. T.; Chatterjee, D.; Hussain, A.; Moiz, M. A.; Polyhedron 1990, 9, 2681.
- 45. Chatterjee, D.; Bajaj, H. C.; J. Chem. Soc., Dalton Trans. 1995, 3415.
- 46. Bajaj, H. C.; Das, A.; van Eldik, R.; J. Chem. Soc., Dalton Trans. 1998, 1563.
- 47. Rocha, R. C.; Araki, K.; Toma, H. E.; Transition Met. Chem. 1998, 23, 13.
- 48. Rein, F. N.; Rocha, R. C.; Toma, H. E.; J. Coord. Chem. 2001, 53, 99.
- 49. Creutz, C.; Kroger, P.; Matsubara, T.; Netzel, T. L.; Sutin, N.; J. Am. Chem. Soc. 1979, 101, 5442.
- 50. Khan, M. M. T.; Hussain, A.; Moiz, M. A.; Polyhedron 1992, 11, 687.
- 51. Chatterjee, D.; Bajaj, H. C.; Das, A.; Inorg. Chem. 1993, 32, 4049.
- 52. Forlano, P.; Cukiernik, F. D.; Poizat, O.; Olabe, J. A.; J. Chem. Soc., Dalton Trans. 1997, 1595.
- 53. Khoshtariya, D. E.; Bajaj, H. C.; Tregloan, P. A.; van Eldik, R.; J. Phys. Chem. A 2000, 104, 5535.
- 54. Bajaj, H. C.; Das, A.; Polyhedron 1997, 16, 3851.
- 55. Das, A.; Bajaj, H. C.; Chatterjee, D.; Polyhedron 1995, 14, 3585.
- 56. Das, A.; Bajaj, H. C.; Polyhedron 1997, 16, 1023.
- 57. Das, A.; Bajaj, H. C.; Bhadbhade, M. M.; J. Organomet. Chem. 1997, 544, 55.
- 58. Rein, F. N.; Toma, H. E.; Polyhedron 1998, 17, 1439.
- 59. Aikawa, M.; Yoshino, Y.; Inorg. Chim. Acta 1998, 282, 38.
- 60. Rocha, R. C.; Araki, K.; Toma, H. E.; Inorg. Chim. Acta 1999, 285, 197.
- 61. Rocha, R. C.; Toma, H. E.; Inorg. Chim. Acta 2000, 310, 65.
- 62. Rocha, R. C.; Toma, H. E.; Quim. Nova 2002, 25, 624.
- 63. Robin, M. B.; Day, P.; Adv. Inorg. Chem. Radiochem. 1967, 10, 247.
- 64. Creutz, C.; Prog. Inorg. Chem. 1983, 30, 1.
- 65. Hush, N. S.; Prog. Inorg. Chem. 1967, 8, 391.
- 66. Rocha, R. C.; Toma, H. E.; Inorg. Chem. Commun. 2001, 4, 230.
- 67. Cameron, B. R.; Bridger, G. J.; Maresca, K. P.; Zubieta, J.; Inorg. Chem. 2000, 39, 3928.
- 68. Toma, H. E.; Araki, K.; J. Chem. Res., Synop. 1990, 82.
- 69. Araki, K.; Santos, P. S.; Toma, H. E.; Spectrosc. Lett. 1993, 26, 1417.
- 70. Araki, K.; Toma, H. E.; Inorg. Chim. Acta 1991, 179, 293.
- 71. Toma, H. E.; Araki, K.; Coord. Chem. Rev. 2000, 196, 307.
- 72. Toma, H. E.; Sernaglia, R. L.; Talanta 1993, 40, 515.
- 73. Toma, H. E.; Santos, P. S.; Camera, S. G.; Sernaglia, R. L.; Spectrosc. Lett. 1997, 30, 507.
- 74. Toma, H. E.; Olive, M. A. L.; Polyhedron 1994, 13, 2647.
- 75. Toma, H. E.; Sernaglia, R. L.; Talanta 1995, 42, 1867.
- 76. Rocha, R. C.; Toma, H. E.; Polyhedron 2002, 21, 2089.
- 77. de Oliveira, L. A. A.; Dellaciana, L.; Haim, A.; Inorg. Chim. Acta 1994, 225, 129.
- 78. Ram, M. S.; Haim, A.; Inorg. Chem. 1991, 30, 1319.
- 79. Haim, A.; Adv. Chem. Ser. 1997, 253, 239.
- 80. Chatterjee, D.; J. Chem. Soc., Dalton Trans. 1996, 4389.
- 81. Chatterjee, D.; Transition Met. Chem. 2000, 25, 227.
- 82. Chatterjee, D.; Polyhedron 1999, 18, 1767.
- 83. Ward, M. S.; Chatterjee, D.; Shepherd, R. E.; Polyhedron 2000, 19, 1339.
- 84. Khan, B. T.; Annapoorna, K.; Inorg. Chim. Acta 1990, 171, 157.
- 85. Khan, B. T.; Annapoorna, K.; Inorg. Chim. Acta 1990, 176, 241.
- 86. Khan, B. T.; Annapoorna, K.; Polyhedron 1991, 10, 2465.
- 87. Chatterjee, D.; Bajaj, H. C.; Das, A.; J. Chem. Soc., Dalton Trans. 1995, 2497.
- 88. Chatterjee, D.; Ward, M. S.; Shepherd, R. E.; Inorg. Chim. Acta 1999, 285, 170.
- 89. Chatterjee, D.; Mitra, A.; Hamza, M. S. A.; van Eldik, R.; J. Chem. Soc., Dalton Trans. 2002, 962.
- 90. Khan, M. M. T.; Hussain, A.; Moiz, M. A.; Chatterjee, D.; Thorat, R. B.; Polyhedron 1993, 12, 1437.
- 91. Vilaplanaserrano, R.; Basallote, M. G.; Ruizvalero, C.; Gutierrezpuebla, E.; Gonzalezvilchez, F.; J. Chem. Soc., Chem. Commun. 1991, 100.
- 92. Marmion, C. J.; Murphy, T.; Nolan, K. B.; Chem. Commun. 2001, 1870.
- 93. Davies, N. A.; Wilson, M. T.; Slade, E.; Fricker, S. P.; Murrer, B. A.; Powell, N. A.; Henderson, G. R.; Chem. Commun. 1997, 47.
- 94. Wanat, A.; Schneppensieper, T.; Karocki, A.; Stochel, G.; van Eldik, R.; J. Chem. Soc., Dalton Trans. 2002, 941.
- 95. Khan, M. M. T.; Rao, A. P.; Bhatt, S. D.; J. Mol. Catal. 1992, 75, 41.
- 96. Khan, M. M. T.; Chatterjee, D.; Siddiqui, M. R. H.; Bhatt, S. D.; Bajaj, H. C.; Venkatasubramanian, K.; Moiz, M. A.; Polyhedron 1993, 12, 1443.
- 97. Khan, M. M. T.; Mirza, S. A.; Bajaj, H. C.; J. Mol. Catal. 1986, 37, 253;
- Khan, M. M. T.; Mirza, S. A.; Bajaj, H. C.; J. Mol. Catal. 1987, 39, 323;
- Khan, M. M. T.; Chatterjee, D.; Kumar, S.; Merchant, R. R.; Bhatt, K. N.; J. Mol. Catal. 1991, 67, 317;
- Khan, N. N. T.; Chatterjee, D.; Bhatt, S. D.; Bajaj, H. C.; Mirza, S. A.; React. Kinet. Catal. Lett. 1991, 44, 505.
- 98. Khan, M. M. T.; Bajaj, H. C.; Chatterjee, D.; J. Mol. Catal. 1992, 71, 177;
- Chatterjee, D.; J. Mol. Catal., A 1997, 127, 57;
- Chatterjee, D.; J. Mol. Catal. A: Chem. 1999, 150, 49.
- 99. Khan, M. M. T.; Mirza, S. A.; Bajaj, H. C.; J. Mol. Catal. 1987, 42, 323;
- Khan, M. M. T.; Shukla, R. S.; J. Mol. Catal. 1988, 44, 85;
- Khan, M. M. T.; Bajaj, H. C.; Shukla, R. S.; Mirza, S. A.; J. Mol. Catal. 1988, 45, 51.
- 100. Bajaj, H. C.; Khan, M. M. T.; React. Kinet. Catal. Lett. 1985, 28, 339;
- Khan, M. M. T.; Chatterjee, D.; Merchant, R. R.; Bhatt, A.; Kumar, S.; J. Mol. Catal. 1991, 66, 289;
- Khan, M. M. T.; Merchant, R. R.; Chatterjee, D.; Bhatt, K. N.; J. Mol. Catal. 1991, 67, 309.
- 101. Khan, M. M. T.; Rao, A. P.; Shukla, R. S.; J. Mol. Catal. 1989, 50, 45.
- 102. Khan, M. M. T.; Rao, N. N.; Chatterjee, D.; J. Photochem. Photobiol., A 1991, 60, 311.
- 103. Khan, M. M. T.; Rao, A. P.; J. Mol. Catal. 1987, 39, 331;
- Khan, M. M. T.; Chatterjee, D.; Bhatt, S. D.; Rao, A. P.; J. Mol. Catal. 1992, 77, 23.
- 104. Khan, M. M. T.; Shukla, R. S.; J. Mol. Catal. 1991, 70, 129.
- 105. Khan, M. M. T.; Halligudi, S. B.; Abdi, S. H. R.; J. Mol. Catal. 1988, 44, 179;
- Khan, M. M. T.; Halligudi, S. B.; Shukla, S.; J. Mol. Catal. 1989, 49, L69;
- Khan, M. M. T.; Halligudi, S. B.; Shukla, S.; Abdi, S. H. R.; J. Mol. Catal. 1989, 51, 129;
- Khan, M. M. T.; Hussain, A.; Moiz, M. A.; Naik, R. M.; Polyhedron 1989, 8, 2199.
- 106. Khan, M. M. T.; Halligudi, S. B.; Shukla, S.; J. Mol. Catal. 1990, 58, 299.
- 107. Khan, M. M. T.; Rao, N. N.; J. Mol. Catal. 1990, 58, 323.
- 108. Khan, M. M. T.; Halligudi, S. B.; Abdi, S. H. R.; J. Mol. Catal. 1988, 45, 215;
- Khan, M. M. T.; Halligudi, S. B.; Abdi, S. H. R.; J. Mol. Catal. 1988, 48, 7.
- 109. Khan, M. M. T.; Siddiqui, M. R. H.; Inorg. Chem. 1991, 30, 1157.
- 110. Udenfriend, S.; Clark, C. T.; Axelrod, J.; Brodie, B. B.; J. Biol. Chem. 1954, 208, 731;
- Udenfriend, S.; Clark, C. T.; Axelrod, J.; Brodie, B. B.; J. Biol. Chem. 1954, 208, 741.
- 111. Khan, M. M. T.; Shukla, R. S.; J. Mol. Catal. 1987, 39, 139;
- Khan, M. M. T.; Shukla, R. S.; J. Mol. Catal. 1988, 44, 73;
- Khan, M. M. T.; Shukla, R. S.; J. Mol. Catal. 1988, 45, 25;
- Khan, M. M. T.; Shukla, R. S.; Rao, A. P.; Inorg. Chem. 1989, 28, 452;
- Khan, M. M. T.; Shukla, R. S.; J. Mol. Catal. 1989, 51, 171;
- Khan, M. M. T.; Shukla, R. S.; J. Mol. Catal. 1990, 58, 405;
- Khan, M. M. T.; Chatterjee, D.; Shukla, R. S.; J. Mol. Catal. 1991, 69, 33;
- Khan, M. M. T.; Shukla, R. S.; J. Mol. Catal. 1992, 71, 157;
- Khan, M. M. T.; Shukla, R. S.; J. Mol. Catal. 1992, 72, 361.
- 112. Shukla, R. S.; Stud. Surf. Sci. Catal. 1998, 113, 897.
- 113. Khan, M. M. T.; Chatterjee, D.; Samad, S. A.; Merchant, R. R.; J. Mol. Catal. 1990, 61, 55.
- 114. Khan, M. M. T.; Chatterjee, D.; Merchant, R. R.; Bhatt, A.; J. Mol. Catal. 1990, 63, 147.
- 115. Khan, M. M. T.; Moiz, M. A.; Bhatt, S. D.; Merchant, R. R.; Chatterjee, D.; J. Mol. Catal. 1991, 67, 1.
- 116. Khan, M. M. T.; Chatterjee, D.; Merchant, R. R.; Paul, P.; Abdi, S. H. R.; Srinivas, D.; Siddiqui, M. R. H.; Moiz, M. A.; Bhadbhade, M. M.; Venkatasubramanian, K.; Inorg. Chem. 1992, 31, 2711.
- 117. Khan, M. M. T.; Rao, N. N.; J. Mol. Catal. 1989, 52, L5;
- Khan, M. M. T.; Rao, N. N.; J. Photochem. Photobiol. A 1991, 56, 101;
- Khan, M. M. T.; Chatterjee, D.; Krishnaratnam, M.; Bala, M.; J. Mol. Catal. 1992, 72, 13;
- Khan, M. M. T.; Bhardwaj, R. C.; Bhardwaj, C.; Rao, N. N.; J. Photochem. Photobiol., A 1992, 68, 137;
- Rao, N. N.; J. Mol. Catal. 1994, 93, 23.
- 118. Chatterjee, D.; J. Mol. Catal. A: Chem. 2000, 154, 1.
- 119. Khan, M. M. T.; Chatterjee, D.; Bala, M.; J. Photochem. Photobiol., A 1992, 67, 349;
- Khan, M. M. T.; Chatterjee, D.; Hussain, A.; Moiz, M. A.; J. Photochem. Photobiol., A 1993, 76, 97.
- 120. Rhodes, M. R.; Meyer, T. J.; Inorg. Chem. 1988, 27, 4772.
- 121. Rhodes, M. R.; Barley, M. H.; Meyer, T. J.; Inorg. Chem. 1991, 30, 629.
- 122. Ramachandraiah, G.; J. Am. Chem. Soc. 1994, 116, 6733.
- 123. Ramachandraiah, G.; Tyagi, B.; Prakash, R.; Bull. Electrochem. 1997, 13, 294.
- 124. Prakash, R.; Tyagi, B.; Chatterjee, D.; Ramachandraiah, G.; Polyhedron 1997, 16, 1235.
- 125. Khan, M. M. T.; Venkatasubramanian, K.; Shirin, Z.; Bhadbhade, M. M.; J. Chem. Soc., Dalton Trans. 1992, 1031.
- 126. Dalal, M. K.; Ram, R. N.; Bull. Mater. Sci. 2001, 24, 237.
- 127. Kobayashi, J.; Anson, F. C.; J. Phys. Chem. 1991, 95, 2595.
- 128. Jiang, R. Z.; Anson, F. C.; J. Phys. Chem. 1992, 96, 452.
- 129. Sabatani, E.; Anson, F. C.; J. Electroanal. Chem. 1995, 386, 111.
- 130. Lindall, C. M.; Crayston, J. A.; Colehamilton, D. J.; J. Mater. Chem. 1995, 5, 955.
- 131. Lindall, C. M.; Crayston, J. A.; ColeHamilton, D. J.; Glidle, A.; Peacock, R. D.; J. Mater. Chem. 1996, 6, 1259.
- 132. Iwaku, M.; Haseba, T.; Tatsuma, T.; Oyama, N.; J. Electroanal. Chem. 1998, 442, 27.
- 133. Gushikem, Y.; Peixoto, C. R. M.; Filho, U. P. R.; Kubota, L. T.; Stadler, E.; J. Colloid Interface Sci. 1996, 184, 236.
- 134. Lazarin, A. M.; Sernaglia, R. L.; Quim. Nova 1999, 22, 342.
- 135. Clarke, M. J.; Gaul, J. B.; Struct. Bond. 1993, 81, 147.
- 136. Fricker, S. P.; Platinum Met. Rev. 1995, 39, 150.
- 137. Merz, A.; Top. Curr. Chem. 1990, 152, 51.
- 138. Kane-Maguire, L. A. P.; Sheridan, P. S.; Basolo, F.; Pearson, R. G.; J. Am. Chem. Soc. 1970, 92, 5865.
- 140. Rocha, R. C.; Rein, F. N.; Toma, H. E.; Inorg. Chem. Commun. 2002, 5, 891.

Datas de Publicação
-
Publicação nesta coleção
09 Mar 2004 -
Data do Fascículo
Fev 2004
Histórico
-
Aceito
15 Maio 2003 -
Recebido
15 Out 2002