Resumo
Microtubules are involved in many aspects of cellular biology and represent an important target of anticancer chemotherapeutics. In the past five years, novel natural products such as epothilones, discodermolide, sarcodictyin, eleutherobin, and laulimalide, all of which have biological activities similar to those of paclitaxel (Taxol<FONT FACE=Symbol>â</FONT>), have been discovered. Taxol<FONT FACE=Symbol>â</FONT> is an important antitumor drug approved by the FDA for the treatment of ovarian, breast and non-small-cell lung carcinomas and became the first natural product described that stabilized microtubules avoiding the cellular replication. The present article reports new natural products that are able to act on the stabilization of microtubules.
natural products; cancer; microtubules
natural products; cancer; microtubules
DIVULGAÇÃO
Novos produtos naturais capazes de atuar na estabilização de microtúbulos, um importante alvo no combate ao câncer
New natural products able to act on the stabilization of microtubules, an important target against cancer
Marcus Vinícius Nora de Souza* * e-mail: mvndesouza@bol.com.br
Fundação Oswaldo Cruz, Instituto de Tecnologia em Fármacos Far-Manguinhos, Rua Sizenando Nabuco, 100, 21041-250 Rio de Janeiro - RJ
ABSTRACT
Microtubules are involved in many aspects of cellular biology and represent an important target of anticancer chemotherapeutics. In the past five years, novel natural products such as epothilones, discodermolide, sarcodictyin, eleutherobin, and laulimalide, all of which have biological activities similar to those of paclitaxel (Taxolâ), have been discovered. Taxolâ is an important antitumor drug approved by the FDA for the treatment of ovarian, breast and non-small-cell lung carcinomas and became the first natural product described that stabilized microtubules avoiding the cellular replication. The present article reports new natural products that are able to act on the stabilization of microtubules.
Keywords: natural products; cancer; microtubules.
INTRODUÇÃO
Câncer é um conjunto de doenças causadas pela proliferação anormal das células, formando tumores. Existem mais de cem tipos de câncer, como exemplo podemos citar o de pele, pulmão, mama, fígado, estômago, rim, ovário, cérebro, próstata, pâncreas e ossos, sendo responsáveis anualmente pela morte de mais de 4 milhões de pessoas em todo mundo1. Nos Estados Unidos, por exemplo, de cada quatro mortes uma está relacionada a essa doença. Somente em 2003, existe expectativa de que cerca de 555.000 americanos, mais de 1.500 pessoas por dia, irão morrer dessa doença2.
O tratamento para câncer varia de acordo com o tipo e a gravidade da doença. Estes tumores podem ser tratados com cirurgia, radioterapia, quimioterapia ou, ainda, com a combinação dessas técnicas. A quimioterapia, diferente da cirurgia e da radioterapia, é utilizada em tratamento sistêmico, ou seja, que atua em todo corpo, à base de fármacos que impedem a reprodução celular e, conseqüentemente, levam as células malignas à morte. Estes fármacos podem ser ministrados isoladamente (monoquimioterapia) ou combinados (poliquimioterapia), sendo que a última apresenta resultados mais eficazes, pois consegue maior resposta a cada aplicação, diminuindo o risco de resistência aos fármacos e conseguindo atingir as células em diferentes fases do seu ciclo. Atualmente, um importante alvo quimioterápico são os microtúbulos3, de fundamental importância à replicação celular, que se tornam ineficazes em presença de alguns produtos naturais, os quais serão aqui abordados.
MICROTÚBULOS
Em 1979, Horwiz e colaboradores, do "Albert Einstein Medical College"4, demonstraram que o produto natural paclitaxel (Figura 1) era capaz de estabilizar os microtúbulos formados, impedindo sua despolimerização necessária à replicação celular, bloqueando, assim, o processo de divisão celular. Tal produto foi o primeiro composto capaz de inibir a divisão celular pela despolimerização dos microtúbulos, um mecanismo de ação até então desconhecido.
Os microtúbulos são constituídos por proteínas heterodiméricas compostas de duas subunidades: a e b-tubulina, de aproximadamente 440 aminoácidos cada uma, com peso molecular de aproximadamente 50 kD3. A tubulina está presente em todas as células eucariotas, existindo sob a forma heterodimérica ab ou microtubular. As duas subunidades a e b da tubulina são organizadas entre si por ligação de hidrogênio pela região CO2H terminal da subunidade b e a região terminal NH2 da subunidade a, sendo a formação dos microtúbulos realizada pelo rearranjo regular em cilindros flexíveis, obtidos em duas etapas: iniciação e elongação3. Os microtúbulos são absolutamente necessários ao processo de divisão celular, sendo usados pelas células para formar uma estrutura estática chamada de citoesqueleto, o qual dá forma à célula e determina a posição das organelas. As propriedades dinâmicas dos microtúbulos são usadas para transmitir sinais celulares, reorganizar organelas, proporcionar mobilidade às células, intervir no processo de secreção celular e na comunicação neuronal, o que explica sua abundância nos neurônios3. Devido à sua versatilidade, uso e importância no crescimento celular, os microtúbulos têm sido comumente considerados importantes alvos subcelulares para a atuação de agentes quimioterápicos.
Estruturalmente, os microtúbulos são rearranjados em forma cilíndrica, contendo 13 protofilamentos e apresentando um diâmetro de 24 nm (Figura 2)3. A iniciação e elongação dos protofilamentos ocorre em presença de proteínas associadas aos microtúbulos (PAM), Mg+2 e trifosfato de guanosina (TFP), sendo o processo reversível em presença de Ca+2 a 0 oC. Em presença do paclitaxel, ocorre uma estabilização dos núcleos a e b tubulinas pela intercalação deste fármaco resultando, assim, em distúrbio na formação do microtúbulo, que apresenta 12 protofilamentos e um diâmetro de 22 nm (Figura 2), bem como impede que ocorra a despolimerização do microtúbulo3.
Basicamente, o ciclo celular é o programa para o crescimento e a divisão (proliferação) celular. Existem 4 fases no ciclo celular: G1, S, G2 e M. A fase G1 é caracterizada por expressão de genes e síntese de proteínas. Isto permite à célula crescer e produzir todas as proteínas necessárias para a síntese de DNA. Durante a próxima etapa, a fase S, a célula replica seu DNA possuindo, então, 2 fitas completas de DNA, entrando na terceira fase do ciclo celular: G2. Durante a fase G2, a célula novamente cresce e sintetiza proteínas necessárias ao processo de divisão celular. Completada esta fase, a célula finalmente entra na quarta fase do ciclo celular: M. Durante a fase M, a célula passa por um processo denominado citocinese, originando 2 células filhas, estando o ciclo celular completo.
No ciclo de divisão celular, o alvo dos fármacos capazes de atuarem na estabilização dos microtúbulos é a metáfase, impedindo a fase G2-M na anáfase (Figura 3), podendo ocorrer a morte celular3 ou a resistência das células aos fármacos utilizados, sobrevivendo e continuando a se multiplicar.
Após a descoberta das propriedades antitumorais do paclitaxel e de seu original mecanismo de ação, foram descobertos diversos produtos naturais, extraídos de diferentes fontes, mas com as mesmas propriedades antitumorais do paclitaxel e que também serão objeto de discussão deste artigo.
PACLITAXEL
O paclitaxel, um triterpeno poliidroxilado extraído de Taxus brevifolia, uma árvore do Pacífico, foi isolado pela primeira vez em 1971, por Wall e colaboradores, nos EUA, sendo sua configuração absoluta obtida por difração de raios X5. O paclitaxel (Figura 1) possui 11 centros assimétricos e uma conformação rígida, com um ciclo A de conformação do tipo barco deformado, um ciclo B de conformação meio barco, um ciclo C em meia cadeira e ainda um ciclo oxetano D que reforça a rigidez da estrutura, importante para a estabilização dos microtúbulos. O paclitaxel possui também uma cadeia lateral ligada ao C-13 do sistema terpênico, que é essencial para a atividade biológica. Este produto natural mostrou-se eficaz contra uma série de tumores em concentrações nanomolares6, como por exemplo, em tumores de mama e ovário: MAD-MB-435 IC50 = 5,7 nM e SK-OV-3 IC50 = 11,5 nM, respectivamente.
Um grande problema na extração do paclitaxel reside no fato de que a espécie Taxus brevifolia demora cerca de 100 a 200 anos para atingir a maturidade e encontra-se em extinção. Para a obtenção de 1 kg do paclitaxel são necessários aproximadamente 10.000 kg da casca da Taxus brevifolia, sendo necessário abater cerca de 3.000 árvores. Essa quantidade poderia ser utilizada no tratamento de apenas 500 pacientes durante o período de um ano, sendo que um número muito reduzido de pessoas seriam beneficiadas, já que o câncer é uma doença que atinge, somente nos EUA, cerca de 6,5 milhões de pessoas por ano7. Uma solução para este problema foi encontrada nas folhas da árvore Taxus baccata, da qual se pode extrair a molécula 10-desacetilbacatina-III que apresenta o esqueleto básico e as funcionalidades do paclitaxel (Figura 4). Pode-se obter, via semi-síntese e em poucas etapas, o paclitaxel pela acetilação da posição 10 da 10-desacetilbacatina-III e pela introdução da cadeia lateral em posição C-13 (Figura 4). Uma vantagem deste método para a obtenção do paclitaxel é que não é necessário se abater as árvores, sendo uma fonte renovável. Por exemplo, para se obter 1 kg de 10-desacetilbacatina-III são necessários cerca de 3.000 kg das folhas de Taxus baccata7. Esta semi-síntese foi desenvolvida por diferentes grupos de pesquisa, destacando-se os grupos de Potier8, Ojima9 e Holton10. Como exemplo, pode-se mencionar a realizada por Holton e colaboradores10 (Figura 4) que, utilizando o cloreto de trimetilsilila, efetuaram a proteção seletiva na posição C-7 da 10-desacetilbacatina-III, seguida da acetilação da posição C-10 em presença de anidrido acético. Posteriormente, a introdução da cadeia lateral em posição C-13 é realizada, utilizando a b-lactama-N-benzoilada 1 em presença de KHMDS (hexametildissililamideto de potássio) como base, que, após a desproteção dos grupos sililas utilizando ácido fluorídrico em piridina, permite a obtenção do produto natural paclitaxel.
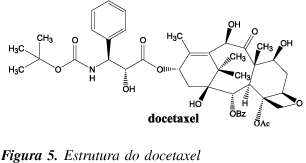
Devido à sua eficácia, este produto natural foi desenvolvido e comercializado pela companhia americana Bristol-Meyer Squibb com o nome de Taxolâ, na década de 90, e atualmente está disponível como medicamento em mais de 60 países. Usado com sucesso em diferentes tipos de tumores sólidos, foi o primeiro medicamento no mercado capaz de impedir a despolimerização da tubulina7,11. Devido ao sucesso terapêutico e econômico do Taxolâ, que gerou um faturamento de 1,6 bilhões de dólares no ano de 20007,11 para a empresa produtora, inúmeros análogos foram sintetizados em laboratório por diferentes grupos de pesquisa, com o objetivo de se identificar os grupos farmacofóricos (responsáveis pela atividade biológica), estabelecendo, assim, a relação estrutura-atividade na tentativa de se obter fármacos mais potentes7. Graças a estes estudos, Potier e colaboradores, do "Centre National de la Recherche Scientifique" (CNRS), em Gif-sur-Yvette França, descobriram, em 1986, o docetaxel8 (Figura 5), que é, em alguns casos, 1,6 vezes mais ativo que o taxol12. Como conseqüência de seus excelentes resultados, o docetaxel foi comercializado em 1994 pela indústria francesa Rhône-Poulenc com o nome de Taxotèreâ, sendo o segundo produto a impedir a despolimerização da tubulina13.
EPOTILONAS
Com o sucesso do paclitaxel, a busca por novos produtos naturais recebeu especial atenção de diferentes grupos de pesquisas. As epotilonas A e B (Figura 6) foram descobertas em 1987, por Hofle, Reichenbach e seus colaboradores da "Gesellschaft fur Biotechnologische Forschung (GBF)", na Alemanha14. Estes compostos foram isolados a partir de extratos de cultura da degradação de celulose pela bactéria Sorangium cellulosum14. Por apresentarem atividade contra Mucor hiemalis15, as epotilonas A e B foram primeiramente testadas como potenciais fungicidas e agrotóxicos14-16. No entanto, experimentos provaram que estes compostos eram muito tóxicos. Importante descoberta foi realizada em 1995, por pesquisadores da Merck nos Estados Unidos que, independentemente, isolaram as epotilonas A e B e comprovaram sua potente atividade antitumoral com valores IC50 próximos ao do paclitaxel17. Outra importante descoberta foi que o mecanismo de ação das epotilonas A e B, no combate ao câncer, era similar ao do taxol, exibindo potentes propriedades na estabilização da tubulina, impedindo a replicação celular. As epotilonas A e B mostraram-se capazes de inibir a glicoproteína-P13, responsável pelo desenvolvimento de resistência aos fármacos. Por esta razão, as epotilonas apresentam atividade superior ao paclitaxel em células resistentes, sendo que estes resultados foram, posteriormente, confirmados por cientistas da GBF15. Após os bons resultados preliminares, a epotilona B passou com sucesso por testes clínicos de fase I, estando atualmente em fase II11,18.
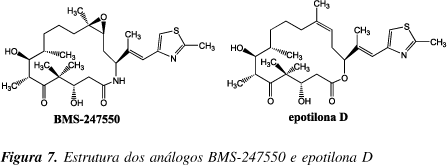
Na tentativa de se obter fármacos mais potentes, inúmeros análogos foram sintetizados por vários pesquisadores19, sendo a primeira síntese da epotilona em estado sólido descrita por Nicolau e colaboradores, o que permitiu a obtenção de uma biblioteca combinatória contendo numerosos análogos20. Graças aos estudos realizados por acadêmicos e industriais, o BMS-24755011, um promissor análogo da epotilona B, foi produzido pela Bristol-Myers Squibb (Figura 7). Este composto mostrou atividade em vários estágios de estudos clínicos de fase I, estando atualmente no início da fase II11,21. Estudos de relações entre estrutura e atividade biológica realizados por Danishefsky e colaboradores forneceram a epotilona D (Figura 7), conhecida também como desoxiepotilona B ou, ainda, KOS-862. Os estudos clínicos de fase I foram iniciados em outubro de 2001, pela companhia Kosan Bioscience e, atualmente, encontra-se em fase II22.
O OCEANO NO COMBATE AO CÂNCER
A medicina moderna tem encontrado no oceano uma vasta fonte de compostos com importante potencial terapêutico. Como exemplo podem-se citar os produtos naturais marinhos: discodermolida, eleuterobina, sarcodictina e laulimalida, que apresentam potente atividade antitumoral e possuem mecanismo de ação similar ao do paclitaxel, sendo compostos promissores na luta contra o câncer23.
Sarcodictina e eleuterobina
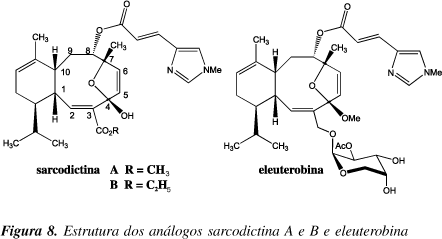
Os diterpenos sarcodictina A e B (Figura 8) foram isolados de corais do Mediterrâneo, Sarcodictyon roseum, pela primeira vez em 1987, por Pietra e colaboradores24. Oito anos mais tarde, a eleuterobina, um diterpeno glicosídico, foi inicialmente isolada na Austrália a partir de corais Eleutherobia sp por Fenical e colaboradores25, da "Scripps Institution of Oceanography", em La Jolla, Califórnia, e também de corais Erythropodium caribaeorum no Caribe26, sendo sua estrutura elucidada por Fenical27, em 1997. Atualmente, já foram identificados cerca de 30 compostos pertencentes à esta classe26. A eleuterobina foi testada pela companhia Bristol-Myers Squibb apresentando potente atividade antitumoral, pois inibe a proliferação de células cancerígenas com IC50 similar à do paclitaxel (10-15 nM)28. A eleuterobina é, também, capaz de estabilizar os microtúbulos, competindo com o paclitaxel pelo sítio ativo, apresentando resistência cruzada29. A eleuterobina foi testada no "National Cancer Institute", nos EUA, em 60 diferentes tipos de células, evidenciando atividade cerca de 100 vezes maior do que o paclitaxel em tumores de mama, rim, ovário e pulmão, sendo sua atividade patenteada e licenciada pela Bristol-Mayers Squibb30. Com o objetivo de se obter esta substância em maiores quantidades para a continuação da avaliação farmacológica e posterior produção industrial, a primeira síntese total da eleuterobina e da sarcodictina A foi realizada por Nicolau e colaboradores30, seguida pela síntese da eleuterobina pelo grupo de Danishefsky31. No entanto, devido à pouca quantidade obtida por estas sínteses, os pesquisadores têm elaborado diferentes estratégias sintéticas, bem como fragmentos análogos e semi-sínteses, no intuito de se obter a eleuterobina em escala industrial.
Discodermolida
O produto natural (+)-discodermolida, uma lactona com uma cadeia alquiltetraeno poliidroxilada (Figura 9), foi isolado de esponjas marinhas da espécie Discodermia dissoluta, encontradas em águas profundas no Caribe. Tais esponjas foram coletadas pela primeira vez em 1987, pelo Dr. S. Pomponi (Diretor da divisão biomédica de pesquisas marinhas), em "Harbor Branch Oceanographic Institution" Flórida32. Porém, somente em 1990, este composto foi isolado e caracterizado por Gunasekera e Longley, em Harbor Branch33. Em seguida, Longley e colaboradores34 demonstraram que a (+)-discodermolida era capaz de atuar como um potente imunossupressor com IC50 = 9 nM, inibindo a proliferação das células T. Em 1996, evidenciou-se que a (+)-discordemolida era capaz de destruir vários tipos de câncer in vitro, com um mecanismo de ação parecido ao do paclitaxel (Figura 1), com IC50 variando entre 3 a 80 nM35. A (+)-discodermolida causa uma desorganização irreversível dos microtúbulos, impedindo a divisão celular, resultando na morte da célula. Este produto natural marinho é mais potente que o paclitaxel em células resistentes, já que é capaz de inibir a glicoproteína-P36. Devido aos excelentes resultados farmacológicos deste produto natural, em 1998, "Harbor Branch" e "Novarthis Pharma" (Basel-Suíça) assinaram contrato para desenvolvimento da (+)-discodermolida como medicamento37.
Com o objetivo de se obter a (+)-discodermolida em maior escala, já que 434 g da esponja fornecem apenas 7 mg desta substância33, bem como a síntese de fármacos mais potentes e simplificados, inúmeros trabalhos têm sido realizados pela comunidade científica. Após a síntese realizada por Schereiber e colaboradores37 da (+)-discodermolida e de seu antípoda, a (-)-discodermolida (Figura 9), a qual exibe também potente atividade antitumoral, próxima à da (+)-discodermolida, vários grupos de pesquisa efetuaram a síntese total deste produto natural marinho38, empregando diferentes estratégias sintéticas, que permitem, também, a síntese de análogos. Graças à síntese total, a (+)-discodermolida tem sido obtida em laboratório em escala de 1 g39, permitindo a continuidade de sua avaliação como possível medicamento.
Laulimalida
O produto natural marinho laulimalida (Figura 10) foi isolado simultaneamente por dois grupos de pesquisa, em 1988, os quais trabalharam de forma independente: Crews e colaboradores40 isolaram a laulimalida da esponja Cacospongia mycofijiensis, enquanto Moore e colaboradores41 obtiveram-na da esponja Hyattella sp, proveniente da Indonésia. A elucidação da estrutura e a determinação da configuração absoluta dos centros estereogênicos foram efetuadas por difração de raios X por Higa e colaboradores42, que também isolaram a laulimalida, em 1996, da esponja Fasciospongia rimosa, proveniente da ilha de Okinawa42. O nome laulimalida é derivado da palavra havaiana laulima, que significa trabalhando junto40, sendo também conhecida como fijianolida B43. Em 1999, Mooberry do "UH Cancer Research Center of Hawaii" (CRCH) e Davidson da "Utah State University"44 demonstraram que a laulimalida possui um mecanismo de ação similar ao paclitaxel. Este composto apresenta um IC50 que varia de 10 a 50 ng/mL contra diferentes tipos de células tumorais, bem como citotoxidade comparada ao paclitaxel contra tumores de mama e ovário, MAD-MB-435 IC50 = 5,7 nM e SK-OV-3 IC50 = 11,5 nM. No entanto, a laulimalida é cerca de 100 vezes mais ativa que o paclitaxel contra células resistentes a diferentes tipos de fármacos, como por exemplo SKVLB-1 (IC50 = 1,2 µM)44, já que inibe também a glicoproteína-P, constituindo-se em fármaco promissor no combate ao câncer. Devido aos resultados promissores, vários pesquisadores têm se interessado na síntese da laulimalida e análogos, com a primeira síntese total da laulimalida realizada por Ghosh e colaboradores45, seguida pelas sínteses de Paterson46, Mulzer43,47, Wender48, Crimmins49, Willians50 e Nelson51. É importante destacar que outros pesquisadores, como por exemplo Davidson52, Nishayama53 e Lee54, também contribuíram para a síntese de fragmentos da laulimalida ou sua síntese parcial, proporcionando informações importantes para a síntese total, bem como para a de diferentes análogos.
Outros compostos pertencentes à classe da laulimalida têm sido isolados, como por exemplo, a isolaulimalida (Figura 11), que também apresenta atividade antitumoral. No entanto, é menos potente que a laulimalida em células KB (IC50>200 nM) e em células resistentes a diferentes tipos de drogas SKVLB-1 (IC50 = 1,6 mM)40. Estes resultados indicam que o epóxido presente em C-16/C-17 na laulimalida é importante para a atividade biológica.
CONCLUSÃO
Após a descoberta e o sucesso do paclitaxel, a busca de novos produtos naturais com mecanismo de ação similar ao deste fármaco recebeu atenção especial por parte de diferentes grupos de pesquisa, fornecendo, assim, uma nova perspectiva para a obtenção de novos e potentes agentes quimioterápicos. Isto pode ser confirmado pelo número de promissores compostos naturais isolados de diferentes fontes, capazes de atuarem na estabilização dos microtúbulos.
REFERÊNCIAS
1. http://www.cancer25.hpg.ig.com.br, acessada em Janeiro 2003.
2. http://www.cancer.org, acessada em Junho 2003.
3. Carvalho, P.; Tirnauer, J. S.; Pellman, D.; Trends Cell. Biol. 2003, 5, 229; Nicolau, K. C.; Roschangar, F.; Vourloumis, D.; Angew Chem., Int. Ed. 1998, 37, 2014; Unger C.; Drugs Fut. 1997, 22, 1337; Nicolaou, K. C.; Dai, W. M.; Guy, R. K.; Angew Chem., Int. Ed. 1994, 33, 15.
4. Schiff, P. B.; Horwitz, S. B.; Proc. Natl. Acad. Sci. U.S.A. 1980, 77, 1561; Horwitz, S. B.; Fant, J.; Schiff, P. B.; Nature 1979, 277, 665.
5. Wani, M. C.; Taylor, H. L.; Wall, M. E.; Coggen, P.; McPhail, A. T.; J. Am. Chem. Soc. 1971, 93, 2325.
6. Rowinsky, E. K.; Cazenave, L. A.; Donehower, R. C.; J. Natl. Cancer Inst. 1990, 82, 1247.
7. Kingston, D. G. I.; J. Nat. Prod. 2000, 63, 726.
8. Denis, J. N.; Greene, A. E.; Guenard, D.; Guerite-Voegelein, F.; Mangatal, L.; Potier, P.; J. Am. Chem. Soc. 1988, 110, 5917; Colin, M.; Guenard, D.; Guerite-Vogelein, F.; Potier, P., U. S. pat. 4924012 1990.
9. Ojima, I.; Habus, I.; Zhao, M.; Georg, G. I.; Jyasinghe, L. R.; J. Org. Chem. 1991, 56, 1681; Ojima, I.; Sun, C. M.; Zucco, M.; Park, Y. H.; Duclos, O.; Kuduk, S.; Tetrahedron Lett. 1993, 34, 4149.
10. Holton R. A; Somoza C.; Kim, H. B.; Liang. F. F.; Biediger, R. J.; Boatman, P. D.; Shindo, M.; Smith, C. C.; Kim, S.; Nadizadeh, H.; Suzuki, Y.; Tao, C.; Vu, P.; Tang, S.; Zhang, P.; Murchi, K. K.; Gentile, L. N.; Liu, J. H.; J. Am. Chem. Soc. 1994, 116, 1597; Holton R. A; Kim, H. B.; Somoza, C.; Liang. F. F.; Biediger, R. J.; Boatman, P. D.; Shindo, M.; Smith, C. C.; Kim, S.; Nadizadeh, H.; Suzuki, Y.; Tao, C.; Vu, P.; Tang, S.; Zhang, P.; Murchi, K. K.; Gentile, L. N.; Liu, J. H.; J. Am. Chem. Soc. 1994, 116, 1599.
11. Toogood, L. P.; Curr. Opin. Chem. Biol. 2002, 6, 472.
12. Ringel, J.; Horwitz, S. B.; J. Natl. Cancer Inst. 1991, 83, 288.
13. Losher, P.; Nagayama, O.; Drugs Fut. 1997, 22, 556.
14. Hofle, G.; Bedorf, N.; Gerth, K.; Reichenbach, H.; Chem. Abstr. 1993, 120, 52841.
15. Hofle, G.; Bedorf, N.; Steinmetz, H.; Schomburg, D.; Gerth, K.; Reichenbach, H.; Angew Chem., Int. Ed. 1996, 108, 1671; Hofle, G.; Bedorf, N.; Steinmetz, H.; Schomburg, D.; Gerth, K.; Reichenbach, H.; Angew Chem. Int. Ed. 1996, 35, 1567.
16. Hofle, G.; Bedorf, N.; Gerth, K.; Reichenbach, H.; Chem. Abstr. 1993, 119, 180598.
17. Bollag, D. M.; McQueney, Z. J.; Hensens, O.; Koupal, K.; Liesch, J.; Goetz, M.; Lazarides, D.; Woods, C. M.; Cancer Res. 1995, 55, 2325.
18. Broker, L. E.; Gioaccone, G.; Eur. J. Canc. 2002, 38, 2347.
19. Nicolau, K. C.; Roschangar, F.; Vourloumis, D.; Angew Chem., Int. Ed. 1998, 37, 2014; Harris, C. R.; Danishefsky, S. J.; J. Org. Chem. 1999, 64, 8434.
20. Nicolau, K. C.; Nature 1997, 387, 268.
21. Florsheimer, A.; Altman, K. H.; Expert Opin. Ther. Pat. 2001, 11, 951.
22. Chou, T. C.; Zhang, X. G.; Balog, A.; Su, D. S.; Meng, D.; Savin, K.; Bertino J. R.; Danishefsky, S. J.; Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 9642; Chou, T. C.; O'Connor O. A.; Tong, W. P.; Guan, Y.; Zhang, X. G.; Stahel, S. J.; Lee, C.; Danishefsky, S. J.; Proc. Natl. Acad. Sci. U.S.A. 2001, 47, 8113.
23. He, L.; Orr, G. A.; Horwitz, S. B.; DDT 2001, 6, 1153; Kavallaris, M.; Verrills, N. M.; Bridget, T. H.; Drug Resist. Up. 2001, 4, 392; Stachel, S. J.; Biswas, K.; Danishefsky, S. J.; Curr. Pharm. Des. 2001, 7, 1277.
24. D'Ambrosio, M.; Guerreiro, A.; Pietra, F.; Helv. Chem. Acta 1987, 70, 2019.
25. Fenical, W.; Jensen, P. R.; Lindel, T.; U. S. pat. 5,473,057 1995; Chem. Abstr. 1996, 124, P194297z.
26. Britton, R.; Roberge, M.; Berisch, H.; Andersen, R. J.; Tetrahedron Lett. 2001, 42, 2953.
27. Lindel, T.; Jensel, P. R.; Fenical, W.; Long, B. H.; Casazza, A. M.; Carboni, J.; Fairchild, C. R.; J. Am. Chem. Soc. 1997, 119, 8744.
28. Nicolau, K. C.; Pfefferkorn, J.; Xu, J.; Winssinger, N.; Ohshima, T.; Kim, S.; Hosakawa, S.; Vourloumis, D.; Van Delft, F.; Li, T.; Chem. Pharm. Bull. 1999, 47, 1199.
29. Long, B. H.; Carbone, J. M.; Wasserman, A. J.; Cornell, L. A.; Casazza, A. M.; Jensen, P. R.; Lindel, T.; Fenical, W.; Fairchild, C. R.; Cancer Res. 1998, 58, 1111.
30. Nicolau, K. C.; Van Delft, F.; P Oshima, T.; Vourloumis, D.; Xu, J.; Hosakawa, S.; Pfefferkorn, J.; Kim, S.; Li, T.; Angew Chem., Int. Ed. 1997, 36, 2520.
31. Chen, X. T.; Gutterideg, C. E.; Bhattacharya, S. K.; Zhou, B.; Pettus, T. R. R.; Hascall, R.; Danishefsky, S. J.; Angew Chem., Int. Ed. 1998, 37, 185; Chen, X. T.; Gutterideg, C. E.; Bhattacharya, S. K.; Zhou, B.; Pettus, T. R. R.; Hascall, R.; Danishefsky, S. J.; Angew Chem., Int. Ed. 1998, 40, 789.
32. Fenwick, S.; DDT 1998, 3, 300.
33. Gunasekera, S. P.; Gunasekera, M.; Longley, R. E.; Schulte, G. K.; J. Org. Chem. 1990, 55, 4912.
34. Longley, R. E.; Caddigan, D.; Harmody, D.; Gunasekera, M.; Gunasekera, S. P.; Transplantation 1991, 52, 650.
35. Hung, D. T.; Chen, J.; Schereiber, S. L.; Chem. Biol. 1996, 3, 287.
36. Martello, L. A.; LaMarche, M. J.; He, L.; Beauchamp, T. J.; Smith, A. B. III; Horwitz, S. B.; Chem. Biol. 2001, 8, 843.
37. Nerenberg, J. B.; Hung, D. T.; Somers, P. K.; Schereiber, S. L.; J. Am. Chem. Soc. 1993, 115, 12621; Hung, L. G.; Nerenberg, J. B.; Schereiber, S. L.; Chem. Biol. 1994, 1, 67.
38. Smith III, A. B.; Qiu, Y. P.; Jones, D. R.; Beauchamp, T. J.; Kobayashi, K.; J. Am. Chem. Soc. 1995, 117, 12011; Nerenberg, J. B.; Hung, D. T.; Schereiber, S. L.; J. Am. Chem. Soc. 1996, 118, 11054; Harried, S. S.; Yang, G.; Strawn, M. A.; Myles, D. C.; J. Org. Chem. 1997, 62, 6098; Marshall, J. A.; Johns, B. A.; J. Org. Chem. 1998, 63, 7885; Smith III, A. B.; Beauchamp, T. J.; LaMarche M. J.; Kaufman, M. D.; Qiu, Y. P.; Jones, D. R.; Kobayashi, K.; J. Am. Chem. Soc. 2000, 122, 8654; Paterson, I.; Florence, G. J.; Gerlach, K.; Scott, J. P.; Angew Chem., Int. Ed. 2000, 39, 377; Paterson, I.; Florence, G. J.; Tetrahedron Lett. 2000, 41, 6935; Paterson, I.; Florence, G. J.; Gerlach, K.; Scott, J. P.; Sereining, N.; J. Am. Chem. Soc. 2001, 123, 9535; Paterson, I.; Delgado, O.; Florence, G. J.; Lyothier, I.; Scott, J. P.; Sereining, N.; Org. Lett. 2003, 1, 35.
39. Smith III, A. B.; Kaufman, M. D.; Beauchamp, T. J.; LaMarche M. J.; Arimoto, H.; Org. Lett. 1999, 11, 1823.
40. Quinoa, E.; Kakou, Y.; Crews, P.; J. Org. Chem. 1988, 53, 3642.
41. Corley, D. G.; Herb, R.; Moore, R. E.; Scheuer, P. J.; Paul, V. J.; J. Org. Chem. 1988, 53, 3644.
42. Jefford, C. W.; Bernardinelli, G.; Tanaka, J. I.; Higa, T.; Tetrahedron Lett. 1996, 37, 159.
43. Mulzer, J.; Tetrahedron Lett. 2002, 43, 3381.
44. Mooberry, S. L.; Tien, G.; Hernandez, A. H.; Plubrukarn, A.; Davidson, B. S.; Cancer Res. 1999, 59, 653.
45. Ghosh, A. K.; Wang, Y.; J. Am. Chem. Soc. 2001, 122, 11027; Ghosh, A. K.; Wang, Y.; Kim, J. T.; J. Org. Chem. 2001, 66, 8973.
46. Paterson, I.; De Savi, C.; Tudge, M.; Org. Lett. 2001, 3, 3149.
47. Mulzer, J.; Ohler, E.; Angew Chem., Int. Ed. 2001, 40, 3842.; Enev, V. S.; Kaehling H.; Mulzer, J.; J. Am. Chem. Soc. 2001, 123, 10764.
48. Wender, P. A.; Hedge, S. G.; Hubbard, R. D.; Zhang, L.; J. Am. Chem. Soc. 2002, 124, 4956.
49. Crimmins, M. T.; Stanton, M. G.; Allwein, S. P.; J. Am. Chem. Soc. 2002, 124, 5958.
50. Willinas, D. R.; Liang, M.; Mullins, J.; Sitites, R. E.; Tetrahedron Lett. 2002, 43, 4841.
51. Nelson, S. G.; Cheng, W. S.; Kassick, A. J.; Hilfiker, M. A.; J. Am. Chem. Soc. 2002, 124, 13654.
52. Davidson, B.; Tetrahedron Lett. 2001, 42, 797; Sivaramakrishnan, A.; Nadolski, G. T.; McAlexander, I. A.; Davidson, B.; Tetrahedron Lett. 2002, 43, 213.
53. Shimizu, A.; Nishiyama, S.; Synlett 1998, 1209.
54. Lee, H. W.; Yoon, S. H.; Lee, I. Y. C.; Chung, B.Y.; Bull. Korean Chem. Soc. 2002, 22, 1179.
Recebido em 30/01/03
aceito em 27/10/03
-
1http://www.cancer25.hpg.ig.com.br, acessada em Janeiro 2003.
-
2http://www.cancer.org, acessada em Junho 2003.
- 3. Carvalho, P.; Tirnauer, J. S.; Pellman, D.; Trends Cell. Biol. 2003, 5, 229;
- Nicolau, K. C.; Roschangar, F.; Vourloumis, D.; Angew Chem., Int. Ed. 1998, 37, 2014;
- Unger C.; Drugs Fut. 1997, 22, 1337;
- Nicolaou, K. C.; Dai, W. M.; Guy, R. K.; Angew Chem., Int. Ed. 1994, 33, 15.
- 4. Schiff, P. B.; Horwitz, S. B.; Proc. Natl. Acad. Sci. U.S.A. 1980, 77, 1561;
- Horwitz, S. B.; Fant, J.; Schiff, P. B.; Nature 1979, 277, 665.
- 5. Wani, M. C.; Taylor, H. L.; Wall, M. E.; Coggen, P.; McPhail, A. T.; J. Am. Chem. Soc. 1971, 93, 2325.
- 6. Rowinsky, E. K.; Cazenave, L. A.; Donehower, R. C.; J. Natl. Cancer Inst. 1990, 82, 1247.
- 7. Kingston, D. G. I.; J. Nat. Prod. 2000, 63, 726.
- 8. Denis, J. N.; Greene, A. E.; Guenard, D.; Guerite-Voegelein, F.; Mangatal, L.; Potier, P.; J. Am. Chem. Soc. 1988, 110, 5917;
- Colin, M.; Guenard, D.; Guerite-Vogelein, F.; Potier, P., U. S. pat. 4924012 1990
- 9. Ojima, I.; Habus, I.; Zhao, M.; Georg, G. I.; Jyasinghe, L. R.; J. Org. Chem. 1991, 56, 1681;
- Ojima, I.; Sun, C. M.; Zucco, M.; Park, Y. H.; Duclos, O.; Kuduk, S.; Tetrahedron Lett. 1993, 34, 4149.
- 10. Holton R. A; Somoza C.; Kim, H. B.; Liang. F. F.; Biediger, R. J.; Boatman, P. D.; Shindo, M.; Smith, C. C.; Kim, S.; Nadizadeh, H.; Suzuki, Y.; Tao, C.; Vu, P.; Tang, S.; Zhang, P.; Murchi, K. K.; Gentile, L. N.; Liu, J. H.; J. Am. Chem. Soc. 1994, 116, 1597;
- Holton R. A; Kim, H. B.; Somoza, C.; Liang. F. F.; Biediger, R. J.; Boatman, P. D.; Shindo, M.; Smith, C. C.; Kim, S.; Nadizadeh, H.; Suzuki, Y.; Tao, C.; Vu, P.; Tang, S.; Zhang, P.; Murchi, K. K.; Gentile, L. N.; Liu, J. H.; J. Am. Chem. Soc. 1994, 116, 1599.
- 11. Toogood, L. P.; Curr. Opin. Chem. Biol. 2002, 6, 472.
- 12. Ringel, J.; Horwitz, S. B.; J. Natl. Cancer Inst. 1991, 83, 288.
- 13. Losher, P.; Nagayama, O.; Drugs Fut. 1997, 22, 556.
- 14. Hofle, G.; Bedorf, N.; Gerth, K.; Reichenbach, H.; Chem. Abstr. 1993, 120, 52841.
- 15. Hofle, G.; Bedorf, N.; Steinmetz, H.; Schomburg, D.; Gerth, K.; Reichenbach, H.; Angew Chem., Int. Ed. 1996, 108, 1671;
- Hofle, G.; Bedorf, N.; Steinmetz, H.; Schomburg, D.; Gerth, K.; Reichenbach, H.; Angew Chem. Int. Ed. 1996, 35, 1567.
- 16. Hofle, G.; Bedorf, N.; Gerth, K.; Reichenbach, H.; Chem. Abstr. 1993, 119, 180598.
- 17. Bollag, D. M.; McQueney, Z. J.; Hensens, O.; Koupal, K.; Liesch, J.; Goetz, M.; Lazarides, D.; Woods, C. M.; Cancer Res. 1995, 55, 2325.
- 18. Broker, L. E.; Gioaccone, G.; Eur. J. Canc. 2002, 38, 2347.
- 19. Nicolau, K. C.; Roschangar, F.; Vourloumis, D.; Angew Chem., Int. Ed. 1998, 37, 2014;
- Harris, C. R.; Danishefsky, S. J.; J. Org. Chem. 1999, 64, 8434.
- 20. Nicolau, K. C.; Nature 1997, 387, 268.
- 21. Florsheimer, A.; Altman, K. H.; Expert Opin. Ther. Pat. 2001, 11, 951.
- 22. Chou, T. C.; Zhang, X. G.; Balog, A.; Su, D. S.; Meng, D.; Savin, K.; Bertino J. R.; Danishefsky, S. J.; Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 9642;
- Chou, T. C.; O'Connor O. A.; Tong, W. P.; Guan, Y.; Zhang, X. G.; Stahel, S. J.; Lee, C.; Danishefsky, S. J.; Proc. Natl. Acad. Sci. U.S.A. 2001, 47, 8113.
- 23. He, L.; Orr, G. A.; Horwitz, S. B.; DDT 2001, 6, 1153;
- Kavallaris, M.; Verrills, N. M.; Bridget, T. H.; Drug Resist. Up. 2001, 4, 392;
- Stachel, S. J.; Biswas, K.; Danishefsky, S. J.; Curr. Pharm. Des. 2001, 7, 1277.
- 24. D'Ambrosio, M.; Guerreiro, A.; Pietra, F.; Helv. Chem. Acta 1987, 70, 2019.
- 25. Fenical, W.; Jensen, P. R.; Lindel, T.; U. S. pat. 5,473,057 1995;
- Chem. Abstr. 1996, 124, P194297z.
- 26. Britton, R.; Roberge, M.; Berisch, H.; Andersen, R. J.; Tetrahedron Lett. 2001, 42, 2953.
- 27. Lindel, T.; Jensel, P. R.; Fenical, W.; Long, B. H.; Casazza, A. M.; Carboni, J.; Fairchild, C. R.; J. Am. Chem. Soc. 1997, 119, 8744.
- 28. Nicolau, K. C.; Pfefferkorn, J.; Xu, J.; Winssinger, N.; Ohshima, T.; Kim, S.; Hosakawa, S.; Vourloumis, D.; Van Delft, F.; Li, T.; Chem. Pharm. Bull. 1999, 47, 1199.
- 29. Long, B. H.; Carbone, J. M.; Wasserman, A. J.; Cornell, L. A.; Casazza, A. M.; Jensen, P. R.; Lindel, T.; Fenical, W.; Fairchild, C. R.; Cancer Res. 1998, 58, 1111.
- 30. Nicolau, K. C.; Van Delft, F.; P Oshima, T.; Vourloumis, D.; Xu, J.; Hosakawa, S.; Pfefferkorn, J.; Kim, S.; Li, T.; Angew Chem., Int. Ed. 1997, 36, 2520.
- 31. Chen, X. T.; Gutterideg, C. E.; Bhattacharya, S. K.; Zhou, B.; Pettus, T. R. R.; Hascall, R.; Danishefsky, S. J.; Angew Chem., Int. Ed. 1998, 37, 185;
- Chen, X. T.; Gutterideg, C. E.; Bhattacharya, S. K.; Zhou, B.; Pettus, T. R. R.; Hascall, R.; Danishefsky, S. J.; Angew Chem., Int. Ed. 1998, 40, 789.
- 32. Fenwick, S.; DDT 1998, 3, 300.
- 33. Gunasekera, S. P.; Gunasekera, M.; Longley, R. E.; Schulte, G. K.; J. Org. Chem. 1990, 55, 4912.
- 34. Longley, R. E.; Caddigan, D.; Harmody, D.; Gunasekera, M.; Gunasekera, S. P.; Transplantation 1991, 52, 650.
- 35. Hung, D. T.; Chen, J.; Schereiber, S. L.; Chem. Biol. 1996, 3, 287.
- 36. Martello, L. A.; LaMarche, M. J.; He, L.; Beauchamp, T. J.; Smith, A. B. III; Horwitz, S. B.; Chem. Biol. 2001, 8, 843.
- 37. Nerenberg, J. B.; Hung, D. T.; Somers, P. K.; Schereiber, S. L.; J. Am. Chem. Soc. 1993, 115, 12621;
- Hung, L. G.; Nerenberg, J. B.; Schereiber, S. L.; Chem. Biol. 1994, 1, 67.
- 38. Smith III, A. B.; Qiu, Y. P.; Jones, D. R.; Beauchamp, T. J.; Kobayashi, K.; J. Am. Chem. Soc. 1995, 117, 12011;
- Nerenberg, J. B.; Hung, D. T.; Schereiber, S. L.; J. Am. Chem. Soc. 1996, 118, 11054;
- Harried, S. S.; Yang, G.; Strawn, M. A.; Myles, D. C.; J. Org. Chem. 1997, 62, 6098;
- Marshall, J. A.; Johns, B. A.; J. Org. Chem. 1998, 63, 7885;
- Smith III, A. B.; Beauchamp, T. J.; LaMarche M. J.; Kaufman, M. D.; Qiu, Y. P.; Jones, D. R.; Kobayashi, K.; J. Am. Chem. Soc. 2000, 122, 8654;
- Paterson, I.; Florence, G. J.; Gerlach, K.; Scott, J. P.; Angew Chem., Int. Ed. 2000, 39, 377;
- Paterson, I.; Florence, G. J.; Tetrahedron Lett. 2000, 41, 6935;
- Paterson, I.; Florence, G. J.; Gerlach, K.; Scott, J. P.; Sereining, N.; J. Am. Chem. Soc. 2001, 123, 9535;
- Paterson, I.; Delgado, O.; Florence, G. J.; Lyothier, I.; Scott, J. P.; Sereining, N.; Org. Lett. 2003, 1, 35.
- 39. Smith III, A. B.; Kaufman, M. D.; Beauchamp, T. J.; LaMarche M. J.; Arimoto, H.; Org. Lett. 1999, 11, 1823.
- 40. Quinoa, E.; Kakou, Y.; Crews, P.; J. Org. Chem. 1988, 53, 3642.
- 41. Corley, D. G.; Herb, R.; Moore, R. E.; Scheuer, P. J.; Paul, V. J.; J. Org. Chem. 1988, 53, 3644.
- 42. Jefford, C. W.; Bernardinelli, G.; Tanaka, J. I.; Higa, T.; Tetrahedron Lett. 1996, 37, 159.
- 43. Mulzer, J.; Tetrahedron Lett. 2002, 43, 3381.
- 44. Mooberry, S. L.; Tien, G.; Hernandez, A. H.; Plubrukarn, A.; Davidson, B. S.; Cancer Res. 1999, 59, 653.
- 45. Ghosh, A. K.; Wang, Y.; J. Am. Chem. Soc. 2001, 122, 11027;
- Ghosh, A. K.; Wang, Y.; Kim, J. T.; J. Org. Chem. 2001, 66, 8973.
- 46. Paterson, I.; De Savi, C.; Tudge, M.; Org. Lett. 2001, 3, 3149.
- 47. Mulzer, J.; Ohler, E.; Angew Chem., Int. Ed. 2001, 40, 3842.;
- Enev, V. S.; Kaehling H.; Mulzer, J.; J. Am. Chem. Soc. 2001, 123, 10764.
- 48. Wender, P. A.; Hedge, S. G.; Hubbard, R. D.; Zhang, L.; J. Am. Chem. Soc. 2002, 124, 4956.
- 49. Crimmins, M. T.; Stanton, M. G.; Allwein, S. P.; J. Am. Chem. Soc. 2002, 124, 5958.
- 50. Willinas, D. R.; Liang, M.; Mullins, J.; Sitites, R. E.; Tetrahedron Lett. 2002, 43, 4841.
- 51. Nelson, S. G.; Cheng, W. S.; Kassick, A. J.; Hilfiker, M. A.; J. Am. Chem. Soc. 2002, 124, 13654.
- 52. Davidson, B.; Tetrahedron Lett. 2001, 42, 797;
- Sivaramakrishnan, A.; Nadolski, G. T.; McAlexander, I. A.; Davidson, B.; Tetrahedron Lett. 2002, 43, 213.
- 53. Shimizu, A.; Nishiyama, S.; Synlett 1998, 1209.
- 54. Lee, H. W.; Yoon, S. H.; Lee, I. Y. C.; Chung, B.Y.; Bull. Korean Chem. Soc. 2002, 22, 1179.
Datas de Publicação
-
Publicação nesta coleção
31 Maio 2004 -
Data do Fascículo
Abr 2004
Histórico
-
Recebido
30 Jan 2003 -
Aceito
27 Out 2003