Abstract
A solid phase extraction procedure using Amberlite XAD-1180/Pyrocatechol violet (PV) chelating resin for the determination of iron and lead ions in various environmental samples was established. The procedure is based on the sorption of lead(II) and iron(III) ions onto the resin at pH 9, followed by elution with 1 mol/L HNO3 and determination by flame atomic absorption spectrometry. The influence of alkaline, earth alkaline and some transition metals, as interferents, are discussed. The recoveries for the spiked analytes were greater than 95%. The detection limits for lead and iron by FAAS were 0.37 µg/L and 0.20 µg/L, respectively. Validation of the method described here was performed by using three certified reference materials (SRM 1515 Apple Leaves, SRM 2711 Montana Soil and NRCC-SLRS-4 Riverine Water). The procedure was successfully applied to natural waters and human hair.
XAD-1180/PV chelating resin; preconcentration; separation
ARTIGO
Solid phase extraction of iron and lead in environmental matrices on amberlite xad-1180/pv
Mustafa SoylakI,* * e-mail: soylak@erciyes.edu.tr ; Mustafa TuzenII; Ibrahim NarinIII
IErciyes University, Faculty of Art and Science, Department of Chemistry, 38039 Kayseri - Turkey
IIGaziosmanpasa University, Faculty of Science and Arts, Chemistry Department, 60250, Tokat - Turkey
IIIErciyes University, Faculty of Pharmacy, 38039 Kayseri, Turkey
ABSTRACT
A solid phase extraction procedure using Amberlite XAD-1180/Pyrocatechol violet (PV) chelating resin for the determination of iron and lead ions in various environmental samples was established. The procedure is based on the sorption of lead(II) and iron(III) ions onto the resin at pH 9, followed by elution with 1 mol/L HNO3 and determination by flame atomic absorption spectrometry. The influence of alkaline, earth alkaline and some transition metals, as interferents, are discussed. The recoveries for the spiked analytes were greater than 95%. The detection limits for lead and iron by FAAS were 0.37 µg/L and 0.20 µg/L, respectively. Validation of the method described here was performed by using three certified reference materials (SRM 1515 Apple Leaves, SRM 2711 Montana Soil and NRCC-SLRS-4 Riverine Water). The procedure was successfully applied to natural waters and human hair.
Keywords: XAD-1180/PV chelating resin; preconcentration; separation.
INTRODUCTION
The need for highly reliable methods and techniques for the determination of trace heavy transition metals has been recognised in analytical chemistry and environmental science. For that purpose, it is necessary to utilize either very sensitive instrumental technique or enrichment/separation methods for the quantification of low concentrations of metals. Because of the higher costs of the instrumental techniques like ICP-MS, generally the separation preconcentration techniques are preferred1,2. Preconcentrative separation techniques including coprecipitation3,4, solvent extraction5,6, membrane filtration7, cloud point extraction8 and ion exchange9 have been used for the traces heavy metal ions in environmental samples.
Solid phase extraction has also emerged as a powerful tool for separation/enrichment of heavy metal ions10,11. Solid phase extraction based on adsorption is a recently used method that compensates solvent extraction disadvantages. The solid phase extraction technique reduces organic solvent usage and exposure, disposal costs, and extraction time for sample preparation. Solid phase extraction allows adsorbing chemical species directly onto a solid phase as an adsorbent provides an effective separation. The relatively high concentration factor and the ability of treating large volume samples compared to the other separation-preconcentration techniques are other advantages of the solid phase extraction. Various solid phase extraction materials including activated carbon12-15, silica disks16-18. Amberlite XAD resins19-21, benzophenone22, naphthalene23 and styrene-ethylene glycol dimethacrylate polymer24 have been used for the preconcentration of traces metal ions as their chelates or complexes. Because both sorption capacities and sorption selectivity of chelating resins are superior to those of ion exchangers and adsorbents in trace levels, the synthesis and characterization of the chelating new resins with the reaction of an Amberlite XAD adsorption resin and a suitable chelating agent in the laboratory conditions are popular in last decade25-31.
In the present work, the separation-preconcentration of iron(III) and lead(II) ions in the various environmental samples by using solid phase extraction on the XAD-1180/ Pyrocatechol violet (PV) chelating resin were performed.
EXPERIMENTAL PART
Apparatus
A Perkin Elmer AAnalyst 700 atomic absorption spectrometer with deuterium background corrector was used in this study. The operating parameters for working elements were set as recommended by the manufacturer. A 10 cm long slot-burner head, a lamp and an air-acetylene flame were used.
Milestone Ethos D closed vessel microwave system (maximum pressure 1450 psi, maximum temperature 300 °C) was used. Mechanical shaker having speed control was used for batch experiments. A pH meter, Sartorius pp-15 Model glass-electrode was employed for measuring pH values in the aqueous phase.
Reagents
All chemicals were reagent-grade and all solutions were prepared in distilled- deionised water. Stock solutions were prepared from appropriate amounts of respective nitrates as 1000 mg/L, solutions in 0.01 mol/L HNO3 and further diluted daily for obtaining reference and working solutions prior to use. The calibration standards were not submitted to the preconcentration procedure.
Sodium phosphate buffer (0.1 mol/L) was prepared by adding an appropriate amount of phosphoric acid to sodium dihydrogen phosphate solution to result in a solution of pH 2. Ammonium acetate buffers (0.1 mol/L) were prepared by adding an appropriate amount of acetic acid to ammonium acetate solutions to result in solutions of pH 4-6. For pH 7, sodium borate (0.1 mol/L) buffer solution was used. Ammonium chloride buffer solutions (0.1 mol/L) were prepared by adding an appropriate amount of ammonia to ammonium chloride solutions to result in solutions of pH 8-10.
The resin used in the present work was XAD-1180/PV that was synthesed and characterized in our previous work31. A column containing 600 mg of chelating resin in water suspension was 10 cm long and 1.0 cm in diameter. The bed height in the column was approximately 2.0 cm. The column was preconditioned with 10-15 mL 1 mol/L HNO3 prior to percolation of the each solution. The standard reference materials used in the experimental studies were SRM 1515 Apple Leaves, SRM 2711 Montana Soil and NRCC-SLRS-4 Riverine Water.
Test procedure
The method was tested with model solutions prior to the determination of trace metal ions. For the metal determinations 40-60 mL of solution containing 10 mg of iron (III) and 20 µg of lead (II) ions was added 10 mL of buffer solution between pH 2-10. The XAD-1180/PV column was preconditioned by passing buffer solution. The buffered metal solution was passed the column at a flow rate of 5 mL/min. After passing of this solution, the column was rinsed with twice 10 mL of water. The adsorbed metals on the XAD-1180/PV column were eluted with 8-10 mL portion of 1 mol/L HNO3. The eluent was analyzed for the determinations of metal concentrations by flame AAS.
Procedures for real samples
A 100 mg amount of apple leaves reference material (SRM 1515) sample was decomposed with the mixture of 4 mL of concentrated HNO3 and 2 mL of H2O2 in Ethos microwave system. Digestion conditions for microwave system were applied as 2 min for 250 W, 2 min for 0 W, 6 min for 250 W, 5 min for 400 W, 8 min for 550 W, vent: 8 min, respectively. All sample solutions were clear. The volume of the samples was completed approximately 25 mL with distilled water. Then this solution was analysed by using the preconcentration procedure described above. The analytes were eluted with 5 mL of 1 mol/L HNO3 and analyzed by AAS.
A 100 mg amount of SRM 2711 Montana Soil was decomposed with the mixture of 4 mL of concentrated HNO3, 1 mL of concentrated H2SO4 and 1 mL of concentrated HClO4 in microwave system. Digestion conditions for microwave system were applied as 6 min for 250 W, 6 min for 400 W, 6 min for 550 W, 6 min for 250 W, vent: 8 min., respectively. The volume of the samples was completed approximately 50 mL with distilled water. Then the separation-preconcentration procedure given above was applied to the final solutions. The analytes were eluted with 5 mL of 1 mol/L HNO3. The same procedure was applied to the blank solution.
One gram of human hair sample was decomposed with the mixture of 4 mL of concentrated HNO3 and 2 mL of H2O2 in Ethos microwave system. Digestion conditions for microwave system were applied as 2 min for 250 W, 2 min for 0 W, 6 min for 250 W, 5 min for 400 W, 8 min for 550 W, vent: 8 min, respectively. All sample solutions were clear. The volume of the samples was completed approximately 25 mL with distilled water. Then this solution was analysed by using the preconcentration procedure described above. The analytes were eluted with 5 mL of 1 mol/L HNO3 and analyzed by AAS.
The water samples analyzed including NRCC-SLRS-4 Riverine Water were filtered through a cellulose membrane filter (Millipore) of 0.45 µm pore size. The pHs of the samples were adjusted to 9. The sample was passed through the column. The analytes adsorbed on column were eluted with 1 mol/L HNO3. The levels of the analyte ions in the samples were determined by AAS.
RESULTS AND DISCUSSION
Influences of pH on the recoveries
The effects of the pH of the test solutions on recoveries of the analyte ions on the chelating resin were investigated in the pH range of 2-10 by using different buffer solutions pointed in the Experimental section. The results are depicted in Figure 1. The recovery values for analytes ions at the acidic pH's were not quantitative. Iron (III) and lead (II) ions were quantitatively recovered on Amberlite XAD-1180/PV chelating resin in the pH range of 8-9. For preconcentration-separation of iron, lead and other metals from real samples by solid phase extraction, at pH 8 or higher pHs is possible32-35. All further work was carried out at pH 9 by using ammonia/ammonium chloride buffer solution. The volume of the relevant buffer added (10 mL) had no effect on the recoveries.
Eluent type and eluent volume
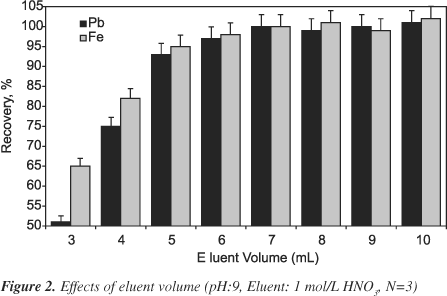
Various eluent solutions for desorption of Pb(II) and Fe(III) ions from the XAD-1180/PV column were evaluated at 5 ml/min flow rate of the each eluent. Both ions were quantitatively recovered (>95%) with 1 mol/L HNO3, 2 mol/L HNO3, 1 mol/L HCl and 2 mol/L HCl. The volumes of 1 mol/L HNO3 as eluent on the recoveries of analytes were also investigated at the eluent volume range of 3-10 mL. This work was performed at pH 9. The results were given in Figure 2. The recoveries of analytes were quantitative in the eluent volume range of 5-10 mL. In all subsequent works, 5.0 or 10.0 mL of 1 mol/L HNO3 was used as eluent.
The column can be reused after regenerated with 10 mL 1 mol/L HNO3 and 10 mL distilled water, respectively, and relatively stable up to 50 runs without appreciable loss of ligating sites. The chelating resin could be stored for at least three months and used repeatedly without any appreciable amount of reagent being lost.
Flow rates
The influences of the flow rates of the sample and eluent solution for the recoveries of analytes were investigated in the flow rate ranges of 1-10 mL/min. The recovery values for analyte ions were quantitative in all the working range of sample flow rates. The quantitative recovery values for eluent solution were obtained at the flow rate range of 1-7 mL/min for lead(II) and 1-8 mL/min for iron. All further studies were performed at the sample and eluent flow rates of 5.0 mL/min.
Influences of resin amounts
Because of the amounts of adsorbent to the glass column is an important point for the recoveries of analytes on the solid phase extraction studies26-28, the effects of amounts of XAD-1180/PV resin were examined by preparing columns filled resin amount range of 300-1000 mg. Then the test procedure given in Experimental was applied. The results were depicted in Figure 3. Pb(II) and Fe(III) ions were quantitatively retained on the resin at the resin amounts of 500-700 mg. In all further studies, the short glass column was filled with 600 mg of resin. If the amount of resin is more than 700 mg, the retained metals cannot be eluted completely with 10 mL of 1 mol/L HNO3.
Sample volume
The effect of the sample solution volume on the metal sorption was studied by passing 25-1000 mL volumes through the column at a 5 mL /min flow rate. The adsorption of the metal ions with 600 mg of chelating resin was not affected by sample volume below 500 mL. Above 500 mL of sample solution, the percent sorption decreased for the analytes (Figure 4). The recoveries of analytes decrease probably due to the excess analytes loaded over the column capacity with increasing sample volume above 500 mL . In the present study 500 mL of sample solution was adopted for the preconcentration of the investigated ions, the adsorbed metals can be eluted with 5 mL of 1 mol/L HNO3 and a preconcentration factor of 100 is achieved.
Matrix effects
The possible interferences of some alkaline and earth alkaline salts and some transition metals as sulfate salts on the retentions of lead and iron ions on XAD-1180/PV resin were also investigated. The quantitative recovery values were obtained for analyte ions in 0.05 mol/L Na3PO4, 0.25 mol/L NaF, 0.90 mol/L NaCl, 0.11 mol/L Na2SO4, 0.14 mol/L KNO3, 0.50 mol/L KCl, 0.40 mol/L CaCl2 and 0.50 mol/L MgCl2. Quantitative recovery values were obtained for lead and iron ions at 0.001 mol/L of Cu2+, Cr3+, Co2+, Mn2+, Ni2+, Al3+, Cd2+ and Zn2+ ions.
Analytical performance of the method
The correctness of results was verified by analyzing the concentration after the addition of known amounts of analytes into a river and an artificial sea water samples. The results were summarized in Table 1. Satisfactory recoveries were obtained for spiked analyte ions. The recoveries were higher than 95%, thus confirming the accuracy of the presented procedure.
The detection limits for the analyte ions were obtained from the signals of twenty blank samples and the slope of the calibration curve. The detection limits were defined as the sample concentration giving signals equal to three times the standard deviation of blank signal. The detection limits are: 0.37 µg/L for Pb(II) and 0.20 µg/L for Fe(III).
Application of the real samples
The presented preconcentration procedure was applied to three different reference materials (SRM 1515 Apple Leaves, SRM 2711 Montana Soil and NRCC-SLRS-4 Riverine Water) for the determination of contents of lead(II) and iron(III) ions. The certified and observed values for SRM samples were given in Table 2. The results are in good agreement with the certified values for the investigated analyte ions.
The proposed method has been also applied to natural water samples including a tap water, a river water, a seawater and a bottled mineral water the enrichment and flame atomic absorption spectrometric determinations of lead and iron ions. The results are given in Table 3.
The proposed preconcentration/separation method has been combined with the microwave assisted digested human hair sample. The levels of lead and iron (N=5) were 1.2±0.1 µg/g and 85±5 µg/g, respectively.
Comparative data from some recent papers on solid phase extraction of traces metal ions on the various adsorbents for the figure of the merits are summarized in Table 4. The method presented in this study is most promising for the analyte ions as the preconcentration factor is 100. The preconcentration factor achieved with presented procedure is superior to solid phase extraction method given in Table 4.
CONCLUSION
The proposed method provides a simple, fairly rapid and reliable technique for preconcentration and determination of iron and lead from the environmental samples. The matrix effects with the method were reasonably tolerable. The elution was easily performed with 1 mol/L HNO3. The XAD-1180/PV resin is quite durable with recycling time >50 cycles, without any major change in its quantitative metal recovery nature. The good features of the proposed method showed that it's a convenient and low cost one. Also the method is relatively rapid as compared with previously reported procedures for the enrichment of traces metal ions. The technique can be applied to environmental samples without the risk of contamination. Results for three reference standard materials confirmed the reliability of the procedures.
ACKNOWLEDGMENT
The authors also would like to thank the Scientific and Technical Research Council of Turkey (TUBITAK) for financial helps (Project No: TBAG-AY/408 (105T010)). The authors are grateful for the financial support of the Unit of the Scientific Research Projects of Gaziosmanpasa University and the Unit of the Scientific Research Projects of Erciyes University.
Recebido em 10/12/04; aceito em 12/8/05; publicado na web em 20/1/06
- 1. Pyrzynska, K.; Chem. Anal. 2003, 48, 781.
- 2. Miró, M.; Estela, J. M.; Cerdà, V.; Talanta 2004, 63, 201.
- 3. Krishna, P. G.; Gladis, J. M.; Rambabu, U.; Rao, T. P.; Naidu, G. R. K.; Talanta 2004, 63,541.
- 4. Liang, P.; Shi, T.; Li, J.; Int. J. Environ. Anal. Chem. 2004, 84, 315.
- 5. Saito, K.; Taninaka, J.; Murakami, S.; Muromatsu, A.; Talanta 1998, 46, 1187.
- 6. Tuzen, M.; Aydemir, E.; Sari, H.; Fresenius Environ. Bull. 2002, 11, 202.
- 7. Soylak, M.; Erdogan, N. D.; Elci, L.; Chin. J. Chem. Soc. 2004, 51, 703.
- 8. Paleologos, E. K.; Stalikas, C. D.; Tzouwara-Karayanni, S. M.; Karayannis, M. I.; Anal. Chim. Acta 2001, 43, 649.
- 9. Bazzi, A.; Kreuz, B.; Wuokila, J.; Maqboul, A.; J. Chem. Educ. 2005, 82, 435.
- 10. Abdullin, I. F.; Turova, E. N.; Budnikov, G. K.; J. Anal. Chem. 2000, 55, 567.
- 11. Kobayashi, J.; Bunseki Kagaku 1994, 43, 727.
- 12. Farias, G. M.; Cerutti, S.; Gasquez, J. A.; Olsina, R. A.; Martinez, L. D.; At. Spectrosc. 2003, 24, 213.
- 13. Daorattanachai, P.; Unob, F.; Imyim, A.; Talanta 2005, 67, 59.
- 14. Ensafi, A. A.; Khayamian, T.; Karbasi, M. H.; Anal. Sci. 2003, 19, 953.
- 15. Karami, H.; Mousavi, M. F.; Yamini, Y.; Shamsipur, M.; Anal. Chim. Acta 2004, 509, 89.
- 16. Shemirani, F.; Mirroshandel, A. A.; Niasari, M. S.; Kozani, R. R.; J. Anal. Chem. 2004, 59, 228.
- 17. Ghiasvand, A. R.; Ghaderi, R.; Kakanejadifard, A.; Talanta 2004, 62, 287.
- 18. Sharma, R. K.; Mittal, S.; Koel, M.; Crit. Rev. Anal. Chem. 2003, 33, 183.
- 19. Venkatesh, G.; Singh, A. K.; Talanta 2005, 67, 187.
- 20. Soylak, M.; Karatepe, A. U.; Elci, L.; Dogan, M.; Turk. J. Chem. 2003, 27, 235.
- 21. Elci, L.; Anal. Lett. 1993, 26, 1025.
- 22. Taher, M. A.; Indian J. Chem. Technol. 2003, 10, 661.
- 23. Pourreza, N.; Mousavi, H.Z.; Anal. Chim. Acta, 2004, 503, 279.
- 24. Praveen, R. S.; Daniel, S.; Rao, T.P.; Talanta 2005, 66, 513.
- 25. Guo, Y.; Din, B.; Liu, Y.; Chang, X.; Meng, S.; Liu, J.; Talanta 2004, 62, 207.
- 26. Guo, Y.; Din, B.; Liu, Y.; Chang, X.; Meng, S.; Tian, M.; Anal. Chim. Acta 2004, 504, 319.
- 27. Singh, D. K.; Gupta, R.; Chem. Anal. 2003, 48, 797.
- 28. Prabhakaran, D.; Subramanian, M.S.; Talanta 2003, 59, 1227.
- 29. Narin, I.; Soylak, M.; Kayakirilmaz, K.; Elci, L.; Dogan, M.; Anal. Lett. 2003, 36, 641.
- 30. Gladis, J. M.; Rao, T. P.; Anal. Bioanal. Chem. 2002, 373, 867.
- 31. Narin, I.; Tuzen, M.; Soylak, M.; Talanta 2004, 63, 411.
- 32. Cerutti, S.; Orsi, R. F.; Gasquez, J. A.; Olsina, R. A.; Martinez, L. D.; J. Trace Microprobe Technol 2003, 21, 421.
- 33. da Silva, E. L.; Martins, A. O.; Valentini, A.; de Fávere, V. T.; Carasek, E.; Talanta 2004, 64, 181.
- 34. Vidal, M. T.; Pascual-Marti, M. C.; Salvador, A.; Llabata, C.; Microchem. J 2002, 72, 221.
- 35. Sarzanini, C.; Abollino, O.; Mentasti, E.; Anal. Chim. Acta 2001, 435, 343.
- 36. Soylak, M.; Narin, I.; Bezerra, M.D.A.; Ferreira, S.L.C.; Talanta 2005, 65, 895.
- 37. Roldan, P. S.; Alcantara, I. L.; Padilha, C. C. F.; Padilha, P. M.; Fuel 2005, 84, 305.
- 38. Korn, M. D. A.; Santos, A. D.; Jaeger, H. V.; Silva, N. M. S.; Costa, A. C. S.; J. Braz. Chem. Soc. 2004, 15, 212.
- 39. Santos, A. D.; Korn, M. D. A.; Jaeger, H. V.; Silva, N. M. S.; Costa, A. C. S.; Quim. Nova 2002, 25, 1086.
- 40. Cassella, R. J.; J. Environ. Monit. 2002, 4, 522.
- 41. Sant'Ana, O. D.; Jesuino, L. S.; Cassella, R. J.; Carvalho, M. S.; Santelli, R. E.; J. Braz. Chem. Soc. 2004, 15, 96.
- 42. Bispo, M. S.; Korn, M. D. A.; Morte, E. S. D.; Teixeira, L. S. G.; Spectrochim. Acta, Part B 2002, 57, 2175.
- 43. Pereira, M. G.; Pereira-Filho, E. R.; Berndt, H.; Arruda, M. A. Z.; Spectroc. Acta, Part B 2004, 59, 515.
- 44. Maltez, H. F.; Melo, L. F. C.; de Queiroz, S. C. D.; Jardim, I. C. S. F.; Curtius, A. J.; Carasek, E.; Microchim. Acta 2004, 144, 17.
- 45. Giacomelli, M. B. O.; Ganzarolli, E. M.; Curtius, A. J.; Spectrochim. Acta, Part B 2000, 55, 525.
- 46. Ferreira, S. L. C.; dos Santos, W. N. L.; Bezerra, M. A.; Lemos, V. A.; Bosque-Sendra, J. M.; Anal. Bioanal. Chem. 2003, 375, 443.
- 47. Ferreira, S. L. C.; Lemos, V. A.; Santelli, R. E.; Ganzarolli, E.; Curtius, A. J.; Microchem J. 2001, 68, 41.
- 48. Tuzen, M.; Parlar, K.; Soylak, M.; J. Hazard. Mater. 2005, 121, 79.
Publication Dates
-
Publication in this collection
03 Apr 2006 -
Date of issue
Apr 2006
History
-
Received
10 Dec 2004 -
Accepted
12 Aug 2005