Resumo
Samples of copper compounds covering all of the XXth century and the end of the XIXth century were submitted to classical and instrumental quantitative analysis. The amount of impurities greatly decreased with time, reaching a constant level since the 1960's. The gravimetric method was suitable for the determination of copper although other procedures also gave good or reasonable results. However, for metal contaminants, atomic absorption spectrometry was the best choice because of its lower detection limits, being able to determine several elements in the oldest samples. Ion chromatography detected several anions in copper salts manufactured before the 1950's. An increasing quality of raw materials and a better sensitivity of analytical methods led to quality improvement of copper compounds with time.
copper salts; chemical analysis; quality control
copper salts; chemical analysis; quality control
ASSUNTOS GERAIS
Análise química de sais de cobre fabricados entre 1877 e 1995
Chemical analysis of copper salts manufactured between 1877 and 1995
Júlio Carlos Afonso* * e-mail: julio@iq.ufrj.br ; Renata de Melo Aguiar; Lôla Maria Braga Gomes
Departamento de Química Analítica, Instituto de Química, Universidade Federal do Rio de Janeiro, CP 68563, 21949-900 Rio de Janeiro - RJ
ABSTRACT
Samples of copper compounds covering all of the XXth century and the end of the XIXth century were submitted to classical and instrumental quantitative analysis. The amount of impurities greatly decreased with time, reaching a constant level since the 1960's. The gravimetric method was suitable for the determination of copper although other procedures also gave good or reasonable results. However, for metal contaminants, atomic absorption spectrometry was the best choice because of its lower detection limits, being able to determine several elements in the oldest samples. Ion chromatography detected several anions in copper salts manufactured before the 1950's. An increasing quality of raw materials and a better sensitivity of analytical methods led to quality improvement of copper compounds with time.
Keywords: copper salts; chemical analysis; quality control.
INTRODUÇÃO
O controle analítico de produtos químicos começou a se tornar uma preocupação em meados do século XIX. Por exemplo, em 1851, a partir da queixa feita por um cliente que duvidara da pureza dos produtos, Heinrich Emanuel Merck (1794-1855) lhe escreveu, em carta a 15 de maio daquele ano: "Garanto-lhe sempre a pureza dos meus preparados e assumo qualquer desvantagem que lhe venha resultar de um produto impuro". Em 1888, foi publicado o livro "O Exame da Pureza dos Reagentes Químicos", por Carl Krauch (1853-1934), que descrevia procedimentos para diversas substâncias. Iniciava-se, assim, a criação de normas sistemáticas para a pureza dos reagentes; a "Chemische Fabrik E. Merck" foi o primeiro fabricante de produtos químicos com garantia de qualidade1 no mundo. Surgiam os reagentes com a expressão até hoje usada "pro analisi" (para análise).
Naquela época, havia uma proliferação indiscriminada de produtos com as expressões latinas "purum", "purissimum" ou "depuratum", mas sem qualquer tipo de controle por parte dos fabricantes2,3. A instalação de um laboratório de controle, separado da área produtiva, tido como a "consciência da fábrica", tinha como meta a inspeção dos produtos fabricados, bem como controlar as matérias-primas2,3. Esse perfil profissional de tratamento dos produtos químicos rapidamente se espalhou às empresas do setor em todo o mundo4,5. Hoje em dia, antes da liberação de um lote de produto químico, este passa por uma série de testes e ensaios, tanto mais quanto mais específico for o emprego em uma determinada área de pesquisa e desenvolvimento.
PERFIL EVOLUTIVO DOS MÉTODOS DE ANÁLISE INORGÂNICA
Na literatura do início do século XX, observam-se muitas citações aos métodos baseados na medida de volume de gases liberados em certas reações; tais métodos eram comumente chamados de métodos volumétricos. Hoje, esses métodos são pouco adotados, e o termo volumetria acabou por ser amplamente utilizado como sinônimo de titrimetria, que consiste na dosagem do volume de uma solução de concentração conhecida com exatidão, necessário para reagir quantitativamente com um determinado volume da solução que contém o analito a se determinar. Como o termo "análise volumétrica", podia ser confundido com a análise de gases liberados durante uma análise, estabeleceu-se o termo "análise titrimétrica", para expressar o processo de titulação6,7. A preocupação com esta distinção foi iniciada por volta de 19508,9, chegando a ter os dois termos no mesmo livro, e ambos continuam a ser utilizados até os dias atuais.
Na literatura do início dos anos 193010,11 já existia referência à titrimetria potenciométrica. Inicialmente tinham-se os eletrodos de hidrogênio, calomelano, antimônio, prata/cloreto de prata e quinidrona (quinona + hidroquinona). Mas após estudos de MacInnes, Dole e Belcher (1931), obteve-se um eletrodo de vidro com ótimos resultados, cuja composição do vidro (SiO2 72%, Na2O 22% e CaO 6%) é utilizada até os dias atuais. Vale lembrar que se conhecia a possibilidade de utilização do eletrodo de vidro desde 1909 (Harber e Klemensiewics), mas somente nessa década os pesquisadores se interessaram pela melhoria dos métodos e eletrodos. Já se tinha conhecimento também das vantagens da determinação com grande precisão do ponto final através da potenciometria, sendo utilizada em soluções coloridas onde os indicadores eram mascarados pela solução-amostra; podia-se agora determinar duas ou mais espécies na mesma amostra (exemplo: I- e Cl-) e a interferência dos contaminantes com o indicador era evitada.
O método polarográfico de análise química foi descrito por Jaroslav Heyrovsky, por volta de 192012,13. Em 1940, Kolthoff e Laitinen chamaram a técnica de voltametria, indicando que voltagem e corrente eram medidas durante uma análise12. Mas somente na década seguinte esta técnica foi introduzida nos livros didáticos. Nos anos 1970, as vantagens do uso de componentes eletrônicos no estado sólido, de baixo custo, e a melhoria da instrumentação impulsionaram grandemente as suas aplicações práticas14.
Em 1945, Barnes, Richardson e Berry construíram o primeiro fotômetro de chama nos Estados Unidos13. Foi um grande avanço na determinação quantitativa de metais alcalinos e alcalino-terrosos.
Dois anos depois, Schwarzenbach estudou a determinação de metais por diversos quelantes e indicadores, resultando na descoberta do EDTA15-18 (sal dissódico do ácido etilenodiaminotetracético). Em 1958, Welcher lançou o livro "The Analytical Uses of Ethylenediaminetracetic Acid", que representou a aplicação em análise quantitativa dos estudos de Schwarzenbach, contendo diversos métodos para 30 metais comuns, mais os elementos das então chamadas "terras raras" (Sc, Y e lantanídios)16. Após uma década já havia cerca de 950 trabalhos publicados sobre o assunto, mostrando o grande interesse dos pesquisadores em aprimorar a técnica, devido à sua versatilidade15,16. Foram feitos diversos estudos também, para a determinação das condições ideais para cada elemento a ser determinado, tais como pH, cinética de formação, estabilidade do complexo, etc. Inicialmente dispunha-se de dois indicadores de uso prático, o negro de eriocromo-T e a murexida9,15, mas após pouco tempo cerca de 40 indicadores já haviam sido relatados16. Aparentemente a grande aplicação inicial foi a determinação da dureza da água (dosagem de cálcio e magnésio).
Esse sucesso levou a um crescente interesse no aprimoramento dos métodos titrimétricos, tornando-os mais práticos e seletivos; crescia o número de métodos propostos conforme caíam em obsolência os métodos gravimétricos.
A partir da década de 1950, observou-se um aumento dos métodos instrumentais mencionados em livros didáticos9,13. Naquela época a análise instrumental já estava bastante avançada, iniciando o aparecimento de literatura especializada, onde eram encontradas a teoria e as aplicações das seguintes técnicas: espectrofotometria no UV-visível, IV, Raman, fluorimetria, espectrometria de emissão atômica, fotometria de chama, potenciometria, condutimetria, polarografia/amperometria, raios-X (difração e fluorescência) e espectrometria de massas13.
Em 1955, surgiu a espectrometria de absorção atômica, com as publicações de Walsh e também de Alkemade e Milatz. No final dos anos 1950, os primeiros aparelhos comerciais começaram a ser vendidos. Em 1959, o russo Boris L'vov propôs o uso de um forno de grafite como elemento atomizador, o qual apresenta a vantagem de ampliar enormemente o limite de detecção de elementos, inclusive refratários, além de permitir mais facilmente a automação19.
Os livros de análise instrumental dos anos 1970 incorporaram os avanços feitos nas décadas anteriores20,21. De lá para cá observou-se um crescimento notável dessa literatura e uma conseqüente redução nas menções aos métodos clássicos, chegando somente a citações4,7,19,22. Este comportamento era de se esperar, já que os métodos instrumentais estão a cada dia sendo aprimorados e modernizados e os métodos clássicos se mantêm quase inalterados. O acoplamento de computadores ao instrumental analítico agilizou sobremodo os resultados, além de facilitar o manuseio, já que os programas são capazes até de controlar todos os parâmetros de análise. Porém, é importante enfatizar que a grande motivação no progresso da análise quantitativa inorgânica sempre foi a necessidade de dosar um dado elemento ou analito com limites de detecção cada vez menores e em tempo o mais curto possível.
DETERMINAÇÃO DE COBRE AO LONGO DO TEMPO
No início do século XX, o método mais aconselhado23,24 era a determinação gravimétrica do metal como sulfeto ou óxido. Na titrimetria, os métodos existentes envolviam KCN (o complexo [Cu(NH3)4]2+, azul intenso, é titulado com o CN- para formar o complexo [Cu(CN)4]3-, incolor), Na2S (titrimetria de precipitação de CuS), KI (titrimetria de precipitação do CuI), e ainda KI + xantato, ácido pícrico e KMnO4. Já se propunham procedimentos espectrofotométricos com base na dosagem do complexo [Cu(NH3)4]2+. A determinação a partir de liberação de gases (método volumétrico) era bastante citada: o sulfato cúprico pode formar diferentes sais hidratados e, desta forma, aquecendo-os a temperaturas determinadas e pesando-os, podia-se determinar a hidratação no sal original.
Na década de 1910, os métodos espectrofotométricos passaram a ser amplamente citados24. Empregava-se para isto, um aparelho conhecido como colorímetro de Duboscq25. Apesar de ser considerado pouco exato, era uma maneira fácil de saber o teor aproximado, quando comparado a soluções de concentração conhecida. É também citada a determinação deste elemento por eletrodeposição (eletrogravimetria)26.
Nos anos 1920, os métodos de precipitação como óxido ou sulfeto ainda eram os mais citados27, mas a eletrogravimetria passou a ser altamente recomendada27-29, já que apresentava alta precisão quando comparada aos outros métodos, além da possibilidade de determinação na presença de metais interferentes. Ainda se mencionava a determinação da água de hidratação do sulfato de cobre27. Na titrimetria, os métodos citados30 para a determinação do cobre empregavam os reagentes: KI, K2Cr2O7, KIO3 e KSCN ou NH4SCN. A década seguinte não registrou grandes inovações nos procedimentos de determinação quantitativa do cobre31,32.
Os anos 1940 apresentam um equilíbrio na menção das técnicas gravimétricas clássicas, titrimétricas e eletrogravimétricas33-35. Afora a dosagem clássica em meio amoniacal, dois novos métodos de determinação espectrofotométrica33 foram propostos, envolvendo o emprego de dietiltiocarbamato de sódio ou do ácido rubeânico (ditiooxamida). Observou-se o aparecimento de uma variante na determinação titrimétrica do cobre através do método iodométrico33,35: após a adição de KI à solução de cobre, o iodo liberado era titulado por uma solução de tiossulfato de sódio.
Na década de 1950, a introdução do EDTA revolucionou a análise titrimétrica16 do cobre. Na década seguinte, a proposta de novos indicadores36 ampliou a versatilidade desta determinação. O método iodométrico, as determinações eletrogravimétrica e espectrofotométrica ainda eram opções8,9,13,15,36 rotineiras. A gravimetria passou a ser muito pouco citada9,15,17,36,37, sendo o método com NH4SCN em meio redutor (precipitação do CuSCN) o mais aconselhado9.
A partir dos anos 1970, a determinação por espectrometria de absorção atômica e polarografia20,21 passou a ganhar espaço, especialmente nas análises de amostras com baixos teores do elemento. Dentre os métodos titrimétricos, o mais recomendado emprega o EDTA6,7,37. O método gravimétrico que ainda subsiste é a precipitação como CuSCN. Os métodos espectrofotométricos mencionam o emprego de dietilditiocarbamato, neocupraína ou biscicloexanona oxalidiidrazona6,7.
Este trabalho analisou a pureza de sais de cobre comerciais fabricados ao longo de mais de 100 anos, empregando métodos quantitativos gravimétricos, titrimétricos e instrumentais de análise. Através da determinação dos níveis de qualidade dos produtos selecionados, buscou-se avaliar as vantagens e limitações de cada um dos métodos empregados, bem como tentar correlacionar a pureza de um dado produto com a rota de síntese do mesmo e a evolução das técnicas de análise. Consideraram-se dois fatores durante a seleção dos produtos: a disponibilidade, na medida do possível, de um mesmo produto fabricado em diversas décadas e a facilidade de analisá-lo pelos métodos clássicos e instrumentais de análise quantitativa. Daí os sais escolhidos para este estudo serem compostos de cobre.
PARTE EXPERIMENTAL
A Tabela 1 apresenta os compostos de cobre utilizados para este trabalho, os quais fazem parte integrante do acervo do Museu da Química Prof. Athos da Silveira Ramos (1906-2002), instalado no IQ/UFRJ. Esses produtos, independentemente do ano de fabricação ou da origem, nunca foram abertos antes. As embalagens, assim, apresentavam seu lacre intacto, bem como a rolha de cortiça ou a tampa rosqueada. Eles passaram por um pré-tratamento de secagem em estufa a 105 ºC, por 2 h, a fim de se eliminar qualquer contaminante volátil presente e prepará-los para as diversas análises quantitativas. A dissolução, sempre que possível, foi feita em água destilada contendo ácido acético (0,1 mol L-1). Para as duas amostras mais antigas e as de CuO, a solubilização foi efetuada em ácido clorídrico (6 mol L-1), a 60 ºC. Durante a solubilização do sal Cu3(AsO4)2. 4H2O (1877), observou-se a presença de um sólido branco, insolúvel em diversos ácidos (nítrico, clorídrico, fluorídrico e água-régia). Então para a continuidade da análise, filtrou-se este precipitado, que representava, em média, 28,9% m/m da amostra em questão.
Métodos clássicos
Análise gravimétrica
Utilizou-se o método da precipitação do cobre por tiocianato em meio redutor (ácido sulfuroso)6. Os precipitados foram filtrados em cadinho filtrante nº 4, secos a 105 ºC em estufa (2 h) e pesados. Foram feitas as precipitações dos sais em triplicata, a partir de três amostragens diferentes.
Análise titrimétrica
Empregou-se o método com EDTA18, utilizando-se o indicador PAR [4-(2-piridil-azo)-resorcinol], em pH 5, obtido após adicionar acetato de amônio (2 g) e ácido clorídrico 12 mol L-1 (0,5 mL) para cada 100 mL de solução de cobre. Foram feitas duas soluções de cada sal, e retiradas duas alíquotas de cada solução, gerando quatro resultados.
Métodos instrumentais
Determinação de metais
O teor de cobre foi avaliado7,19 por espectrometria de absorção atômica, utilizando-se o comprimento de onda a 324,8 nm, que tem faixa linear até 5 mg L-1 e limite de detecção 0,5 mg L-1.
Os contaminantes especificados, quanto ao limite máximo de impurezas, nos rótulos dos reagentes fabricados a partir dos anos 1940 foram determinados por espectrometria de absorção atômica (Fe, Pb, Zn, Ni) e fotometria de chama (Na, Ca, K). Nestas análises foram preparadas soluções bastante concentradas, sendo explorado o método de adição de padrão19,21. Os parâmetros utilizados nas determinações dos elementos contaminantes estão apresentados na Tabela 2.
Análise de ânions por cromatografia de íons
Os produtos foram dissolvidos em ácido clorídrico (0,33 mol L-1). As amostras de CuO não foram analisadas, já que estas não apresentam ânions provenientes do composto original, somente os provenientes do ácido empregado para solubilização.
Fluorescência de raios-X
As duas amostras mais antigas foram analisadas, para verificação de contaminantes presentes. Empregou-se uma fonte de radiação Cu-Ka, varredura angular (2q) 12-70º, com passo de 0,020º e intervalo de 1 segundo por passo.
RESULTADOS E DISCUSSÃO
Perda por dessecação
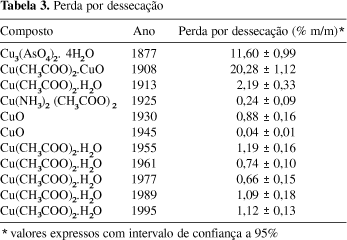
Os resultados obtidos após a secagem (Tabela 3) mostraram altos valores para os três produtos mais antigos. Deve-se levar em conta que eles não se apresentavam sob forma pulverulenta, mas como blocos maciços de formato irregular. Esse fato pode justificar as altas perdas por dessecação, já que pode ocorrer a presença de água ocluída no sólido.
Fluorescência de raios-X
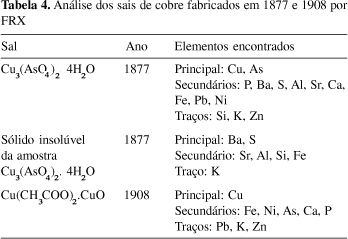
Os elementos encontrados nas duas amostras mais antigas estão listados na Tabela 4. O precipitado da amostra mais antiga, Cu3(AsO4)2.4H2O (1877), insolúvel nos ácidos clorídrico, nítrico e fluorídrico e água-régia, era basicamente sulfato de bário. Mesmo a amostra global apresentava uma grande quantidade de elementos, indicativo de um grau de pureza abaixo dos padrões hoje estabelecidos (é de se notar que esta amostra é anterior à publicação do primeiro livro sobre a pureza dos produtos químicos). Já na amostra de Cu(CH3COO)2.CuO (1908) o número de elementos detectados era sensivelmente inferior, o que sugere um aprimoramento do controle de qualidade do produto e de suas matérias-primas.
Determinação dos contaminantes metálicos
Houve uma significativa redução da quantidade de elementos contaminantes até os anos 1920, continuando de maneira mais lenta até os anos 1960, e mantendo-se em um mesmo patamar a partir dessa época, como mostra a Tabela 5. As amostras mais recentes (a partir dos anos 1940) estão dentro das especificações de limites de impurezas contidas nos seus respectivos rótulos.
O elevado teor de impurezas na amostra de Cu3(AsO4)2.4H2O se acha em conformidade com os resultados de FRX (Tabela 4). Do mesmo modo, os dados para a amostra de Cu(CH3COO)2.CuO também são compatíveis.
A grande diferença no teor de impurezas metálicas entre as duas primeiras amostras e as demais tem explicação no modo de obtenção do cobre, com a popularização da técnica de refinamento deste metal obtido de seus minérios por via eletrolítica: o metal impuro é imerso (como ânodo) num banho ácido contendo sulfato de cobre, tendo uma barra de cobre puro no cátodo38. Ao passar a corrente, metais com ferro, zinco, chumbo e níquel dissolvem-se e permanecem em solução (bem como os metais alcalinos e alcalino-terrosos), enquanto que o cobre se deposita no cátodo e metais como prata e ouro se depositam como lama anódica no fundo da célula de eletrólise39.
Um fato interessante foi que, de modo geral, os métodos gravimétricos e titrimétricos, e mesmo a maioria dos testes qualitativos em via úmida aplicáveis aos metais da Tabela 5, não foram bem sucedidos, devido aos baixos teores existentes. Isso mostra a grande vantagem da análise instrumental, que é seu menor limite de detecção em relação aos procedimentos convencionais. Esse fato veio a ser uma grande conquista na evolução do controle analítico das matérias-primas e dos produtos.
Determinação do cobre
A pureza do arseniato de cobre (1877) é inferior à dos demais sais, tendo em vista a presença do resíduo branco de BaSO4, aliás também em consonância com o elevado teor de contaminantes listados nas Tabelas 4 e 5. Contudo, o teor de cobre dessa amostra determinado por titrimetria apresentou um resultado bastante superior àqueles obtidos pela gravimetria e pela espectrometria de absorção atômica (Tabela 6). Esse aumento deve ser atribuído à interferência das impurezas no método de dosagem titrimétrico escolhido.
O sal de cobre fabricado em 1908 era efetivamente acetato básico de cobre, Cu(CH3COO)2.CuO, que apresenta maior teor de cobre por mol de amostra que o acetato normal, o que se acha em conformidade com os resultados obtidos, embora com valores de 5 a 10% abaixo do teórico, talvez por conta da presença de contaminantes (Tabelas 4 e 5).
Na amostra de CuO (1945), observou-se uma quantidade de cobre superior à teórica. As explicações possíveis são a presença de cobre metálico ou de óxido cuproso (Cu2O) na amostra. Os demais resultados concordam bastante bem com as previsões estequiométricas.
Apesar de ser um método trabalhoso, a gravimetria (Tabela 6) prestou-se bem à determinação de um componente principal da amostra, com a vantagem de ser um método absoluto, isto é, não dependente de uma calibração.
Os resultados obtidos pela titrimetria mostraram valores superiores aos da gravimetria e da espectrometria de absorção atômica. Excetuando-se a amostra do século XIX, os dados obtidos na análise de contaminantes (Tabela 5), mesmo considerando que todos fossem complexados pelo EDTA nas condições da análise, não justificam os maiores valores para o teor de cobre obtidos pelo método titrimétrico escolhido. Outros indicadores foram testados (cloroazurol S, murexida), concluindo-se que esse desvio é inerente ao indicador: o PAR possui um tempo de viragem um pouco maior que os demais, o que pode justificar o aumento uniforme dos resultados. A sua escolha recaiu na melhor visualização do ponto de final em relação aos demais indicadores.
O emprego da espectrometria de absorção atômica levou a resultados com uma dispersão maior em relação aos procedimentos clássicos, o que pode ser explicado pelo elevado fator de diluição necessário para enquadrar a solução na faixa de resposta do equipamento. Isto não é de se admirar pois esta técnica, bem como a fotometria de chama e outras técnicas similares são mais adequadas para a análise de constituintes menores presentes numa amostra4,8,19.
Determinação dos ânions
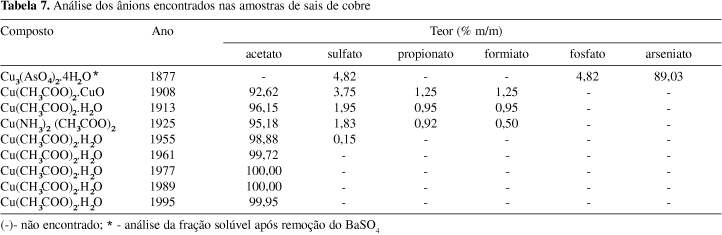
Esta análise gerou um resultado interessante (Tabela 7): observou-se a presença dos ânions propionato e formiato nas amostras de acetato comercializadas até os anos 1920. A explicação mais provável deve-se à forma de obtenção do ácido acético que, até esta época, era feita principalmente através da destilação seca da madeira (feita em retortas a cerca de 250 ºC, em ausência de ar); obtinha-se o ácido pirolenhoso, que consistia numa mistura líquida de água, ácido acético, metanol, acetona, e outros produtos (tais como ácido fórmico, ácido propiônico, etc)40.
Observou-se também que estes teores foram decrescendo com o tempo, provavelmente, devido a melhorias nas técnicas de refino do ácido pirolenhoso, obtendo-se uma solução de ácido acético mais puro. A extinção destes contaminantes deu-se pela mudança na técnica de obtenção do ácido acético, através da oxidação do etanol40, que permite obter ácido acético livre dos contaminantes propionato e formiato e mesmo outros, gerando um acetato de cobre isento dos mesmos. Trata-se de um caso em que a melhoria da qualidade de um produto foi conseguida através da alteração de rota de síntese de uma de suas matérias-primas.
CONCLUSÕES
Percebeu-se uma clara melhoria da qualidade dos sais de cobre ao longo do tempo, constatada tanto pelas melhorias na preparação de suas matérias-primas como pelo controle de impurezas dos produtos. O menor limite de detecção obtido pelos métodos instrumentais de análise permitiu verificar a existência de consideráveis quantidades de contaminantes nas amostras mais antigas, referentes a uma época em que o controle de qualidade era inexistente ou incipiente. Como era de se esperar, a análise de macro e microconstituintes das amostras pelos métodos utilizados neste trabalho correspondeu às vantagens e limitações de cada um deles. Para a continuidade desta pesquisa, sugere-se a aplicação da técnica de análise por ativação de nêutrons para dosar quantitativamente os elementos presentes nos sais de cobre estudados neste trabalho, posto que esta técnica é de grande sensibilidade, precisão e exatidão19.
A melhoria, diversificação e busca de novos métodos analíticos é uma resposta constante à necessidade de certificar um produto, qualificando-o para suas várias aplicações, uma exigência da química moderna em suas múltiplas interações com outras áreas do conhecimento humano. Em outras palavras, há uma clara conexão da evolução dos métodos de análise com a qualidade exigida dos produtos químicos. A interação contínua qualidade-análise permanece em curso até hoje; com o advento da nanotecnologia, e a especialização cada vez maior da química no campo de suas aplicações, a análise instrumental terá papel extraordinário no controle de qualidade ao longo do século XXI.
AGRADECIMENTOS
À Fundação Universitária José Bonifácio (FUJB) e ao CNPq pelo auxílio financeiro. Ao Instituto de Engenharia Nuclear (IEN/CNEN) pelas análises de fluorescência de raios-X.
REFERÊNCIAS
1. Ebner, F.; Lerch, L.; Da Antiga Farmácia do Anjo à Grande Empresa Químico-Farmacêutica, Merck: Rio de Janeiro, 1968.
2. Possehl, I.; Modern by Tradition - The History of the Chemical-Pharmaceutical Factory E. Merck Darmstadt, Merck: Darmstadt, 1994.
3. http://www.merck.de/history, acessada em Dezembro 2002.
4. Harris, D. C.; Quantitative Chemical Analysis, 5ª ed., W.H. Freeman and Company: Nova Iorque, 1998.
5. Hupe, K. G. M.; 75 Anos Merck no Brasil, Merck: Rio de Janeiro, 1998.
6. Jeffery, G. H.; Bassett, J.; Mendham, J.; Denney, R. C.; Vogel-Análise Química Quantitativa, Guanabara Koogan: São Paulo, 1988.
7. Mendham, J.; Denney, R. C.; Barnes, J. D.; Thomas, M.; Vogel-Análise Quimica Quantitativa, Livros Técnicos e Científicos: Rio de Janeiro, 2002.
8. Kolthoff, I. M.; Sandell, E. B.; Textbook of Quantitative Inorganic Analysis, 3ª ed., The MacMillan Company: Nova Iorque, 1952.
9. Pierce, W. C.; Haenisch, E. L.; Sawyer, D. T.; Quantitative Analysis, 4ª ed., John Wiley & Sons: Nova Iorque, 1958.
10. Kolthoff, I. M.; Furman, N. H.; Potenciometric Titrations, 2ª ed., John Wiley & Sons: Nova Iorque, 1931.
11. Sutton, F.; A Systematic Handbook of Volumetric Analysis, 20ª ed., P. Blakiston's Son & Co: Filadélfia, 1935.
12. Kolthoff, I. M.; Lingane, J. J.; Polarography, 2ª ed., Interscience Publishers: Nova Iorque, 1952.
13. Harley, J. H.; Wiberley, S. E.; Instrumental Analysis, John Wiley & Sons: Nova Iorque, 1954.
14. Bond, A. M.; J. Electroanal. Chem. 1981, 118, 381.
15. Belcher, R.; Wilson, C. L.; New Methods in Analytical Analysis, Reinhold Publishing Corporation: Nova Iorque, 1953.
16. Welcher, F. J.; The Analytical Uses of Ethylenediaminetetracetic Acid, D. van Nostrand Company: Nova Jersei, 1958.
17. Ayres, G. H.; Análisis Químico Cuantitativo, 2ª ed., Harper & Row Publishers Inc.: Cidade do México, 1968.
18. Métodos Complexometricos de Valoración con Titriplex, 3ª ed., Merck AG.: Darmstadt, 1972.
19. Skoog, D. A.; Holler, F. J.; Nieman, T. A.; Princípios de Análise Instrumental, 5ª ed., Bookman: São Paulo, 2002.
20. Brumblay, R. U.; A First Course in Quantitative Analysis, Addison-Wesley Publishing Company: Wausan, 1970.
21. Ewing, G. W.; Métodos Instrumentais de Análise Química, Edgard Blücher: São Paulo, 1972; Day, R. A. Jr.; Underwood, A. L.; Quantitative Analysis, 6ª ed., Prentice-Hall: Nova Iorque, 1991.
22. Skoog, D. A.; West, D. M.; Holler, F. J.; Fundamentals of Analytical Chemistry, 7ª ed., Sanders College Publishing: Boston, 1996.
23. Barral, E.; Précis D'Analyse Chimique Quantitative, Librairie J.B. Bailliére et Fils: Paris, 1905, vol. 1.
24. Blasdale,W. C.; Principles of Quantitative Analysis, 2ª ed., D. van Nostrand Company: Nova Iorque, 1918.
25. Teixeira, J. M.; Noções de Chimica Geral, 11ª ed., Livraria Francisco Alves: Rio de Janeiro, 1918.
26. Olsen, J. C.; A Textbook of Quantitative Chemical Analysis, 5ª ed., D. van Nostrand Company: Nova Iorque, 1920.
27. Lincoln, A. T.; Walton, J. H.; Exercises in Elementary Quantitative Chemical Analysis, The MacMillan Company: Nova Iorque, 1920.
28. Blasdale, W. C.; The Fundamentals of Quantitative Analysis, 3ª ed., D. van Nostrand Company: Nova Iorque, 1928.
29. Kolthoff, I. M.; Volumetric Analysis, John Wiley & Sons: Nova Iorque, 1929.
30. Mahin, E. G.; Quantitative Analysis, 4ª ed., McGraw-Hill Book Company: Nova Iorque, 1932.
31. Rieman, W.; Neuss, J. D.; Quantitative Analysis - A Theoretical Aproach, McGraw-Hill Book Company: Nova Iorque, 1937.
32. Willard, H. H.; Diehl, H.; Advanced Quantitative Analysis, D. van Nostrand Company: Nova Iorque, 1943.
33. Booth, H. S.; Damerell, V. R.; Quantitative Analysis, 2ª ed., McGraw-Hill Book Company: Nova Iorque, 1944.
34. Triebold, H. O.; Quantitative Analysis, D. van Nostrand Company: Nova Iorque, 1946.
35. Day, R. A. Jr.; Underwood, A. L.; Quantitative Analysis, Prentice-Hall: Nova Iorque, 1958.
36. Alexeév, V.; Analyse Quantitative, Mir: Moscou, 1966.
37. Ohlweiler, O. A.; Química Analítica Quantitativa, 3ª ed., Livros Técnicos e Científicos: Rio de Janeiro, 1981, vol. 2.
38. Ostwald, W.; Éléments de Chimie Inorganique, Gauthier-Villards: Paris, 1913, vol. 2.
39. Ohlweiler, O. A.; Química Inorgânica, Edgard Blücher: São Paulo, 1971, vol. 1.
40. Rothe, O.; Tecnologia Orgânica, Instituto Nacional do Livro: Rio de Janeiro, 1947.
Recebido em 12/8/03; aceito em 7/11/03; publicado na web em 27/05/04
- 1. Ebner, F.; Lerch, L.; Da Antiga Farmácia do Anjo à Grande Empresa Químico-Farmacêutica, Merck: Rio de Janeiro, 1968.
- 2. Possehl, I.; Modern by Tradition - The History of the Chemical-Pharmaceutical Factory E. Merck Darmstadt, Merck: Darmstadt, 1994.
- 4. Harris, D. C.; Quantitative Chemical Analysis, 5Ş ed., W.H. Freeman and Company: Nova Iorque, 1998.
- 5. Hupe, K. G. M.; 75 Anos Merck no Brasil, Merck: Rio de Janeiro, 1998.
- 6. Jeffery, G. H.; Bassett, J.; Mendham, J.; Denney, R. C.; Vogel-Análise Química Quantitativa, Guanabara Koogan: São Paulo, 1988.
- 7. Mendham, J.; Denney, R. C.; Barnes, J. D.; Thomas, M.; Vogel-Análise Quimica Quantitativa, Livros Técnicos e Científicos: Rio de Janeiro, 2002.
- 8. Kolthoff, I. M.; Sandell, E. B.; Textbook of Quantitative Inorganic Analysis, 3Ş ed., The MacMillan Company: Nova Iorque, 1952.
- 9. Pierce, W. C.; Haenisch, E. L.; Sawyer, D. T.; Quantitative Analysis, 4Ş ed., John Wiley & Sons: Nova Iorque, 1958.
- 10. Kolthoff, I. M.; Furman, N. H.; Potenciometric Titrations, 2Ş ed., John Wiley & Sons: Nova Iorque, 1931.
- 11. Sutton, F.; A Systematic Handbook of Volumetric Analysis, 20Ş ed., P. Blakiston's Son & Co: Filadélfia, 1935.
- 12. Kolthoff, I. M.; Lingane, J. J.; Polarography, 2Ş ed., Interscience Publishers: Nova Iorque, 1952.
- 13. Harley, J. H.; Wiberley, S. E.; Instrumental Analysis, John Wiley & Sons: Nova Iorque, 1954.
- 14. Bond, A. M.; J. Electroanal. Chem 1981, 118, 381.
- 15. Belcher, R.; Wilson, C. L.; New Methods in Analytical Analysis, Reinhold Publishing Corporation: Nova Iorque, 1953.
- 16. Welcher, F. J.; The Analytical Uses of Ethylenediaminetetracetic Acid, D. van Nostrand Company: Nova Jersei, 1958.
- 17. Ayres, G. H.; Análisis Químico Cuantitativo, 2Ş ed., Harper & Row Publishers Inc.: Cidade do México, 1968.
- 18. Métodos Complexometricos de Valoración con Titriplex, 3Ş ed., Merck AG.: Darmstadt, 1972.
- 19. Skoog, D. A.; Holler, F. J.; Nieman, T. A.; Princípios de Análise Instrumental, 5Ş ed., Bookman: São Paulo, 2002.
- 20. Brumblay, R. U.; A First Course in Quantitative Analysis, Addison-Wesley Publishing Company: Wausan, 1970.
- 21. Ewing, G. W.; Métodos Instrumentais de Análise Química, Edgard Blücher: São Paulo, 1972;
- Day, R. A. Jr.; Underwood, A. L.; Quantitative Analysis, 6Ş ed., Prentice-Hall: Nova Iorque, 1991.
- 22. Skoog, D. A.; West, D. M.; Holler, F. J.; Fundamentals of Analytical Chemistry, 7Ş ed., Sanders College Publishing: Boston, 1996.
- 23. Barral, E.; Précis D'Analyse Chimique Quantitative, Librairie J.B. Bailliére et Fils: Paris, 1905, vol. 1.
- 24. Blasdale,W. C.; Principles of Quantitative Analysis, 2Ş ed., D. van Nostrand Company: Nova Iorque, 1918.
- 25. Teixeira, J. M.; Noções de Chimica Geral, 11Ş ed., Livraria Francisco Alves: Rio de Janeiro, 1918.
- 26. Olsen, J. C.; A Textbook of Quantitative Chemical Analysis, 5Ş ed., D. van Nostrand Company: Nova Iorque, 1920.
- 27. Lincoln, A. T.; Walton, J. H.; Exercises in Elementary Quantitative Chemical Analysis, The MacMillan Company: Nova Iorque, 1920.
- 28. Blasdale, W. C.; The Fundamentals of Quantitative Analysis, 3Ş ed., D. van Nostrand Company: Nova Iorque, 1928.
- 29. Kolthoff, I. M.; Volumetric Analysis, John Wiley & Sons: Nova Iorque, 1929.
- 30. Mahin, E. G.; Quantitative Analysis, 4Ş ed., McGraw-Hill Book Company: Nova Iorque, 1932.
- 31. Rieman, W.; Neuss, J. D.; Quantitative Analysis - A Theoretical Aproach, McGraw-Hill Book Company: Nova Iorque, 1937.
- 32. Willard, H. H.; Diehl, H.; Advanced Quantitative Analysis, D. van Nostrand Company: Nova Iorque, 1943.
- 33. Booth, H. S.; Damerell, V. R.; Quantitative Analysis, 2Ş ed., McGraw-Hill Book Company: Nova Iorque, 1944.
- 34. Triebold, H. O.; Quantitative Analysis, D. van Nostrand Company: Nova Iorque, 1946.
- 35. Day, R. A. Jr.; Underwood, A. L.; Quantitative Analysis, Prentice-Hall: Nova Iorque, 1958.
- 36. Alexeév, V.; Analyse Quantitative, Mir: Moscou, 1966.
- 37. Ohlweiler, O. A.; Química Analítica Quantitativa, 3Ş ed., Livros Técnicos e Científicos: Rio de Janeiro, 1981, vol. 2.
- 38. Ostwald, W.; Éléments de Chimie Inorganique, Gauthier-Villards: Paris, 1913, vol. 2.
- 39. Ohlweiler, O. A.; Química Inorgânica, Edgard Blücher: São Paulo, 1971, vol. 1.
- 40. Rothe, O.; Tecnologia Orgânica, Instituto Nacional do Livro: Rio de Janeiro, 1947.
Datas de Publicação
-
Publicação nesta coleção
02 Ago 2004 -
Data do Fascículo
Ago 2004
Histórico
-
Aceito
07 Nov 2003 -
Recebido
12 Ago 2003