Resumo
This work starts with a historical perspective of the social and scientific progress related to the understanding of the atmospheric aerosol. Its origin, physical, chemical and optical characteristics, as well as its environmental behaviour are described, retracing the evolution of the concepts related to this subject over the last centuries. The main sources that contribute to atmospheric particulate matter and the modern understanding of its formation processes and constitution, focusing on the chemical pathways leading to it and on its organic components are presented. This discussion is complemented with recent evaluations of the quantities emitted by primary, secondary, biogenic and anthropogenic sources and the effects due to accumulation or dispersion of aerosols, justifying the chemical and environmental interest they engender.
atmospheric aerosol; sources; organic components
atmospheric aerosol; sources; organic components
REVISÃO
Aerossóis atmosféricos: perspectiva histórica, fontes, processos químicos de formação e composição orgânica
Atmospheric aerosols: historical perspective, sources, chemical formation processes and organic composition
Célia Alves* * e-mail: celia@estg.ipvc.pt
Escola Superior de Tecnologia e Gestão, Instituto Politécnico de Viana do Castelo, Av. do Atlântico, 4900-348 Viana do Castelo, Portugal
ABSTRACT
This work starts with a historical perspective of the social and scientific progress related to the understanding of the atmospheric aerosol. Its origin, physical, chemical and optical characteristics, as well as its environmental behaviour are described, retracing the evolution of the concepts related to this subject over the last centuries. The main sources that contribute to atmospheric particulate matter and the modern understanding of its formation processes and constitution, focusing on the chemical pathways leading to it and on its organic components are presented. This discussion is complemented with recent evaluations of the quantities emitted by primary, secondary, biogenic and anthropogenic sources and the effects due to accumulation or dispersion of aerosols, justifying the chemical and environmental interest they engender.
Keywords: atmospheric aerosol; sources; organic components.
INTRODUÇÃO
Criado por Schumauss em 19201, o termo aerossol designa as suspensões relativamente estáveis de partículas sólidas ou gotículas dispersas num gás com dimensões inferiores a 100 µm, mas tamanhos superiores aos das moléculas individualizadas. Tais suspensões são freqüentemente designadas como aerocolóides, matéria particulada ou simplesmente partículas e incluem poeiras, fumos, cinzas, nevoeiros e "sprays". As principais propriedades que caracterizam um sistema aerocoloidal são: (i) deposição gravitacional negligenciável; (ii) efeitos inerciais desprezáveis; (iii) movimentos Brownianos significativos, devidos à agitação térmica das moléculas do gás e (iv) elevada área superficial específica2,3.
Incluídos entre os principais vectores da poluição e da radioatividade atmosféricas, os aerossóis participam em diversos fenomenos naturais que abrangem a formação de nuvens e de precipitação, o balanço radiativo da atmosfera, a visibilidade, as permutas entre o oceano e a troposfera e as eventuais modificações na camada de ozono. Os aerossóis intervêm igualmente em numerosos domínios industriais, incluindo as tecnologias de despoeiramento do ar e a climatização, e os seus efeitos são considerados nas directrizes dos programas de higiene e segurança laborais. Pelas características qualitativas e importância quantitativa da sua presença na atmosfera, as partículas constituem ainda um dos principais poluentes monitorizados nas redes de medida da qualidade do ar.
É sabido que as dispersões aéreas variam enormemente nas suas propriedades físicas e químicas dependendo da natureza das partículas suspensas, da sua concentração no gás, do tamanho e forma, e da homogeneidade espacial da dispersão. Quer o material líquido, quer o sólido pode estar suspenso no ar por uma grande variedade de mecanismos. Os aerossóis produzidos em condições laboratoriais, ou em dispositivos de geração especiais, podem ter propriedades uniformes que permitem uma investigação relativamente fácil a partir de métodos conhecidos. Contudo, os aerossóis naturais são misturas de materiais resultantes de várias fontes, as quais são grandemente heterogéneas na composição química e propriedades físicas, tornando ainda hoje muito difícil a sua caracterização. Não obstante a avultada acumulação de conhecimentos, fruto do notável empenho da comunidade científica nas últimas décadas, persistem numerosas lacunas cuja colmatação é imperiosa para uma percepção abrangente da participação dos aerossóis na química da atmosfera. O entendimento detalhado dos mecanismos de formação, magnitude das emissões, reactividade, composição e impacto ambiental da matéria particulada exige ainda a conjugação de esforços a nível internacional e uma aposta na experimentação.
EVOLUÇÃO DE CONCEITOS DA ANTIGUIDADE AOS DIAS ACTUAIS
No século XX, a ciência e a tecnologia dos aerossóis conheceram avanços significativos decorrentes do interesse crescente no estudo das suas características físico-químicas e dos seus efeitos ambientais. Mas a história dos aerocolóides remonta a tempos recônditos.
É possível que os fumos dos incêndios florestais, as erupções vulcânicas e as emanações associadas ao fogo doméstico fossem prejudiciais ou mesmo letais em localidades e momentos específicos, mesmo antes dos nossos ancestrais se organizarem em comunidades fixas, e que os odores provocados pelos resíduos animais e vegetais resultantes da actividade diária constituíssem, já nessa altura, causa de protesto e discussão entre residentes. No entanto, o mais certo é que esses fenómenos ambientais fossem encarados como problemas com origem exclusivamente natural e fortuita, constituindo motivo para a transumância. Com o desenvolvimento da agricultura e a fixação do Homem em aglomerados permanentes, a actividade humana passou a produzir efluentes que representavam agressões ambientais com repercussões a nível social. O conhecimento empírico na Idade Média da noção de "miasmas" e "ar empestado" como agentes etiológicos de certas doenças e pestes, fornece-nos a informação de que a avaliação da composição da atmosfera já era reconhecida como um problema importante. Um exemplo ilustrativo provém do Reino Unido, onde o consumo do carvão se encontrava generalizado. São conhecidos os protestos da nobreza contra o uso deste combustível, durante o reinado de Eduardo I (1272-1307). Mais tarde, nos reinados de Ricardo II (1307-1377) e Henrique V (1377-1422) editaram-se regulamentações e foram impostas taxas com o objectivo de restringir o consumo de carvão na cidade de Londres. Recorde-se também que, em 1382, o rei Carlos VI proibiu, na cidade de Paris, toda a emissão de "fumos nauseabundos". Desde o século XVI até meados do século XX, as emissões resultantes da queima de carvão, que progressivamente passou a ser utilizado como combustível em substituição da madeira, ocuparam quase exclusivamente o centro das atenções no respeitante à poluição atmosférica. Note-se que, em Londres, apesar das regulamentações referidas, o problema dos fumos persistiu, sendo relatados vários episódios de nevoeiros sulfurosos em documentos do século XVII. A situação agravou-se de tal forma que, no início do século XIX, o parlamento inglês criou um comité ao qual foi atribuída a competência de produzir medidas mitigadoras do problema4,5.
O interesse científico pelos aerossóis atmosféricos começou a manifestar-se a partir do século XVIII, na era do Iluminismo, acompanhando o rápido desenvolvimento verificado em diversas áreas das ciências naturais. Surgiram, neste período, diversas teorias focalizadas essencialmente na origem da matéria particulada e nas consequências da sua presença na atmosfera. No final do século XIX, Udden relata os estudos de vários geólogos, nos quais se estabelece a conexão entre a formação do solo e a presença dos aerossóis na atmosfera. Simultaneamente, os meteorologistas reconhecem as múltiplas influências que os aerossóis exercem na formação da precipitação, na visibilidade atmosférica e no balanço térmico e radiativo6. Pode referir-se que a dissertação de doutoramento de Kempf7 representa a resenha histórica mais significativa onde se teorizam as doutrinas sobre a origem dos aerossóis e se apresenta a literatura científica publicada sobre a matéria até 1870. Kempf agrupa as fontes emissoras em seis categorias:
(i) Emissões gasosas terrestres De acordo com esta teoria, os aerossóis têm a sua origem nas emanações gasosas produzidas em consequência dos tremores de terra ou resultantes da libertação de gases pelos ecossistemas. Por ex., Kant (1756) observou em Locarno, Itália, a formação e a dispersão pelo vale de um nevoeiro avermelhado após o registo de alguma actividade sísmica na região. O fenómeno culminou com uma intensa chuvada cuja formação foi atribuída à intrusão no vale de uma nuvem de poeiras transportada a longa distância, desde o deserto do Sahara, a qual teria actuado como núcleo de condensação6. Marcorelle8 verificou que o aumento da temperatura do ar durante a primavera libertava "produtos de fermentação" para a atmosfera. Segundo o investigador, a evaporação do conteúdo em água por acção solar produziria aquilo a que chamou "nevoeiro seco primaveril". Conceptualmente, esta teoria representa uma aproximação à moderna explicação das emissões biogénicas e da formação de aerossóis secundários.
(ii) Electricidade A formação de aerossóis a partir das descargas eléctricas durante as trovoadas constituiu uma base teórica sustentada até meados do século XIX. Verdeil9 definia o nevoeiro como "um conjunto de gotículas preenchidas com um fluido eléctrico, cuja sedimentação seria dificultada pelo facto de serem ascencionalmente atraídas pela electricidade da alta atmosfera". Schreiber10 atribuía o cheiro peculiar do nevoeiro ao ozono produzido por descargas eléctricas.
(iii) Poeiras de meteoritos As poeiras resultantes dos meteoritos foram consideradas uma fonte significativa de aerossóis atmosféricos. Por ex., Benjamin Franklin (1784) invocou a teoria meteórica para explicar as condições climatéricas observadas no frígido e nublado ano de 1783. Na mesma época, outros investigadores argumentavam que os meteoritos contribuíam com "mais de 1 polegada/século de matéria sólida para o Globo". Contudo, esta elevada taxa de acumulação não é suportada por evidências de natureza geológica. De facto, o impacto global das poeiras cósmicas e meteóricas no balanço global dos aerossóis atmosféricos foi considerado insignificante6.
(iv) Emissões vulcânicas Já na Antiguidade o filósofo Seneca (60 a.C.) reconhecia que as cinzas libertadas nas erupções vulcânicas constituíam a causa da turvação atmosférica. No entanto, só no século XVIII se descobriu que os aerossóis vulcânicos podiam ser transportados a longas distâncias. Invocando o ano de 1783, Benjamin Franklin escreveu que "a época sem Verão" podia dever-se a uma erupção de um vulcão na Islândia.
(v) Poeiras transportadas pelo vento Desde longa data que o vento é considerado veículo transportador e simultaneamente causador, por erosão eólica, de poeiras responsáveis pela turvação da atmosfera. Curiosamente, o binómio vento/poeira adquiriu nomes distintos em várias partes do mundo6: harmattan (África ocidental), sciroco ou calina (Espanha), gobar (África oriental), haboob (Arábia), kosa (Japão), whangsa (Coreia), huangsha (China), etc.
(vi) Processos de combustão A turvação atmosférica pode também ser causada por produtos sólidos e líquidos em processos de combustão. Em particular, o fumo libertado nos incêndios florestais e nas queimadas tem sido reconhecido como uma fonte significativa de aerossóis atmosféricos. É de salientar o trabalho de revisão de Brimblecombe11 onde se abordam as conseqüências da utilização de combustíveis na época da revolução industrial em Londres.
Os métodos científicos para estabelecer as causas e origens dos aerossóis atmosféricos foram mais claramente delineados por Egen12. Segundo o cientista, a causalidade pode ser evidenciada por: (1) observação directa, por ex. de uma pluma de fumo; (2) olfactação do ar; (3) variação temporal; (4) decaimento com a distância a partir da fonte; (5) variação das concentrações com a direcção do vento, hoje em dia definida como rosa de poluição; (6) análise das trajectórias das massas de ar. A metodologia proposta por Egen é virtualmente idêntica à utilizada nas modernas ciências atmosféricas com base no estabelecimento de relações fonte-receptor.
A dispersão local dos aerossóis atmosféricos e o transporte destes a longas distâncias constituiu, desde cedo, assunto de debate científico. De acordo com Kempf7, o primeiro relato de transporte troposférico transfronteiriço deve-se a Sir Francis Bacon, por volta de 1600. O nobre reporta as reclamações dos Gasgogners, uma abastada família do sul de França, apresentadas ao monarca inglês após os fumos oriundos da queima de algas em Sussex, Inglaterra, terem atingido na altura da floração as vinhas de que eram proprietários, comprometendo a colheita desse ano.
Em 1767, Wargentin aponta os incêndios florestais da Rússia e Finlândia como causas dos nevoeiros e neblinas observados na Europa Central e considera a possibilidade de cartografar a dispersão dos fumos a partir do registo simultâneo da intensidade e direcção dos ventos6.
Desde finais do século XVIII e até meados do século XIX, as planícies a norte dos Alpes, numa extensa região que se estendia desde Paris até Varsóvia, eram frequentemente cobertas por uma espessa camada de nevoeiro que motivou muitos estudiosos a debruçarem-se sobre as suas causas, transporte e efeitos. A origem do fenómeno atmosférico foi atribuída à queima dos depósitos de turfa resultantes da drenagem de terrenos pantanosos para conquista de áreas agrícolas no noroeste da Alemanha e Holanda. As objecções públicas durante mais de um século foram apenas consumadas por volta de 1870, altura em que a prática das queimadas cessou e o problema atmosférico desapareceu. Prestel13 estudou o comportamento espacial e temporal dos "fumos de turfa" recorrendo a observações de visibilidade em dezenas de cidades localizadas na trajectória dos ventos dominantes transportadores das plumas poluentes. Com os registos efectuados, o investigador cartografou a propagação das nuvens de fumo desde a sua origem no noroeste da Alemanha e Holanda em direcção a este e a sul.
Segundo Danckelman14, os nevoeiros e fumos provenientes dos fogos nas savanas africanas e observados em várias regiões da Europa seriam já conhecidos desde a Época Romana. O investigador calculou a massa de ervas queimadas em aproximadamente 600 milhões de toneladas e considerou esta estimativa equivalente ao consumo de 300 milhões de t de carvão. Comparando estes valores com as emissões de fumos associadas às máquinas a vapor e à queima de turfa na Europa, julgou estas últimas insignificantes relativamente às quantidades produzidas nos fogos atrás referidos.
É de destacar também o trabalho de Dinkage15 sobre a propagação espaço-temporal das poeiras do Sahara efectuado a partir da análise dos diários de bordo compilados pelo Instituto de Meteorologia Inglês e pela Capitania de Hamburgo. Com base em dados relativos a 13 anos, concluiu que as nuvens de pó relatadas pelos marinheiros representavam extrusões com origem no norte de África e penetração no Atlântico até uma distância por vezes superior a 1400 milhas. Notou ainda que as "chuvas de poeiras" ocorriam com mais frequência nos meses de janeiro e fevereiro.
Em 1883 foram observados com bastante frequência ocasos anormalmente avermelhados em vários pontos do Globo. Em face da ausência de qualquer explicação plausível para o entendimento do fenómeno, a Sociedade Real Britânica organizou uma competição para colmatar esta lacuna científica. O prémio foi ganho por Kiessling16 que considerou o "pôr do sol avermelhado" um evento atribuível aos aerossóis estratosféricos emitidos após a erupção do Cracatoa, na Indonésia. O estudo de Kiessling representou também uma importante contribuição para a meteorologia dinâmica, dado que evidenciou, pela primeira vez, a existência de circulações atmosféricas globais. As explicações apresentadas serviram ainda para demonstrar que os aerossóis podem ser utilizados como "traçadores" nos processos de transporte atmosférico.
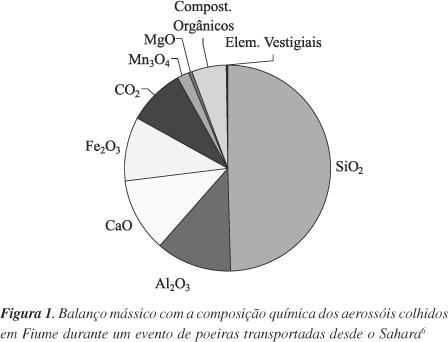
Segundo Husar6, o primeiro balanço mássico com a composição química dos aerossóis é da autoria de Barac, datando de 1901 (Figura 1). As partículas analisadas foram colhidas em Fiume, actualmente conhecida como Rijeka, na Croácia, durante um evento de poeiras no mar Adriático oriundas do Sahara. Recorrendo a microscopia óptica, Barac observou que as poeiras possuíam uma tonalidade avermelhada. Contudo, sob luz polarizadora grande parte dos aerossóis mostravam-se incolores, apresentando ainda fragmentos de cristal irregulares, juntamente com esqueletos de microrganismos e pequenas partículas de fuligens. Após análise dos dados de Barac, Husar6 sugere que a sedimentação de poeiras em Fiume poderá ter variado entre 260 e 1400 g m-2, considerando tratar-se do mesmo material que é transportado desde o norte de África até ao Atlântico. Husar faz ainda referência à importância atribuída no início do século XX aos estudos de Barac. O editor da prestigiada revista Monthy Weather Review escreveu que as partículas em suspensão na atmosfera seriam as responsáveis pela cor avermelhada do nevoeiro observado em março de 1901 e pela diminuição da radiação solar na superfície terrestre. O editor acrescentou ainda que, em consequência destes factos, as partículas contribuiriam indirectamente para o aquecimento das camadas altas da atmosfera.
Deve referir-se que os estudos e debates científicos sobre os aerossóis atmosféricos não se centralizaram apenas na sua origem primária e na distribuição espacial e temporal. Nos últimos séculos surgiram teorias fundamentadas em vários trabalhos de investigação com abordagens científicas mais elaboradas sobre diversos processos de formação, mecanismos de remoção e propriedades físico-químicas dos aerossóis atmosféricos. É fascinante notar que entre a miríade de teorias propostas, a extraordinária clareza e o teor das explicações científicas tornam, algumas delas, ferramentas de análise obrigatória 200 anos após a sua divulgação! São, por ex., notáveis as publicações de Rafinesque17,18 sobre o comportamento atmosférico dos aerossóis. Na obra do cientista são, pela primeira vez, teorizados os processos de remoção da matéria particulada na atmosfera, abordando-se com discernimento os mecanismos de sedimentação seca e húmida. Rafinesque é também pioneiro no reconhecimento de que "uma parte dos aerossóis atmosféricos é formada quimicamente a partir da combinação de gases e partículas elementares dissolvidos no ar". A verificação experimental das suas teorias sobre a formação de aerossóis secundários ocorreu apenas 80 anos depois. As experiências ópticas de Tyndall em 1870, seguidas da apresentação da teoria da dispersão da luz de Rayleigh em 1871, marcam o início da moderna ciência dos aerossóis6. Seguiram-se os trabalhos desenvolvidos por Aitken19 sobre os mecanismos de nucleação. Segundo o cientista, "a radiação solar pode produzir algumas alterações nos constituintes atmosféricos fotoquimicamente activos dando lugar à formação de núcleos com dimensões moleculares". Estes últimos são, hoje em dia, designados como partículas ou núcleos de Aitken. A ele se deve também o desenvolvimento de um método, actualmente ainda válido, para analisar a relação entre a concentração das partículas e a visibilidade atmosférica. As medições de longo termo por ele realizadas permitiram-lhe calcular constantes de proporcionalidade entre os dois parâmetros em condições de humidade atmosférica distintas. No virar do século, Eintein20 propõe a teoria dos movimentos Brownianos, estabelecendo a ponte entre a aproximação microscópica macromolecular e a moderna física dos fluidos que encara a atmosfera como um meio contínuo. Desde então, a ciência progrediu rapidamente, tal como é descrito nos trabalhos de revisão de Fuchs21 e Hidy e Brock22.
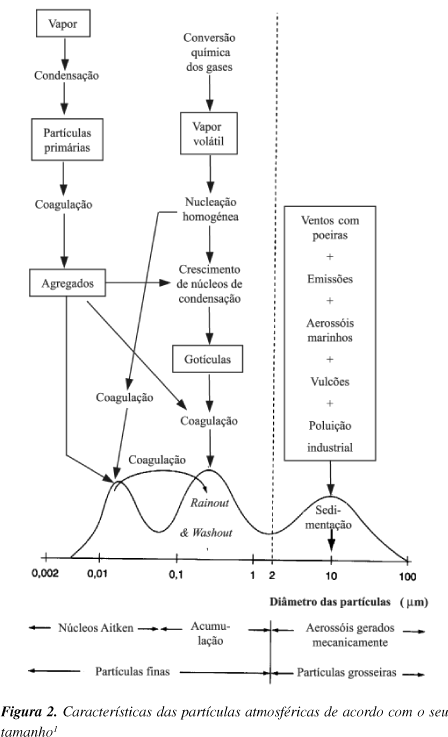
É de destacar o primeiro trabalho publicado sobre distribuições contínuas de aerossóis separados por classes de tamanhos, abrangendo raios de 0,01 a 10 µm. Junge23 concebeu uma classificação geográfica, dividindo os aerossóis em marinhos, continentais e "de fundo", e classificou-os por tamanhos em partículas de Aitken (0,001-0,1 µm), grosseiras (0,1-1 µm) e gigantes (> 1 µm). Deve referir-se que tamanho da partícula é, em geral, associado ao diâmetro aerodinâmico equivalente, ou seja, ao diâmetro de uma esfera com densidade unitária e a mesma velocidade de queda.
Em 1973, Whitby24 iniciou a representação das distribuições por tamanhos em gráficos com eixos logarítmicos e introduziu os termos modo de nucleação (0,001-0,1 µm), modo de acumulação (0,1-1 µm) e modo das partículas grosseiras ou modo de sedimentação (> 1 µm), na tentativa de relacionar o tamanho das partículas com os processos de formação25. Assim, no modo de nucleação incluem-se os aerossóis que resultam da conversão gás-partícula; o modo de acumulação é formado por coagulação e condensação heterogénea; e o modo de sedimentação engloba as partículas produzidas por processos mecânicos (Figura 2).
A ciência dos aerossóis tornou-se, desde a década de 70, uma componente dinâmica no domínio científico e tecnológico, conhecendo progressos importantes para a compreensão da atmosfera, como adiante se verá.
ORIGEM, ESTIMATIVAS DE PRODUÇÃO E EFEITOS DOS AEROSSÓIS ATMOSFÉRICOS
A classificação mais simplista aplicada, na actualidade, aos aerossóis atmosféricos categoriza-os como primários ou secundários, atendendo à sua origem e processos de formação. Na Figura 3 representam-se, de forma esquemática, as fontes de emissão directas e os mecanismos subsidiários da formação de aerossóis.
Fontes primárias
As partículas primárias são emitidas por múltiplas fontes naturais ou antropogénicas, abrangendo os processos de combustão, as erupções vulcânicas, os fogos florestais, as emanações derivadas de certas actividades industriais e viárias, o "spray" marinho e alguns materiais biológicos. Na Tabela 1 compilam-se as estimativas mais recentes para a emissão anual de partículas de natureza primária.
Aerossóis carbonosos (carbono orgânico e carbono negro)
O carbono presente nos aerossóis representa uma mistura de compostos inorgânicos (CI), carbono orgânico (CO) e carbono negro (CN), este último também designado como carbono elementar (CE). Os compostos de CN e CO constituem, em parte, produtos primários de origem antropogénica, representando no seu conjunto aquilo a que se chama o aerossol carbonoso ou carbonáceo. Uma fracção do carbono orgânico é, no entanto, resultante da condensação de moléculas orgânicas sobre a superfície de partículas durante a permanência destas na atmosfera. Os CI são fundamentalmente compostos primários de origem natural, embora seja possível que uma pequena percentagem seja formada por reacções atmosféricas. As partículas de CN são formadas por cadeias de átomos de carbono apresentando uma estrutura microcristalina semelhante à da grafite36. Na estrutura grafítica, cada átomo C apresenta electrões p com elevada mobilidade, os quais são responsáveis pelas características de absorção luminosa do CN37. Uma das definições usuais considera CN como todo o carbono que apresenta cor negra, sendo o restante, à excepção dos carbonatos, carbono orgânico. A capacidade de adsorção de poluentes gasosos pelos aerossóis carbonosos deriva das características químicas e da larga área específica das partículas. Devido aos defeitos estruturais, nos microcristais grafíticos existem muitos electrões desemparelhados que funcionam como centros activos na adsorção e reacção catalítica dos poluentes gasosos. As propriedades catalíticas dos aerossóis carbonosos têm sido evidenciadas na formação de sulfatos acídicos que podem provocar o ataque dos tecidos pulmonares, causar danos na vegetação e atacar as construções38,39. À presença destes aerossóis na atmosfera associa-se também o escurecimento de monumentos devido à deposição de fuligem e a redução da visibilidade como consequência das propriedades de absorção da radiação luminosa40-46.
As principais fontes de aerossóis carbonosos são representadas pela queima de biomassa e de combustíveis fósseis e pela oxidação atmosférica de compostos orgânicos voláteis (COV) biogenica ou antropogenicamente produzidos. As emissões globais de aerossóis orgânicos resultantes da combustão de biomassa e combustíveis foram estimadas em 45-80 e 10-30 Tg ano-1, respectivamente26-28. Os processos de combustão constituem também a fonte dominante de CN, calculando-se as massas anualmente produzidas a partir da biomassa em 6-9 Tg e em 6-8 Tg as provenientes dos carburantes26-29.
Poeiras do solo
As principais fontes deste material particulado estão sediadas no Hemisfério Norte, particularmente na África, na Península Arábica e nos desertos asiáticos de Gobi e Taklamakan. No Hemisfério Sul destaca-se apenas o deserto australiano. Apesar da contribuição natural ser considerada preponderante, têm vindo a ser demonstrado que o revolvimento dos solos nas actividades agrícolas pode constituir 50% das emissões globais, principalmente em anos de estiagem e ventosos47. A investigação mais recente recorre a imagens satélite e a modelos meteorológicos complexos na tentativa de explicitar os padrões de circulação intercontinentais30. As propriedades ópticas das poeiras constituem também uma área de estudo com importância crescente, dado que a dispersão ou a absorção da radiação solar pelos constituintes silicatados podem contribuir para as alterações climáticas globais48-50.
Emissões vulcânicas
Os vulcões são fontes esporádicas relativamente importantes de aerossóis. Estima-se que a actividade vulcânica seja responsável pelo lançamento para a atmosfera de 15-90 Tg ano-1 de partículas51. Uma fracção importante das partículas produzidas por estas fontes apresenta dimensões bastante reduzidas (» 1 µm), permanecendo por longos períodos na atmosfera. As erupções vulcânicas têm ainda como consequência o lançamento a elevadas altitudes de enormes quantidades de compostos sulfurosos, principalmente SO42- e H2S52. Em poucos dias, este último oxida-se a SO2, o qual é posteriormente recoberto com gotículas de ácido sulfúrico, num processo que se desenrola na estratosfera nas semanas e meses seguintes à erupção. Os aerossóis assim formados constituem uma camada duradoura que se espalha por todo o Globo e funciona como filtro energético. Julga-se que o arrefecimento registado em 1993 na superfície terrestre tenha sido uma consequência indirecta da erupção do Pinatubo. Dados obtidos com satélites dois anos mais tarde permitiram concluir que a camada de aerossóis vulcânicos permanecia praticamente inalterada na estratosfera53. Os aerossóis vulcânicos podem ainda actuar como núcleos de condensação de partículas de gelo na alta troposfera, interferindo desta outra forma no balanço radiativo terrestre54. Calcula-se que as erupções vulcânicas das últimas décadas tenham provocado um arrefecimento global de 0,1-0,5 ºC55.
Partículas de sal marinho
A formação de aerossóis marinhos é consequência do rebentamento de bolhas de ar nas cristas espumosas das ondas56-58. Tal como sucede com as partículas com origem edáfica, o sal marinho apresenta tamanhos variáveis, pelo que os tempos de residência na atmosfera podem oscilar entre alguns minutos e uma semana59.
O sal marinho contribui positivamente para a dispersão da luz e para o aumento dos núcleos de condensação de nuvens (NCN) em atmosferas remotas de regiões marinhas onde são desprezáveis outras fontes de matéria particulada60,61. Especialmente em áreas com ventos fortes, as partículas do modo de acumulação são mais numerosas do que o que anteriormente se supunha62.
Recorrendo a uma formulação semi-empírica baseada em princípios físicos, Gong et al.34 estimaram o fluxo anual de sal marinho para a atmosfera em 3300 Tg. Esta estimativa está em consonância com os cálculos apresentados por Tegen et al.63, 5900 Tg ano-1, e por Erickson e Duce64, 1000-3000 Tg ano-1.
Partículas resultantes de actividades tecnológicas e industriais
Entre as actividades tecnológicas e industriais responsáveis pela produção de aerossóis primários contam-se os transportes, a combustão de carvão, o processamento de cimentos, a metalurgia e a incineração de resíduos. Pelo facto de estas fontes representarem o impacto ambiental mais conspícuo, a generalidade das emissões antropogénicas têm sido objecto de regulamentações cada vez mais restritivas. As estimativas mais recentes para estas emissões oscilam entre 100 Tg ano-1 [32] e 200 Tg ano-1 [31], prevendo-se que possam atingir 300 Tg ano-1 nas próximas décadas31, em consequência do crescimento industrial no continente asiático não sujeito a monitorização rigorosa.
Não devem esquecer-se ainda os quantitativos produzidos nas actividades humanas do quotidiano. Tome-se como exemplo uma família americana constituída por 4 pessoas. O aquecimento doméstico, a utilização do automóvel e a incineração de resíduos associados a esta família contribuem individualmente para a emissão anual de 5 kg de partículas; o processamento de alimentos produz 30 kg ano-1; a jardinagem e a queima dos resíduos vegetais originam 25 kg ano-1; das actividades de lazer resultam 2 kg ano-1[65].
Partículas biogénicas
Os aerossóis primários biogenicamente emitidos são constituídos por componentes vegetais (ceras cuticulares, fragmentos foliares, etc.), matéria húmica e partículas microbianas (bactérias, fungos, vírus, algas e esporos). Infelizmente, a informação existente é insuficiente para validar qualquer estimativa da representatividade destes constituintes do aerossol atmosférico. Sabe-se, no entanto, que mesmo nas áreas urbanas das regiões temperadas representam 10-30% da distribuição numérica das partículas66. Estas percentagens sugerem que a contribuição biogénica primária pode ser substancial em áreas com coberto vegetal denso, particularmente na zona tropical húmida. A análise da fracção lipídica de aerossóis amazónicos permitiu concluir que 10-20% dos constituintes orgânicos estavam associados à matéria microbiana e às ceras vegetais67.
O número imenso de fontes de aerossóis biológicos primários e a utilização de métodos de amostragem e análise muito distintos tornam as medições até agora efectuadas representativas apenas do local de colheita, das condições ambientais particulares e da fenologia específica. Por exemplo, a concentração bacteriana no ar circundante a uma estação de tratamento de águas residuais foi estimada em 104-105 bacilos m-3. As determinações efectuadas com amostras de ar de um jardim público e de uma rua movimentada produziram valores de 290 e 7500 bactéria m-3, respectivamente68.
A absorção da luz, particularmente na região UVB, é potenciada com a presença de substâncias húmicas nos aerossóis. Suspeita-se também que as partículas biológicas primárias possam actuar como núcleos de condensação de nuvens ou de gelo30. Além das repercussões a nível climático, está também estabelecido o vínculo dos bioaerossóis com a saúde pública. As características alérgicas dos pólenes e a patogenicidade de alguns microrganismos atmosféricos constituem exemplos que justificam o interesse epidemiológico por estas partículas biogénicas.
Fontes secundárias
Uma parte importante dos aerossóis resulta de mecanismos de nucleação e condensação de produtos gasosos, podendo constituir-se três categorias principais: aerossóis de sulfato, aerossóis de nitrato e aerossóis orgânicos secundários (Tabela 2).
Os aerossóis de sulfato são formados pela oxidação a SO2 e posteriormente a ácido sulfúrico de H2S emitido pelos processos biológicos, por vulcões ou pela transformação deste e de outros compostos sulfurosos resultantes de actividades antropogénicas. A nucleação homogénea heteromolecular do ácido sulfúrico é uma das vias possíveis para a produção de aerossóis. Em condições de supersaturação do precursor, a colisão entre moléculas origina núcleos estáveis que se combinam com outras moléculas ou núcleos. Os movimentos brownianos potenciam a coagulação dos núcleos de Aitken, dando origem a partículas com cerca de 0,1 µm de diâmetro. A condensação de compostos gasosos com baixa pressão de vapor à superfície destas partículas provoca o seu crescimento e a formação de aerossóis com dimensões de 0,1 a 1 µm. Os aerossóis de sulfato podem também ser constituídos por sais de enxofre formados nas gotículas das nuvens e nevoeiros. Nestas gotículas, o ácido sulfúrico poderá estar total ou parcialmente neutralizado por substâncias alcalinas igualmente solúveis em meio aquoso. Os sais de enxofre dissolvidos podem permanecer em suspensão na atmosfera após a evaporação da água das gotículas. Compilações bibliográficas extensivas sobre a investigação mais recente no âmbito da química atmosférica do enxofre e sobre os aerossóis de sulfato podem ser encontradas nas refs. 30, 54, 71 e 72. Grande parte da investigação desenvolvida sobre os aerossóis de sulfato focaliza o cálculo, a partir de medições reais ou da aplicação de modelos matemáticos, da produção destas partículas secundárias. A importância dos compostos de enxofre na química da atmosfera e sua influência nas alterações climáticas são motivos que suscitam o interesse científico por este assunto. O papel dos aerossóis de enxofre no clima do Globo desde a era pré-industrial até ao presente é descrito nas publicações de Charlson et al.73,74. Devido à complexidade dos processos físico-químicos envolvidos na produção e remoção atmosféricas das partículas sulfatadas, os vários modelos utilizados pela comunidade científica partem de pressupostos distintos, tornando as estimativas demasiado dependentes do suporte matemático. Assim, podem encontrar-se na literatura avaliações da produção de aerossóis de sulfato bastante discordantes. Os valores incluídos na Tabela 2 são, por isso, meramente indicativos.
Os aerossóis de nitrato derivam da oxidação e neutralização de compostos de NOx e NH3 naturalmente emitidos ou resultantes de actividades humanas75,76. O NH3 desempenha um papel importante na neutralização do ácido sulfúrico, transformando-o em sulfato de amónia. Os óxidos de azoto participam em múltiplas reacções de competição, nas quais também intervêm o radical OH, dando origem ao ácido nítrico. A condensação deste último é promovida pelas partículas pré-existentes, as quais actuam como núcleos. As gotículas acídicas formadas deste modo podem ser neutralizadas pelo NH3 biogénico, produzindo-se sais de nitrato de amónio. Tal como sucede com os aerossóis de sulfato, também as estimativas para os aerossóis de nitrato são discordantes. Andreae32 avaliou a produção anual de NH4NO3 atribuível a fontes naturais e antropogénicas em 0,24 e 0,4 Tg, respectivamente. Adams et al.77 obtiveram uma estimativa global de 0,17 Tg ano-1. Por sua vez, no último relatório do IPCC são apresentados valores de 12,7 Tg ano-1 para a formação com origem antrópica e de 2,9 Tg ano-1 para o aerossol secundário biogénico. Uma possível explicação para as discrepâncias encontradas relaciona-se com o facto dos modelos de cálculo nem sempre incluírem os mecanismos de remoção e de deposição. Apesar de actualmente a influência dos aerossóis de nitrato nas alterações climáticas ser considerada de somenos importância relativamente ao papel exercido pelos sulfatos, supõe-se que adquirirá destaque com o triplicar das emissões de NOx previsto para este século30.
Os aerossóis orgânicos são formados por processos de condensação de compostos voláteis emitidos pela exudação das plantas78-81, transportes e indústrias. Dado que os compostos terpénicos emitidos pelas plantas (Figura 4) são quimicamente semelhantes às olefinas de massa molecular inferior libertadas pelos escapes dos automóveis, supõe-se a ocorrência do mesmo tipo de reacções fotoquímicas, sendo a formação de matéria particulada um resultado subsequente. As reacções iniciam-se com a formação de oxigénio atómico (O) a partir de óxidos de azoto (NO2) com a participação de radiação ultravioleta (hn) da luz solar. O oxigénio atómico pode reagir com o oxigénio molecular (O2) presente na atmosfera e formar ozono (O3). Quer o O3, quer o oxigénio atómico podem reagir com as olefinas (C = C) e produzir vários produtos orgânicos (P). O radical hidroxilo (OH), formado durante o ciclo fotoquímico dos óxidos de azoto, representa outra espécie reactiva capaz de atacar as ligações duplas das olefinas e formar novos produtos orgânicos. As moléculas destes produtos podem agregar-se, originando polímeros de elevada massa molecular (Pn). Por sua vez, os processos de conversão gás-partícula contribuem para a formação de compostos ainda maiores envolvendo o crescimento polimérico. Quando o tamanho é suficientemente grande, os agregados moleculares são reconhecidos como partículas. A sequência descrita pode ser esquematizada da seguinte forma82:

A formação de novas partículas resulta quer de mecanismos de nucleação homogénea ou heterogénea, quer da condensação dos compostos sobre partículas pré-existentes. Este último processo é controlado pela área superficial dos aerossóis já existentes. Como a superfície apresenta um máximo para as partículas com tamanhos compreendidos entre 0,1 e 1 µm, a condensação processa-se sobretudo na fracção submicrométrica.
A condensação de compostos orgânicos pode ocorrer mesmo quando os precursores gasosos se encontram em concentrações abaixo do nível de saturação. Isto é possível porque o aerossol orgânico é constituído por um filme líquido, no qual vários hidrocarbonetos se encontram em solução. Assim, devido à solubilidade da substância condensável na fase orgânica líquida podem estabelecer-se condições de equilíbrio termodinâmico, para níveis inferiores aos da saturação, de acordo com a lei de Raoult83,84. A solubilidade aumenta com o incremento de hidrocarbonetos orgânicos menos voláteis na fase condensada. Os produtos particulados resultantes da condensação de gases orgânicos possuem um estado de oxidação elevado e são muito mais polares que os seus precursores.
Guenther et al. (1995)85 efectuaram uma estimativa das emissões globais de monoterpenos e outros COV reactivos a partir da inventariação das espécies vegetais representativas e da identificação e quantificação dos compostos emitidos em cada ecossistema. A combinação desta informação com mecanismos reaccionais e de transporte em modelos matemáticos específicos permitiu apresentar estimativas de 13-24[69], 8-40[70] e 2,5-44,5 Tg ano-1[80] para os aerossóis derivados das emissões biogénicas. Contudo, deve impor-se uma análise cautelosa aos valores apresentados. As emissões antropogénicas, especialmente de NOx, são responsáveis pelo aumento das concentrações de O3 e NO3, interferindo igualmente no processo de formação de núcleos poliméricos. Estudos recentes conduzidos na Amazónia demonstraram que a produção de novas partículas a partir da oxidação de COV é escassa em áreas impolutas86-88. Considerando os elevados quantitativos de COV emitidos e os aumentos esperados nas emissões antropogénicas decorrentes do desenvolvimento tecnológico crescente, é também expectável um incremento significativo na produção de aerossóis secundários. Kanakidou et al.89 recorreram a um modelo tridimensional global com o objectivo de estudar a contribuição do aerossol orgânico secundário (AOS), derivado da ozonólise de COV biogénicos, para as concentrações observadas de aerossol orgânico e, simultaneamente, avaliar a evolução desde a época pré-industrial até ao presente. Os investigadores calcularam que o AOS terá aumentado de 17-28 para 61-79 Tg ano-1. O incremento da formação de aerossóis orgânicos a partir de COV naturais foi atribuído ao acréscimo sucessivo nas concentrações de ozono e de aerossóis antropogénicos. De acordo com os autores, os aerossóis produzidos por deposição de produtos de oxidação condensáveis em partículas pré-existentes representam aproximadamente 75% dos AOS com origem em COV biogénicos, sendo os restantes 25% representativos de processos de nucleação e condensação homogéneas. Deve referir-se que os estudos atrás citados, designadamente o de Griffin et al.69, consideravam apenas mecanismos de auto-nucleação dos COV naturais e partição com o AOS, não assumindo a possibilidade de deposição sobre outros aerossóis orgânicos. Assim, não são de estranhar as diferenças nas estimativas publicadas pelos dois grupos de trabalho.
Apesar dos COV antropogénicos poderem ser oxidados para formar matéria particulada, só os compostos aromáticos produzem quantidades significativas de aerossóis. Estima-se que, em ambiente urbano, a oxidação de 1 kg de compostos aromáticos contribua para a formação de 30 g de partículas90. As emissões globais de COV antropogénicos foram calculadas em 109 Tg ano-1, das quais aproximadamente 60% são atribuíveis à utilização de combustíveis fósseis e as restantes à queima de biomassa91. No cômputo das emissões antrópicas obteve-se o valor de 19 Tg ano-1 como representativo dos compostos aromáticos, estimandose a produção de aerossóis deles originários em 0,6 Tg ano-1. Esta fraca representatividade é suportada pelas medições efectuadas em áreas altamente urbanizadas com emissões significativas de COV antropogénicos, como Los Angeles. De facto, constatou-se que apenas 15% da fracção orgânica das partículas finas tinham origem secundária e que, destas, menos de 50% derivavam das emissões antropogénicas92,93.
A constituição de poluentes orgânicos secundários, especialmente o nevoeiro fotoquímico, a partir de reacções dos COV com oxidantes atmosféricos, tem grande influência sobre a qualidade do ar. A formação de material particulado tem numerosos efeitos, tais como o da redução da visibilidade e o da interferência na dispersão da luz. Por outro lado, a fracção inalável das partículas atmosféricas (dp < 2,5 µm) pode contribuir para o agravamento das doenças pulmonares obstrutivas e, inclusivamente, ser veiculada para a corrente sanguínea ou sistema linfático. Além disto, este material, ou os seus componentes solúveis extraíveis, pode ser transportado para os órgãos mais afastados dos pulmões e exercer uma acção prejudicial sobre eles. Partículas transportadas a partir do tracto respiratório são, em larga extensão, absorvidas pelo tracto gastrointestinal. Assim, o material particulado, ou os compostos solúveis extraíveis que fazem parte da sua constituição, pode desencadear graves efeitos fisiológicos94.
PRINCIPAIS GRUPOS ORGÂNICOS NOS AEROSSÓIS ATMOSFÉRICOS
Até à data, já foram identificadas várias centenas de compostos orgânicos nas emissões primárias dos aerossóis orgânicos. Todavia, nos inúmeros estudos foi possível identificar constituintes que representam apenas 10 a 40% da massa de carbono orgânico, dependendo das fontes emissoras. Apesar do conhecimento da composição molecular da matéria orgânica particulada ter aumentado significativamente, a complexidade da mistura é tal que compostos traçadores ou marcadores são ainda necessários para avaliar as várias fontes.
As medições de compostos orgânicos particulados mostram que em áreas rurais ou florestais a fracção maioritária é constituída por espécies oxigenadas e derivados terpénicos característicos da vegetação81,88,96-98. Nas atmosferas urbanas predominam os compostos com origem petrogénica e pirogénica, destacando-se os hidrocarbonetos alifáticos e aromáticos, constituintes cíclicos associados às emissões dos veículos motorizados, ácidos n-alcanóicos e ácidos dicarboxílicos alifáticos97,99-103. Na Tabela 3 representam-se os principais grupos funcionais detectados nos aerossóis atmosféricos.
Na tentativa de estabelecer conexões entre os constituintes orgânicos detectados e as fontes emissoras, diversos outros trabalhos de investigação têm sido direccionados para a pesquisa de traçadores naturais ou antropogénicos que caracterizem a origem da fracção carbonosa da matéria particulada67,93,104-114. Um grupo importante de traçadores naturais é constituído por sesquiterpenos, diterpenos, fitoesteróis, ácidos triterpénicos e outros triterpenos. Os compostos sesquiterpénicos possuem na sua estrutura o esqueleto do cadinano, um constituinte predominante das plantas resínicas115. Os componentes diterpénicos são originados a partir do esqueleto esteróide dos precursores vegetais abietano e pimarano. Estas duas estruturas instáveis sofrem reacções de oxidação microbiana ou térmica, polimerização, condensação, isomerização e aromatização, dando origem a vários ácidos resínicos116. O ácido desidroabiético é um dos biomarcadores diterpénicos mais importantes detectados nos aerossóis atmosféricos, podendo ser directamente emitido pelas coníferas ou formado durante a combustão da lenha de espécies resínicas67,96,112,117-119. Os fitoesteróis são esteróis das plantas fotossintéticas, bioprocessados a partir do esqualeno96,119-122. Os compostos mais comuns encontrados nos extractos orgânicos dos aerossóis abrangem o ergosterol (C27), campesterol (C28), sitosterol (C29) e estigmasterol (C29) Os constituintes triterpénicos são derivados oxidados de precursores naturais, como o esqualeno-2,3-epóxido109. Como exemplos de compostos integrados neste grupo podem citar-se o amirinol e seus derivados, friedolean-3-ona, lup-1-en-3-ona, colesta-3,5-dien-7-ona, lup-20(29)-en-3-ol e estigmast-4-en-4-ona123. Outros traçadores biogénicos frequentemente encontrados nos aerossóis incluem os n-alcanos ímpares C27 a C33, característicos das plantas vasculares124. O levoglucosano tem sido utilizado como traçador da celulose nas partículas resultantes da combustão de biomassa97,113,125. Os ácidos n-tetradecanoíco (mirístico), n-hexadecanóico (palmítico), n-octadecanóico (esteárico) e cis-9-octadecenóico (oleico), o nonanal, a 2-decanona e o colesterol foram relacionados com as emissões resultantes da cozedura de carnes126,127. A presença de esteranos e triterpanos pentacíclicos na matéria particulada foi considerada indicadora das emissões automóveis128-130. Os iso- e anteiso-alcanos foram correlacionados com o fumo do tabaco131.
CONCLUSÕES
Os aerossóis atmosféricos são de grande interesse nos estudos do clima e importantes indicadores de poluição natural ou induzida pelas actividades antrópicas. Representam componentes importantes dos ciclos biogeoquímicos e participam na química heterogénea que afecta os gases vestigiais na troposfera e estratosfera. Nos últimos séculos e sobretudo nas últimas décadas, registrou-se um avanço científico significativo na caracterização dos aerossóis atmosféricos, esclarecendo-se as fontes, transformações, destinos e interacções com a radiação. A monitorização continuada tem permitido a obtenção de informações sobre a distribuição da matéria particulada e realizar estimativas das suas concentrações, dependendo dos processos físico-químicos que estão na sua origem. Estudos recentes provaram que uma parte significativa da massa dos aerossóis atmosféricos pode ser atribuída a constituintes orgânicos. A fracção carbonácea das partículas atmosféricas é composta por carbono elementar, também designado de carbono negro ou grafítico, e por carbono orgânico. O primeiro é emitido directamente para a atmosfera durante os processos de combustão. O segundo pode constituir as emissões primárias ou ser formado in situ por condensação de produtos de baixa volatilidade da foto-oxidação de hidrocarbonetos. Um conjunto de compostos traçadores foi proposto para identificar e quantificar as contribuições das várias fontes emissoras e processos de formação da matéria particulada. As dificuldades analíticas e a complexidade dos componentes presentes têm, contudo, limitado a obtenção de resultados esclarecedores. É, pois, importante contribuir, através de trabalhos de campo, para um melhor conhecimento da composição orgânica dos aerossóis atmosféricos, considerando simultaneamente a contribuição primária de fontes biogénicas e antropogénicas e a formação secundária de produtos por oxidação de compostos orgânicos voláteis.
AGRADECIMENTOS
Ao Prof. Dr. C. Pio, do Departamento de Ambiente e Ordenamento da Universidade de Aveiro, pela oportunidade concedida, ensinamentos e incentivos demonstrados ao longo da execução do trabalho. A autora é também reconhecida à Fundação para a Ciência e a Tecnologia pela atribuição da bolsa PRAXIS XXI/BD/9622/96.
53. http://oea.larc.nasa.gov/PAIS/Aerosols.html, acessada em Abril 2004.
72. http://www.agu.org/revgeophys/kreide01/kreide01.html, acessada em Abril 2004.
Recebido em 26/5/04; aceito em 5/1/05; publicado na web em 13/4/05
- 1. Renoux, A.; Boulard, D.; Les aérosols. Physique et Métrologie, Lavoisier Technique & Documentation: Paris, 1998.
- 2. Götz, G.; Mészáros, E.; Vali, G.; Atmospheric particles and nuclei, Akadémiai Kiadó: Budapest, 1991.
- 3. Witschger, O.; Theoretical and Experimental Modeling of Particulate Flow, Von Karman Institute for Fluid Dynamics: Brussels, 2000.
- 4. Brimblecombe, P. Em Urban air polution: European aspects; Fenger, J.; Hertel, O.; Palmgren, F. eds.; Kluwer Academic Publishers: The Netherlands, 1998, p. 7-21.
- 5. Lutgens, F. K.; Tarbuck, E. J.; The Atmosphere An Introduction to Meteorology, 7th ed., Prentice Hall: New Jersey, p. 303-318.
- 6. Husar, R. B.; Meeting "History of Aerosol Science", Viena, Austria, 1999.
- 7. Kempf, N.; Die Enwicklung der Theorien ueber den Hoehenrauch, Doctors Dissertation vor der Kgl. Technichen Hochschule zu Muenchen, Verlag von F.C.W. Vogel: Leipzig, 1914.
- 8. Marcorelle, J. F.; J. Phys. 1784, 24, 3.
- 9. Verdeil, A.; Mémoires de la Société des Sciences Physiques de Lausanne, 1783, vol. I, p. 110-114.
- 10. Schreiber, F.; Versuch einer neuen Theorie der Hoehenrauchbildung, Dissertion, Marbug, 1844.
- 11. Brimblecombe, P.; The Big Smoke. A History of Air Pollution in London since Medieval Times, Methuen: London/New York, 1987.
- 12. Egen, P. N.C.; Der Haarrauch, Essen, 1835.
- 13. Prestel, M. A. F.; Kleine Schrifte der Naturforschenden Geselschaft in Emden, Emden Schnellpressen Druck von Th. Hahn Wwe, Emden, 1861.
- 14. Danckelman, V.; Meteorologische Zeitschrift 1884, 8, 301.
- 15. Dinkage, L. E.; Annalen der Hydrographie 1891, 313.
- 16. Kiessling, J.; Untersuch. ueber Daemmerungs-Erscheing. zur Erklaerung d. nach d. Krakatauausbrush beobact atmosphaer., Opstich Stoerung: Hamburg-Leipiz, 1888.
- 17. Rafinesque, C.; Am. J. Sci. 1819, 1, 4.
- 18. Rafinesque, C.; Silliman's Am. J. 1820, 1, 134.
- 19. Aitken, J.; Nature 1894, 49, 544.
- 20. Eintein, A.; Annalen der Physik 1905, 17, 549.
- 21. Fuchs, N. A.; The mechanics of Aerosols, Pergamon: Oxford, 1964.
- 22. Hidy, G. M.; Brock, J. R.; The Dinamics of Aerocolloidal System, Pergamon Press: Oxford, 1970.
- 23. Junge, C. E.; Air Chemistry and Radioactivity, Academic Press: London, 1963.
- 24. Whitby, K. T.; IIIth International Conference on Nucleation, Leningrad, URSS, 1973.
- 25. Renoux, A.; Boulard, D.; Les aérosols. Physique et Métrologie, Lavoisier Technique & Documentation: Paris, 1998.
- 26. Liousse, C.; Penner, J. E.; Chuang, C.; Walton, J. J.; Eddleman, H.; Cachier, H.; J. Geophys. Res. Atmos. 1996, 101, 19411.
- 27. Scholes, R. J.; Andreae, M. O.; Geophys. Res. Lett. 1999, 20, 29.
- 28. Cooke, W. F.; Wilson, J. J. N.; J. Geophys. Res. Atmos. 1996, 101, 19395.
- 29. Penner, J. E.; Eddleman, H.; Novakov, T.; Atmos. Environ. 1993, 27A, 1277.
- 30. IPCC Em Climate Change 2001 IPCC WG1 Third Assessment Report; Houghton, J. T.; Ding, Y.; Griggs, D. J.; Noguer, M.; van der Linden, P. J.; Xiaosu, D. eds.; Cambridge University Press: Cambridge, UK, 2001, cap. 5.
- 31. Wolf, M. E.; Hidy, G. M.; J. Geophys. Res. Atmos. 1997, 102, 11113.
- 32. Andreae, M. O. Em World Survey of Climatology; Henderson-Sellers, A.; ed.; Elsevier: Amsterdam, 1995, vol. 16, p. 341-392.
- 33. Penner, J. E. Em Aerosol Forcing of Climate; Charlson, R. J.; Heintzenberg, J., eds.; John Wiley and Sons: Chichester, 1995, p. 91-108.
- 34. Gong, S. L.; Barrie, L. A.; Blanchet, J.-P.; Spacek, L. Em Air Pollution Modelling and its Applications XII; Gryning, S.-E.; Chaumerliac, N., eds.; Plenum Press: New York, 1998.
- 35. Ginoux, P.; Chin, M.; Tegen, I.; Prospero, J. M.; Holben, B; Dubovik, O; Lin, S. J.; J. Geophys. Res.- Atmos. 2001, 106, 20255.
- 36. Tartarelli, R.; Giorgini, M.; Davini, P.; Marcolini, P. F.; Riv. Combust. 1973, 27, 27.
- 37. Wolff, G. T.; J. Air Pollut. Control Assoc. 1981, 31, 935.
- 38. Chang, S. G.; Novakov T.; Atmos. Environ. 1979, 9, 495.
- 39. Chang, S. G.; Novakov, T.; Soot catalyzed oxidation of sulfur dioxide, NASA, Nş 79 10644, 1979.
- 40. Fassina, V.; Atmos. Environ. 1978, 12, 2205.
- 41. Groblicki, P. J.; Wolff, G. T.; Countess, R. J.; Atmos. Environ. 1981, 15, 2473.
- 42. Hegg, D. A.; Livingston, J.; Hobbs, P. V.; Novakov, T.; Russel, P.; J. Geophys. Res. 1997, 10, 25293.
- 43. Novakov, T.; Hegg, D. A.; Hobbs, P. V.; J. Geophsy. Res. Lett. 1997, 23, 2141.
- 44. Hand, J. L.; Kreldenweis, S. M.; Sherman, D. E.; Collett, J. L.; Hering, S. V.; Day, D. E.; Malm, W. C.; Atmos. Environ 2002, 36, 5043.
- 45. Chan, Y. C.; McTainsh, G. H; Simpson, R. W.; Vowles, P. D.; Cohen, D. D.; Bailey, G. M.; Aerosol Sci. Technol 2002, 36, 890.
- 46. Garcia-Neto, P. J.; Aerosol Sci. Technol 2002, 36, 814.
- 47. Tegen, I.; Lacis, A.; Fung, I.; Nature 1996, 380, 419.
- 48. Hansen, A. D. A.; Kapustin, V. N.; Kopeilin, V. M.; Gillette, D. A.; Bodhaine, B. A.; Atmos. Environ. 1993, 27A, 2527.
- 49. Penner, J. E.; Charlson, R. J.; Hales, J.M.; Laulainen, N.; Leifer, R.; Novakov, T.; Ogren, J.; Radke, L. F.; Schwartz, S. E.; Travis, L.; Quantifying and minimizing uncertainty of climate forcing by anthropogenic aerosols, U.S. Department of Energy, 1993.
- 50. Sokolik, I. N.; Andronova, A. V.; Johnson, T. C.; Atmos. Environ. 1993, 27A, 2495.
- 51. Jaenicke, R. Em Aerosol-Cloud-Climate Interactions; Hobbs, P. V., ed.; Academic Press Inc.: U.K., 1993, p. 1-31.
- 52. Graf, H. F.; Langmann, B.; Feitcher, J.; Chem. Geol. 1998, 147, 131.
- 54. Graf, H. F.; Feitcher, J.; Langmann, B.; J. Geophys. Res. 1997, 102, 10727.
- 55. Salstein, D. Em Composition, Chemistry, and Climate of the Atmosphere; Sigh, H. B., ed.; Van Nostrand Reinhold: U.S.A., 1995, p. 19-170.
- 56. Monahan, E. C.; Spiel, D. E.; Davidson, K. L. Em Oceanic whitecaps and their role in air-sea exchange processes; Monaham, E. C.; Niocaill, G. M., eds.; D. Reidel Publishing: Dordrecht, Holland, 1986, p. 167-174.
- 57. Ricard, V.; Jaffrezo, J. L.; Kerminen, V. M.; Hillamo, R. E.; Sillanpaa, M.; Ruellan, S; Liousse, C.; Cachier, H.; J. Geophys. Res. Atmos. 2002, 107, 10.
- 58. Piazzola, J.; Forget, P.; Despiau, S.; Ann. Geophys. Germany 2002, 20, 121.
- 59. Alves, C.; Tese de Doutoramento, Universidade de Aveiro, Portugal, 2001.
- 60. Quinn, P. K.; Coffman, V. N.; Kapustin, T. S.; Covert, D. S.; J. Geophys. Res. 1998, 103, 16547.
- 61. Russell, P. B.; Redemann, J.; Schmid, B; Bergstrom, R. W.; Livingston, J. M.; McIntosh, D. M.; Ramirez, S. A.; Hartley, S.; Hobbs, P. V.; Quinn, P. K.; Carrico, C. M.; Rood, M. J.; Ostrom, E.; Noone, K. J.; von Hoyningen-Huene, W.; Remer, L.; J. Atmos. Sci. 2002, 59, 609.
- 62. Murphy, D. M.; Anderson, J. R.; Quinn, P. K.; McInnes, L. M.; Brechtel, F. J.; Kreidenweis, S. M.; Middlebrook, A. M.; Posfai, M.; Thomson, D. S.; Buseck, P. R.; Nature 1998, 392, 62.
- 63. Tegen, I.; Hollrig, P.; Chin, M.; Fung, D.; Jacob, D.; Penner, J. E.; J. Geophys. Res. - Atmos. 1997, 102, 23895.
- 64. Erickson, D. J. III; Duce, R. A.; J. Geophys. Res. 1988, 93, 14079.
- 65. Renoux, A.; Pollut. Atmos. 1990, 125, 10.
- 66. Matthias-Maser, S.; Jaenicke, R.; Atmos. Res. 1995, 39, 279.
- 67. Simoneit, B. R. T.; Cardoso, J. N.; Robinson, N.; Chemosphere 1990, 21, 1285.
- 68. Matthias-Maser, S. Em Atmospheric Particles; Harrison, R. M.; Briecken, R. V., eds.; John Wiley & Sons: England, 1998.
- 69. Griffin, R. J.; Cocker, D. R.; Seinfeld, J. H.; Dabdud, D.; Geophys. Res. Lett. 1999, 26, 2721.
- 70. Penner, J. E.; Chuang, C. C.; Grant, K.; Climate change and radiative forcing by anthropogenic aerosols: A review of research during the last five years, La Jolla International School of Science, The Institute for Advanced Physics Studies, La Jolla, CA: U.S.A., 1999.
- 71. Cerqueira, M. M. A.; Tese de Doutoramento, Universidade de Aveiro, Portugal, 1999.
- 73. Charlson, R. J.; Langner, J.; Rodhe, H.; Leovy, C. B.; Warren, S. G.; Tellus 1991, 43AB, 152.
- 74. Charlson, R. J.; Schwartz, S. E.; Hales, J. M.; Cess, R. D.; Coakley, J. A.; Hansen, J. E.; Hoffmann, D. J.; Science 1992, 255, 423.
- 75. Lee, G.; Merrill, J. T.; Huebert, B. J.; J. Geophys. Res. 1994, 99, 12821.
- 76. Stockwell, W. R.; Watson, J. G.; Abstr. Pap. Am. Chem. - Phys. 2002, Part 2, S224.
- 77. Adams, P. J.; Seinfeld, J. H.; Koch, D. M.; J. Geophys. Res. 1999, 104, 13791.
- 78. Bramley, P. M. Em Plant biochemistry; Dey, P. M.; Harborne, J. B., eds.; Academic Press: Bristol, 1997.
- 79. Tsigaridis, K.; Kanakidou, M.; Atmos. Environ 2002, 36, 3137.
- 80. Tsigaridis, K.; Kanakidou, M.; Atmos. Chem. Phys 2003, 3, 1849.
- 81. Spirig, C.; Guenther, A.; Greenberg, J. P.; Calanca P.; Tarvainen, V.; Atmos. Chem. Phys 2004, 4, 215.
- 82. Zenchelsky, S.; Youssefi, M.; Rev. Geophys. Space Phys. 1979, 17, 459.
- 83. Pandis, S. H.; Harley, R. A.; Cass, G. R.; Seinfeld, J. H.; atmos. Environ. 1992, 26A, 2269
- 84. Pankow, J. F.; Atmos. Environ. 1987, 21, 2275.
- 85. Guenther, A.; Hewitt, C.; Erickson, D.; Fall, R.; Geron, C.; Graedel, T.; Harley, P.; Klinger, L.; Lerdau, M.; McKay, W.; Pierce, T.; Scholes, B.; Steinbrecker, R.; Tallamjau, R.; Taylor, J.; Zimmerman, P.; J. Geophys. Res. Atmos. 1995, 100, 8873.
- 86. Artaxo, P.; Swietlicki, E.; Zhou, J.; Hansson, H.-C.; Maenhaut, W.; Claeys, M.; Andreae, M. O.; Ström, J.; Martins, J. V.; Yamasoe, M. A.; van Grieken, R.; Eos Trans. AGU 1998, 79, F155.
- 87. Roberts, G.; Andreae, M.O.; Maenhaut, W.; Artaxo, P.; Martins, J.V.; Zhou, J.; Swietlicki, E.; Eos Trans. AGU 1998, 79, F159.
- 88. Graham, B.; Guyon, P.; Taylor, P. E.; Artaxo, P.; Maenhaut, W.; Glovsky, M. M.; Flagan, R. C.; Andreae, M. O.; J. Geophys- Res. Atmos 2003, 108, 4766.
- 89. Kanakidou, M.; Tsigaridis, K.; Dentener, F. J.; Crutzen, P. J.; J. Geophys. Res. 2000, 105, 9243.
- 90. Odum, J. R.; Hoffmann, T.; Bowman, F.; Collins, D.; Flagan, R. C.; Seinfeld, J. H.; Environ. Sci. Technol. 1996, 30, 2580.
- 91. Piccot, S. D.; Watson, J. J.; Jones, J. W.; J. Geophys. Res. 1992, 97, 9897.
- 92. Pandis, S. N.; Paulson, S. E.; Seinfeld, J. H.; Flagan, R. C.; Atmos. Environ. 1991, 27A, 997.
- 93. Schauer, J. J.; Rogge, W. F.; Hildemann, L. M.; Mazurek, M. A.; Cass, G. R.; Simoneit, B. R. T.; Atmos. Environ. 1996, 30, 3837.
- 94. Nunes, F. M. N.; Pereira, P. A. de P.; de Andrade, J. B.; Quim. Nova 2000, 23, 794.
- 95. Sicre, M. A.; Peltzer, E. T.; Atmos. Environ 2004, 38, 1615.
- 96. Pio, C.; Alves, C.; Duarte, A.; Atmos. Environ., 2001, 35, 389.
- 97. Alves, C.; Carvalho, A.; Pio, C.; J. Geophys. Res. Atmos 2002, 107, 8345.
- 98. Nolte, C. G.; Schauer, J. J.; Cass, G. R.; Simoneit, B. R. T.; Environ. Sci. Technol 2002, 36, 4273.
- 99. Braun, A.; Shah, N.; Huggins, F. E.; Huffman, G. P.; Wirick, S.; Jacobsen, C.; Kelly, K.; Sarofim, A. F.; Fuel 2004, 86, 997.
- 100. Fine, P. M.; Chakrabarti, B.; Krudysz, M.; Schauer, J. J.; Sioutas, C.; Environ. Sci. Technol 2004, 38, 1296.
- 101. Peterson, R. E.; Tyler B. J.; Atmos. Environ 2002, 36, 6041.
- 102. Santos, C. Y. M.; Azevedo, D. D.; Neto, F. R. D.; Atmos. Environ 2004, 38, 1247.
- 103. Azevedo, D. D.; Santos, C. Y. M.; Neto, F. R. D.; Atmos. Environ 2002, 36, 2383.
- 104. Marty, J.-C.; Saliot, A.; Nature 1982, 298, 144.
- 105. Cass, G. R.; Trends Anal. Chem. 1998, 17, 356.
- 106. Gogou, A.; Apostolaki, M.; Stephanou, E.; J. Chromatogr., A 1998, 799, 215.
- 107. Gogou, A.; Stephanou, E. G.; Stratigakis, N.; Grimalt, J. O.; Simo, R.; Aceves, M.; Albaiges, J.; Atmos. Environ. 1994, 28, 1301.
- 108. Li, C. K.; Kamens, R. M.; Atmos. Environ. 1993, 27A, 523.
- 109. Simoneit, B. R. T.; J. Atmos. Chem. 1989, 8, 251.
- 110. Simoneit, B. R. T.; Environ. Sci. Pollut. Res. 1999, 6, 159.
- 111. Simoneit, B. R. T.; Mazurek, M. A.; Aerosol Sci. Technol. 1989, 10, 267.
- 112. Standley, L.; Simoneit, B. R. T.; J. Atmos. Chem. 1994, 18, 1.
- 113. Simoneit, B. R. T.; Schauer, J. J.; Nolte, C. G.; Oros, D. R.; Elias, V. O.; Fraser, M. P.; Rogge, W. F.; Cass, G. R.; Atmos. Environ. 1999, 33, 173.
- 114. Fraser, M. P.; Yue, Z. W.; Buzcu, B.; Atmos. Environ 2003, 37, 2117.
- 115. Simoneit, B. R. T.; Mazurek, M. A.; Atmos. Environ. 1982, 16, 2139.
- 116. Monteiro, S.; Tese de Mestrado, Universidade de Aveiro, Portugal, 1998.
- 117. Gijzen, M.; Lewinsohn, E.; Savage, T.;Croteau, R. Em Bioactive Volatile Compounds from Plants; Teranishi, R.; Buttery, R.; Sugisawa, H., eds.; ACS Symposium Series, American Chemical Society: Washington, 1993.
- 118. Simoneit, B. R. T.; Cox, R. E.; Standley, L. J.; Atmos. Environ. 1988, 22, 983.
- 119. Pio, C.; Alves, C.; Duarte, A.; Atmos. Environ. 2001, 35, 1365.
- 120. Ikan, R.; Natural Products, 2nd ed., Academic Press: San Diogo, California, 1991.
- 121. Kozlowski, T. T.; Pallardy, S.; Physiology of woody plants, 2nd ed., Academic Press: USA, 1997.
- 122. Simoneit, B. R. T.; Sheng, G.; Chen, X.; Fu, J.; Zhang, J.; Xu, Y.; Atmos. Environ. 1991, 25A, 2111.
- 123. Simoneit, B. R. T.; Crisp, P. T.; Mazurek, M. A.; Standley, L. J.; Environ. Int. 1991, 17, 405.
- 124. Simoneit, B. R. T.; Atmos. Environ. 1984, 18, 51.
- 125. Santos, C. Y. M.; Azevedo, D. A.; Neto, F. R. A.; Atmos. Environ 2002, 36, 3009.
- 126. Nolte, C. G.; Schauer, J. J.; Cass, G. N.; Simoneit, B. R. T.; Environ. Sci. Technol. 1999, 33, 3313.
- 127. Rogge, W. F.; Hildemann, L. M.; Mazurek, M. A.; Cass, G. R.; Simoneit, B. R. T.; Environ. Sci. Technol 1991, 25, 1112.
- 128. Rogge, W. F.; Hildemann, L. M.; Mazurek, M. A.; Cass, G. R.; Simoneit, B. R. T.; Environ. Sci. Technol. 1993 , 27, 636.
- 129. Zheng, M.; Wan, T. S. M.; Fang, M.; Wang, F.; Atmos. Environ. 1997, 31, 227.
- 130. Fraser, M. P.; Cass, G. R.; Simoneit, B. R. T.; Atmos. Environ. 1999, 33, 2715.
- 131. Kavouras, I. G.; Stratigakis, N.; Stephanou, E. G.; Environ. Sci. Technol. 1998, 32, 1369.
Datas de Publicação
-
Publicação nesta coleção
23 Set 2005 -
Data do Fascículo
Out 2005
Histórico
-
Aceito
05 Jan 2005 -
Recebido
26 Maio 2004