Resumo
Thermal decomposition of [Bu4N]2[Zn(imnt)2] and [M(NH3)2(imnt)] complexes with M = Zn and Cd, and imnt = (bis 1,1-dicyanoethylene-2,2 dithiolate) in inert atmosphere was investigated by thermogravimetric analysis (TG) and differential scanning calorimetry (DSC). Pyrolysis studies at different temperatures, 300, 400, 500, and 600 ºC, in N2 atmosphere were performed and the products were characterized by X-ray diffraction (XRD), infrared and Raman spectroscopy, and scanning electron microscopy (SEM). The products were identified as sulfide sub-micron particles, along with amorphous carbon. Particle sizes estimated by SEM were ca. 50 nm for the cationic complexes and 500 nm for the neutral complexes.
single-source precursor; thermal decomposition; II-VI semiconductor
single-source precursor; thermal decomposition; II-VI semiconductor
ARTIGO
Decomposição térmica de complexos de Zn e Cd com isomaleonitriladitiolato (imnt)
Thermal decomposition of Zn and Cd complexes with isomaleonitriledithiolate (imnt)
Cristiane M. L. Barbosa; Maria Terezinha Caruso Sansiviero* * e-mail: mtcaruso@dedalus.lcc.ufmg.br
Departamento de Química, Instituto de Ciências Exatas, Universidade Federal de Minas Gerais, Av. Antônio Carlos, 6627, 30270-960 Belo Horizonte - MG
ABSTRACT
Thermal decomposition of [Bu4N]2[Zn(imnt)2] and [M(NH3)2(imnt)] complexes with M = Zn and Cd, and imnt = (bis 1,1-dicyanoethylene-2,2 dithiolate) in inert atmosphere was investigated by thermogravimetric analysis (TG) and differential scanning calorimetry (DSC). Pyrolysis studies at different temperatures, 300, 400, 500, and 600 ºC, in N2 atmosphere were performed and the products were characterized by X-ray diffraction (XRD), infrared and Raman spectroscopy, and scanning electron microscopy (SEM). The products were identified as sulfide sub-micron particles, along with amorphous carbon. Particle sizes estimated by SEM were ca. 50 nm for the cationic complexes and 500 nm for the neutral complexes.
Keywords: single-source precursor; thermal decomposition; II-VI semiconductor.
INTRODUÇÃO
Semicondutores do grupo 12-16, especialmente os sulfetos de Zn e Cd apresentam importantes propriedades elétricas e ópticas, tais como semicondutividade e fotocondutividade1. Nanopartículas e clusters de semicondutores mostram uma forte dependência das propriedades eletrônicas e ópticas com o tamanho das partículas2-5. Propriedades ópticas não lineares encontram aplicações importantes em dispositivos como chaveadores ópticos e fibras ópticas de material altamente não linear6-9.
A obtenção de semicondutores a partir de precursores organometálicos para deposição química de vapor (OMCVD) vem sendo investigada desde o início da década de 80, tendo um grande desenvolvimento ocorrido em meados da década de 90. Podem ser citados alguns artigos de revisão, como ex. o de Alves et al.10 que trata da busca de precursores organometálicos para obtenção de filmes de semicondutores binários dos grupos III-V (GaAs), II-VI (CdS) por OMCVD. Mais recentemente, Gleizes11 enfoca alguns precursores organometálicos que já estão sendo utilizados com sucesso na obtenção desses filmes. Complexos de metais do grupo 12-16 contendo enxofre, tais como tiolatos12, ditioxamida13, ditiocarbamatos14 e complexos de tiouréia15 vêm sendo utilizados com sucesso na produção dos respectivos sulfetos. Precursores tipo fonte única vêm sendo aplicados na obtenção de filmes finos de sulfetos de zinco e de cádmio16.
Em contraste, a pirólise de complexos de ditiolatos quelatos foi pouco investigada. Esta investigação mostra-se importante, uma vez que os mecanismos de decomposição de precursores variam. Por ex., na obtenção de filmes por deposição química de vapor Parkin et al.17 objetivando obter SnS partiram de dois precursores diferentes, sendo eles Sn(SR4) (onde R = radical orgânico), um tiolato monodentado, e Sn(S-CH2-CH2-S)2, um ditiolato quelato. No primeiro caso houve necessidade de se efetuar a deposição na presença de um fluxo de H2S para garantir a formação do sulfeto e evitar que o estanho metálico, que se forma inicialmente na superfície, conduza à formação de óxido de estanho na presença de oxigênio. Já no caso do precursor quelato as etapas do mecanismo de decomposição são diferentes, de forma que não ocorre a formação inicial do estanho metálico, obtendo-se sulfeto de estanho sem utilização do fluxo de H2S.
Neste trabalho nosso objetivo é investigar a decomposição térmica de complexos de Zn e Cd com o ligante imnt, onde imnt = [C2S2(CN)2]2- (1,1-dicianoetileno-2,2-ditiolato), Esquema 1, em atmosfera inerte. Anteriormente investigamos a pirólise ao ar de [Bu4N]2[Zn(imnt)2] , que levou à obtenção de partículas submicrométricas de ZnO18. Complexos do tipo [Bu4N]2[Zn(imnt)2] têm a vantagem de serem estáveis ao ar, são fáceis de serem sintetizados pois são obtidos pela adição do ligante a um sal do metal geralmente em meio aquoso ou água/etanol e, além disso, apresentam boa solubilidade em solventes orgânicos como clorofórmio e acetona. Investigamos inicialmente a decomposição térmica de [Bu4N]2[Zn(imnt)2] que levou à obtenção de uma mistura de nanopartículas de ZnS e carbono amorfo. Incluímos no trabalho os compostos [Zn(NH3)2(imnt)] and [Cd(NH3)2(imnt)], a fim de verificar se diminuía a quantidade de carbono no produto da decomposição, uma vez que estes possuem menor quantidade de carbono na sua estrutura.
PARTE EXPERIMENTAL
Sínteses
Os reagentes para as sínteses foram obtidos da Aldrich Co.
O ligante na forma de sal de sódio Na2imnt foi preparado de acordo com o descrito na literatura, segundo o método de Söderbäck19.
À uma solução de 0,05 mol (1,15 g) de Na° em 20 ml de etanol adicionou-se uma solução de 0,05 mol (3,3 g) de malononitrila em 15 ml de etanol à temperatura ambiente (20 °C), resfriou-se a 5 °C e adicionaram-se 3,1 mL de CS2, gota a gota, com agitação. Nesta etapa há desenvolvimento de calor e forma-se uma solução amarela-esverdeada. Em seguida adicionou-se mais 0,05 mol (1,15 g) de Na° dissolvidos em 20 mL de etanol. Adicionaram-se então 5,8 mL de H2O, que causou uma rápida cristalização. Após repouso, colocou-se na geladeira por 2 dias. Filtra-se e lava-se 2 vezes com porções de 5 mL de etanol e, a seguir, 3 vezes com éter. Deixou-se secar ao ar por algumas horas.
Para purificação do sal obtido dissolveu-se 0,01 mol (2 g) de Na2imnt em 100 mL de etanol. Adicionaram-se 4,5 mL de H2O, resfriou-se. Formaram-se cristais amarelos claro, os quais devem ser lavados conforme descrito anteriormente.
Análise elementar calculada para Na2imnt.3H2O, M.M. = 240 g. C = 20,00%, H = 2,50%, N = 11,60%. Encontrada: C = 19,86%, H = 2,50%, N = 11,25%. Principais frequências no IV: 3400-3200 cm-1n(OH) ; 2200 cm-1n(CºN), 1625 cm-1n(C=C), e 875 cm-1n(C-S).
Síntese do complexo [Bu4N]2[Zn(imnt)2]
[Bu4N]2[Zn(imnt)2 ] policristalino foi preparado segundo o método descrito por Fackler e Coucovanis20. À uma solução de 0,005 mol (1,1 g) de ZnCl2 em 100 mL de H2O adicionou-se uma solução de 0,02 mol (3,22g) de Bu4NBr dissolvido em 3,4 mL de H2O. Aqueceu-se a solução até próximo do ponto de ebulição e adicionou-se sob agitação uma solução de 0,012 mol (2,23 g) de Na2imnt em 37,5 mL de H2O. Observou-se a formação de um precipitado amarelo claro. Para purificação dissolveu-se o [Bu4N]2[Zn(imnt)2] em acetona quente e, em seguida, adicionou-se água até que a solução se turvasse. Deixou-se em repouso. Houve formação de cristais amarelos claro. Análise elementar calculada para [Bu4N]2 [Zn(C2S2(CN)2)2], M.M. = 829 g. Calculado: C = 57.90%, H = 8.68%, N = 10.13%, S = 15.44%. Encontrada: C = 57.96%, H = 8.60%, N = 10.36%, S = 14.83%. Ponto de Fusão: 148,5-150,5 °C. Principais frequências no IV: 2850 cm-1n(C-H), 2200 cm-1n(CºN), 1400 cm-1n(C=C), e 875 cm-1n(C-S).
Síntese dos complexos [Zn(NH3)2imnt] e [Cd(NH3)2(imnt)2]
Foram sintetizados de acordo com o método descrito por Singh21,22. A uma solução de 0,005 mol (1,2 g) de Na2imnt em 50 mL de H2O adicionou-se, sob agitação, uma solução de 0,005 mol (0,7 g) de ZnCl2 (ou de Cd(NO3)2.4H2O) dissolvido em 50 mL de H2O, contendo um excesso de NH4OH. Observou-se a formação de um precipitado amarelo claro, tanto para o complexo de Zn como para o de Cd. Os complexos obtidos foram lavados repetidas vezes com água, álcool e éter e secos à vácuo sobre o CaCl2. [Zn(NH3)2(imnt)]: Dec = 160 ºC. Principais frequências no IV: 3100-3400 cm-1n(N-H); 2200 cm-1n(CºN); 1350 cm-1n(C=C) and 875 cm-1n(C-S). [Cd(NH3)2(imnt)]: Dec = 130 ºC. Principais frequências no IV: 3100-3400 cm-1n(N-H); 2200 cm-1n(CºN); 1375 cm-1n(C=C); 875 cm-1n(C-S)
Medidas físicas
As análises termogravimétricas (TG) foram efetuadas num equipamento Schimadzu TGA-50H em atmosfera de N2 (50,0 mL min-1) e taxa de aquecimento de 10 ºC min-1 entre 25 e 750 ºC. A calorimetria diferencial de varredura (DSC) foi efetuada num equipamento Schimadzu DSC-50H em atmosfera de N2 (50,0 mL min-1) e taxa de aquecimento de 10 ºC min-1 entre 20 e 600 ºC.
Os difratogramas de Raios-X (DRX) foram registrados num equipamento Rigaku Geigerflex Diffractomer, na região de 2q entre 5 e 75º, usando-se radiação CuKa, l = 1.5418 Å .
Os espectros no infravermelho foram registrados num equipamento FTIR Bomen MB-102 entre 4000 e 200 cm-1. As amostras foram preparadas na forma de pastilhas de CsI.
Os espectros Raman foram obtidos num espectrômetro Micro-Raman Dilor XY usando a linha 514,5 nm de um laser de íons argônio (Coherent model Innova 90).
Micrografias de microscopia eletrônica de varredura (MEV) foram obtidas num equipamento JEOLSM-8404. As amostras foram preparadas na forma de pó disperso e recobertas com ouro.
Tratamento térmico das amostras
A decomposição térmica das amostras foi efetuada num forno tubular programável sob fluxo de N2 (30 mL min-1). A taxa de aquecimento foi de 10 °C min-1 até atingir a temperatura desejada e mantida nesta temperatura por 1 h.
RESULTADOS E DISCUSSÃO
Resultados para [Bu4N]2[Zn(imnt)2]
Comportamento térmico
A Figura 1 mostra as curvas TG e DSC em atmosfera inerte. Uma perda de massa de 71,5% ocorre entre 201 e 491 ºC. Uma perda total de massa de 80% ocorre em 743 ºC. O valor teórico esperado para obtenção de ZnS puro seria de 88,3%. Esta diferença, como será apresentado nas caracterizações, é devida a carbono amorfo que não é eliminado com o aquecimento em atmosfera inerte.
A curva DSC mostra dois picos. Um primeiro pico endotérmico ocorrendo em 150 ºC, correspondente ao ponto de fusão do precursor. O segundo pico endotérmico, em 275 ºC, é atribuído à decomposição do complexo. Ao ar este processo de decomposição é exotérmico e ocorre em ca de 300 ºC, ou seja a combustão da parte orgânica da molécula19.
A decomposição térmica do precursor em atmosfera inerte foi efetuada nas temperaturas de 300, 400, 500 e 600 ºC. As amostras assim obtidas foram caracterizadas por DRX, espectroscopia no IV e Raman e MEV.
Difração de raios X (DRX)
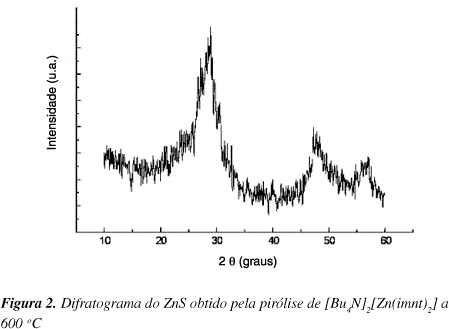
A Figura 2 mostra o DRX para a amostra a 400 ºC. Os DRXs obtidos para as amostras a 300, 500 e 600 ºC são análogos ao mostrado na Figura 2. Foram observadas reflexões com d = 3,12 Å (2q = 28,5º); 1,91 Å (2q = 45,3); 1,63 Å (2q = 57,3) características de ZnS cúbico (esfalerita) relativos aos planos [111], [220] e [311], respectivamentere, segundo a ficha 5-566 Card 1, Philips Eletronic Instruments. Estas reflexões apresentam picos muito largos, o que reflete o pequeno tamanho dos cristalitos. O tamanho médio dos cristalitos foi estimado por análise da largura de linha correspondente ao plano [111], em 2q = 28,5º usando-se a equação de Scherrer23,  sem nenhuma correção padrão, onde g é o tamanho médio do microcristalito, b é a largura da linha a meia altura, qb é o ângulo de Bragg (radianos) e l o comprimento de onda da radiação incidente (1.5418 Å). Sabe-se que o valor de g é proporcional à distância dhkl e que K é uma constante que depende da forma dos cristalitos com valores próximos a um. Neste trabalho, assumimos K= 0,9 e o valor calculado de g foi de 4,0 nm.
sem nenhuma correção padrão, onde g é o tamanho médio do microcristalito, b é a largura da linha a meia altura, qb é o ângulo de Bragg (radianos) e l o comprimento de onda da radiação incidente (1.5418 Å). Sabe-se que o valor de g é proporcional à distância dhkl e que K é uma constante que depende da forma dos cristalitos com valores próximos a um. Neste trabalho, assumimos K= 0,9 e o valor calculado de g foi de 4,0 nm.
Espectroscopia vibracional
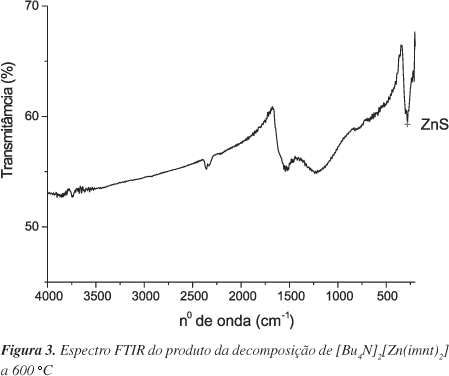
As amostras foram caracterizadas por espectroscopia Raman e no infravermelho. A Figura 3 mostra o espectro no infravermelho para a amostra aquecida a 600 ºC. A presença de ZnS foi confirmada através de uma banda em 280 cm-1, atribuída ao modo vibracional A1(TO), E1(TO)7,24, onde TO é fónon óptico transversal, juntamente com a presença de carbono amorfo25 caracterizado por bandas largas em 1550 e 1350 cm-1.
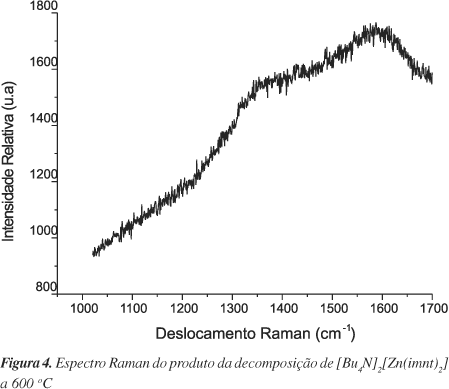
Este comportamento foi também confirmado por espectroscopia Raman. O espectro Raman, Figura 4, apresenta duas bandas muito largas, uma com máximo em ca 1350 e outra em 1590 cm-1 atribuídas, respectivamente, à banda D (1200-1400 cm-1) e à banda G (1500-1600 cm-1) de carbono amorfo.
Microscopia eletrônica de varredura (MEV)
A Figura 5 mostra a micrografia obtida para a amostra decomposta à 600 ºC. Observa-se a formação de aglomerados contendo partículas da ordem de aproximadamente 50 nm.
Resultados para Zn(NH3)2(imnt) e Cd(NH3)2(imnt)
Comportamento térmico
As curvas TG em atmosfera inerte para os complexos de Cd e Zn são mostradas na Figura 6. A primeira perda de massa foi de 14% e corresponde à perda esperada para as duas moléculas de amônia (teórico 14,3%) e ocorreu na faixa de temperatura entre 40 e 245 ºC para o complexo de Zn, acompanhado por um evento endotérmico em 151 ºC na DSC, e entre 71 e 286 ºC para o complexo de Cd, com um pico endotérmico observado na DSC em 130 ºC. Após a perda das moléculas de amônia, os complexos perderam massa lentamente até atingir uma perda total de 50% a 800 ºC. A perda total esperada para que o produto final seja ZnS puro deveria ser de 60%.
Difração de raios X
As Figuras 7 e 8 mostram os difratogramas obtidos para as amostras tratadas a 300 e 600 ºC para o complexo de Zn, e a 300 e 500 ºC para o de Cd, respectivamente. A Figura 7 exibe três picos mais proeminentes com d = 3,12 Å (2q = 28,5º); 1,91 Å (2q = 45,3) e 1,63 Å (2q =57,3) característicos de ZnS cúbico (mineral esfalerita) relativos aos planos [111], [220] e [311]. A Figura 8 mostra três picos com d = 3,36 Å (2q = 26,8º); 2,06 Å (2q = 44,5º) e 1,75 Å (2q = 52,0º) característicos de CdS cúbico relativos aos planos [111], [220] e [311].
O tamanho médio dos microcristalitos de ZnS foi estimado por análises da largura de linha a meia altura usando a equação de Scherrer para a linha referente ao plano [111]. O tamanho dos cristalitos obtido foi de 2,3 nm para as amostras em 300, 400 e 600 ºC. O tamanho médio dos microcristalitos estimado para CdS, usando-se a reflexão referente ao plano [111], foi de 2,0 nm.
Espectroscopia vibracional
Para todas as amostras obtidas pela pirólise de [Zn(NH3)2(imnt)] e [Cd(NH3)2(imnt)], o espectro no infravermelho indicou a presença dos respectivos sulfetos com alguma quantidade de carbono amorfo.
Os produtos de decomposição do [Zn(NH3)2(imnt)] apresentaram no espectro no infravermelho uma banda em 280 cm-1 característica de ZnS e bandas largas em ca 1350 e 1550 cm-1 atribuídas a presença de carbono amorfo. Os espectros foram análogos ao obtido para o produto da decomposição do [Bu4N]2[Zn(imnt)2], mostrado na Figura 3.
O espectro no IV do produto da decomposição do [Cd(NH3)2(imnt)], Figura 9, apresentou uma banda em 248 cm-1, atribuída ao fónon TO do CdS24 e bandas referentes à presença de carbono amorfo.
Os espectros Raman obtidos foram análogos ao apresentado na Figura 4, onde foi confirmada a presença de carbono amorfo.
Microscopia eletrônica de varredura (MEV)
A Figura 10 mostra as micrografias obtidas para os produtos da decomposição térmica de [Cd(NH3)2imnt] a 300 ºC e [Zn(NH3)2imnt] a 600 ºC por 1 h. O tamanho das partículas é de aproximadamente 500 nm para o CdS e ZnS, respectivamente. As partículas de CdS exibem uma morfologia em forma de bastão, enquanto as partículas de ZnS exibem uma forma quase esférica.
Caracterização quantitativa do carbono amorfo no produto de decomposição do [Bu4N]2[Zn(imnt)2]
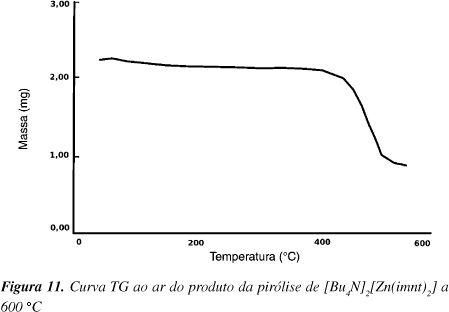
Através de uma análise elementar CHN determinou-se a presença de 46,19% de carbono no produto de decomposição do [Bu4N]2[Zn(imnt)2] a 600 ºC. A análise termogravimétrica ao ar do produto de decomposição, Figura 11, mostra uma perda total de massa de 64% no intervalo de temperatura entre 450 e 750 ºC. O resíduo assim obtido foi caracterizado por DRX como sendo ZnO. Como 16,5% de perda de massa deve-se ao processo de ZnS passando a ZnO, o restante é devido à oxidação do carbono (44,5%), obtendo-se um resultado muito próximo ao de análise elementar para quantidade de carbono.
CONCLUSÃO
Os precursores investigados [Bu4N]2[Zn(imnt)2] e [M(NH3)2(imnt)] onde M = Zn e Cd formaram como produto final da decomposição os respectivos sulfetos, identificados por análise de DRX e do espectro no infravermelho, junto com carbono amorfo como foi observado por dados de espectroscopia Raman e no infravermelho. Carbono amorfo está presente também nos produtos da pirólise dos complexos neutros, onde não se tem o cátion contendo os quatro grupos tetrabutilamônio, o que provavelmente se deve à presença da ligação dupla (C=C) na molécula do ligante. Neste trabalho optamos por um tempo de aquecimento de 1 h. No momento, estamos testando efetuar a decomposição numa temperatura de 600 ºC e mantendo-se nesta temperatura por um tempo maior, na tentativa de verificar se ocorre a eliminação ou diminuição do carbono amorfo. Será testada também a pirólise em temperaturas mais elevadas.
AGRADECIMENTOS
Ao CNPq pelas bolsas concedidas. Ao Prof. Dr. O. L. Alves pela sugestão do tema e proveitosas discussões. Ao Prof. Dr. Rochel M. Lago pelas discussões. À Profa. Mª I. Yoshida pelas análises térmicas. Ao Prof. Dr. M. Pimenta, do Departamento de Física da UFMG, por nos facilitar o uso do espectrômetro Micro-Raman.
REFERÊNCIAS
1. Ray, B.; II-VI Compounds, Pergamon Press: Oxford, 1969, p. 229.
2. Marcus, M. A.; Flood, W.; Stiegerwald, M.; Brus, L.; Bawendi, M.; J. Phys. Chem. 1991, 95, 1572.
3. Fragnito, H. L.; Rios, J. M. M.; Duarte, A. S.; Palange, E.; Medeiros Neto, J. A.; Cesar, C. L.; Barbosa, L. C.; Alves, O. L.; Brito Cruz, C. H.; J. Phys.: Condens. Matter 1993, 5 A, 179.
4. Brus, L. E.; J. Chem. Phys. 1984, 80, 4403.
5. Kayanuma, Y.; Phys. Rev. B. Condens. Matter Mater. Phys. 1988, B38, 9797.
6. Khairutdinov, R. F.; Russ. Chem. Rev. 1998, 67, 109.
7. Scholz, S. M.; Vacassy, R.; Lemaire, L.; Dutta, J.; Hofmann, H.; Appl. Organomet. Chem. 1998, 12, 327.
8. Medeiros Neto, J. A.; Barbosa, L. C.; Cesar, C. L.; Alves, O. L.; Galembeck, F.; Appl. Phys. Lett. 1991, 59, 2715.
9. Yang, Y.; Huang, J.; Liu, S.; Shen, J.; J. Mater. Chem. 1997, 7, 131.
10. Zarbin, A. J. G.; Alves, O. L.; Vargas, M. D.; Quim. Nova 1995, 18, 274.
11. Gleizes, A. N.; Chem. Vap. Deposition 2000, 6, 155
12. Osakada, K.; Yamamoto, T.; J. Chem. Soc. Chem. Commun. 1987, 14, 1117; Inorg. Chem. 1999, 30, 2328; Shaw, R. A.; Woods, W. K.; J. Chem. Soc. A 1971, 1569; Rees Jr., W. S.; Krauter, G.; J. Mater. Res. 1996, 11, 3005.
13. Abboudi, M.; Mosset, A.; J. Solid State Chem. 1994, 109, 70.
14. Cui, H.; Pike, R. D.; Kershaw, R.; Dwight, K.; Wold, A.; J. Solid State Chem. 1992, 101, 115.
15. Wold, A.; Dwight, K.; J. Solid State Chem. 1992, 96, 53; Karanjai, M. K.; Dasgupta, D.; Mater. Lett. 1986, 4, 368.
16. Lee, E. Y. M.; Tran, N. H.; Russell, J. J.; Lamb, R. N.; J. Phys. Chem. B 2004, 108, 8355; Barreca, D.; Tondello, E.; Lydon, D.; Spalding, T. R.; Fabrizio, M.; Chem. Vap. Deposition 2003, 9, 93.
17. Parkin, I. P.; Price, L. S.; Hibbert, T. G.; Mollov, K. C.; J. Mater. Chem. 2001, 11, 1486.
18. Sansiviero, M. T. C.; Alves, O. L.; Mater. Lett. 2000, 45, 217.
19. Söderbäch, E.; Acta Chem. Scand. 1963, 17, 362.
20. Fackler Jr., J. P.; Coucovanis, D.; J. Am. Chem. Soc. 1966, 88, 3913.
21. Singh, M. K. A. M.; J. Indian Chem. Soc. 1999, 76, 148.
22. Singh, M.K. A. M.; Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem. 1999, 38, 1294.
23. Cullity, B. D.; Elements of X- ray Diffraction, 2nd ed.; Addison-Wesley: USA, 1978, series in Metallurgy and Materials.
24. Milekhin, A. G.; Sveshnikova, L. L.; Repinskii, S. M.; Gutakovsii, A. K.; Friedrich, M.; Zahn, D. R. T.; Phys. Solid State 2002, 44, 1976.
25. Ferrari, A. C.; Rodil, S. E.; Robertson, J.; Phys. Rev. B 2003, 64, 1.
Recebido em 13/4/04; aceito em 6/1/05; publicado na web em 13/4/05
- 1. Ray, B.; II-VI Compounds, Pergamon Press: Oxford, 1969, p. 229.
- 2. Marcus, M. A.; Flood, W.; Stiegerwald, M.; Brus, L.; Bawendi, M.; J. Phys. Chem. 1991, 95, 1572.
- 3. Fragnito, H. L.; Rios, J. M. M.; Duarte, A. S.; Palange, E.; Medeiros Neto, J. A.; Cesar, C. L.; Barbosa, L. C.; Alves, O. L.; Brito Cruz, C. H.; J. Phys.: Condens. Matter 1993, 5 A, 179.
- 4. Brus, L. E.; J. Chem. Phys. 1984, 80, 4403.
- 5. Kayanuma, Y.; Phys. Rev. B. Condens. Matter Mater. Phys. 1988, B38, 9797.
- 6. Khairutdinov, R. F.; Russ. Chem. Rev. 1998, 67, 109.
- 7. Scholz, S. M.; Vacassy, R.; Lemaire, L.; Dutta, J.; Hofmann, H.; Appl. Organomet. Chem. 1998, 12, 327.
- 8. Medeiros Neto, J. A.; Barbosa, L. C.; Cesar, C. L.; Alves, O. L.; Galembeck, F.; Appl. Phys. Lett. 1991, 59, 2715.
- 9. Yang, Y.; Huang, J.; Liu, S.; Shen, J.; J. Mater. Chem. 1997, 7, 131.
- 10. Zarbin, A. J. G.; Alves, O. L.; Vargas, M. D.; Quim. Nova 1995, 18, 274.
- 11. Gleizes, A. N.; Chem. Vap. Deposition 2000, 6, 155
- 12. Osakada, K.; Yamamoto, T.; J. Chem. Soc. Chem. Commun. 1987, 14, 1117;
- Inorg. Chem. 1999, 30, 2328;
- Shaw, R. A.; Woods, W. K.; J. Chem. Soc. A 1971, 1569;
- Rees Jr., W. S.; Krauter, G.; J. Mater. Res. 1996, 11, 3005.
- 13. Abboudi, M.; Mosset, A.; J. Solid State Chem. 1994, 109, 70.
- 14. Cui, H.; Pike, R. D.; Kershaw, R.; Dwight, K.; Wold, A.; J. Solid State Chem. 1992, 101, 115.
- 15. Wold, A.; Dwight, K.; J. Solid State Chem. 1992, 96, 53;
- Karanjai, M. K.; Dasgupta, D.; Mater. Lett. 1986, 4, 368.
- 16. Lee, E. Y. M.; Tran, N. H.; Russell, J. J.; Lamb, R. N.; J. Phys. Chem. B 2004, 108, 8355;
- Barreca, D.; Tondello, E.; Lydon, D.; Spalding, T. R.; Fabrizio, M.; Chem. Vap. Deposition 2003, 9, 93.
- 17. Parkin, I. P.; Price, L. S.; Hibbert, T. G.; Mollov, K. C.; J. Mater. Chem. 2001, 11, 1486.
- 18. Sansiviero, M. T. C.; Alves, O. L.; Mater. Lett. 2000, 45, 217.
- 19. Söderbäch, E.; Acta Chem. Scand. 1963, 17, 362.
- 20. Fackler Jr., J. P.; Coucovanis, D.; J. Am. Chem. Soc. 1966, 88, 3913.
- 21. Singh, M. K. A. M.; J. Indian Chem. Soc. 1999, 76, 148.
- 22. Singh, M.K. A. M.; Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem. 1999, 38, 1294.
- 23. Cullity, B. D.; Elements of X- ray Diffraction, 2nd ed.; Addison-Wesley: USA, 1978, series in Metallurgy and Materials.
- 24. Milekhin, A. G.; Sveshnikova, L. L.; Repinskii, S. M.; Gutakovsii, A. K.; Friedrich, M.; Zahn, D. R. T.; Phys. Solid State 2002, 44, 1976.
- 25. Ferrari, A. C.; Rodil, S. E.; Robertson, J.; Phys Rev. B 2003, 64, 1.
Datas de Publicação
-
Publicação nesta coleção
23 Set 2005 -
Data do Fascículo
Out 2005
Histórico
-
Aceito
06 Jan 2005 -
Recebido
13 Abr 2004