Resumo
Here we present an overview of electroluminescent devices that use conjugated polymers as the active media. The principal components of the devices are described and we show some examples of conjugated polymers and copolymers usually employed in polymeric light emitting devices (PLED). Some aspects of the photo and electroluminescence properties as well as of the energy transfer processes are discussed. As an example, we present some of the photophysical properties of poly(fluorene)s, a class of conjugated polymers with blue emission.
polymer; photoluminescence; electroluminescence
polymer; photoluminescence; electroluminescence
REVISÃO
Dispositivos poliméricos eletroluminescentes
Polymeric light emitting devices
Hueder P. M. de OliveiraI; Rafael F. CossielloI; Teresa D. Z. AtvarsI, * * e-mail: tatvars@iqm.unicamp.br ; Leni AkcelrudII
IInstituto de Química, Universidade Estadual de Campinas, CP 6154, 13084-971 Campinas - SP
IIDepartamento de Química, Universidade Federal do Paraná, CP 19081, 81531-990 Curitiba - PR
ABSTRACT
Here we present an overview of electroluminescent devices that use conjugated polymers as the active media. The principal components of the devices are described and we show some examples of conjugated polymers and copolymers usually employed in polymeric light emitting devices (PLED). Some aspects of the photo and electroluminescence properties as well as of the energy transfer processes are discussed. As an example, we present some of the photophysical properties of poly(fluorene)s, a class of conjugated polymers with blue emission.
Keywords: polymer; photoluminescence; electroluminescence.
INTRODUÇÃO
Os primeiros trabalhos sobre condução eletrônica em polímeros conjugados datam de 19701,2 sendo que a eletroluminescência (EL) em polímeros foi relatada pela primeira vez em 19893. O uso de polímeros conjugados com um sistema de elétrons p delocalizados em dispositivos optoeletrônicos, conhecidos como OPLEDs ("Organic Polymeric Light Emission Devices"), tem crescido desde então. Na atualidade, monitores coloridos na forma de matriz ativa, fabricados com polímeros orgânicos emissores de luz (OPLEDs) estão na fronteira de serem comercializados. As razões para o sucesso dos OPLEDs devem-se às múltiplas possibilidades de estruturas químicas dos compostos possíveis, às várias possibilidades de formas de montagem dos dispositivos, aliadas à alta performance e ao menor custo de produção4. A tecnologia de exibição não é, entretanto, o único campo em que materiais orgânicos em geral, e poliméricos conjugados em particular, aparecem com um futuro promissor. Entre esses novos campos aparecem as novas tecnologias de células solares5-8, transistores9-11, emissores tipo laser, sistemas de armazenamento e circuitos integrados poliméricos12-17.
O uso de materiais orgânicos, poliméricos ou não, nos vários tipos de dispositivos tem, entre suas principais vantagens, a imensa possibilidade de preparação de estruturas químicas diferentes, além de poderem ser misturados, formando sistemas com outras propriedades. Têm, entretanto, alguns problemas tecnológicos importantes ainda não resolvidos, relacionados com a menor estabilidade química quando comparados com materiais inorgânicos ou organo-metálicos. Entretanto, a possibilidade de mistura física entre componentes permitindo a obtenção de sistemas com diferentes propriedades é, sem dúvida, uma forma atraente e de custo reduzido de preparação de novos materiais. Essa flexibilidade nas formas de preparação de sistemas é particularmente interessante no caso de polímeros eletroluminescentes para os quais pequenas alterações nas rotas de síntese podem levar a materiais com diferentes estruturas e, o fato de apresentarem diferentes estruturas químicas, leva a que os materiais passem a emitir em diferentes regiões do espectro18-21. As diferenças nos processos sintéticos podem, também, envolver reações de copolimerização, permitindo a geração de materiais com propriedades projetadas para fornecer dispositivos mais eficientes11,13-16.
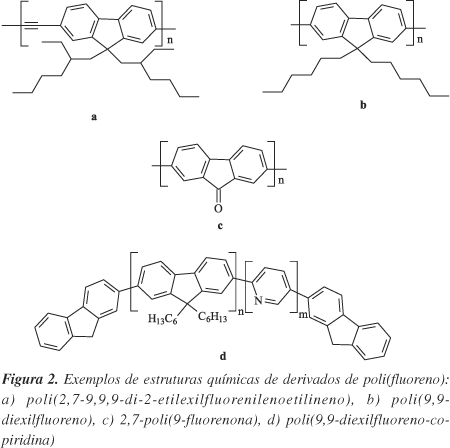
Existem diversos polímeros conjugados sendo empregados como materiais eletroluminescentes. Entre esses, algumas classes vem sendo mais estudadas: poli(p-vinilenofenilenos); poli(fluorenos), poli(N-vinilcarbazóis), poli(tiofenos), polímeros derivados das fenotiazinas, derivados das triazinas, etc18. O poli(fluoreno) é um polímero conjugado, com estrutura química mostrada na Figura 1. Ele e seus derivados, normalmente com substituição na posição 9 do anel central, formam uma classe de polímeros conjugados que emitem na região azul do espectro da radiação eletromagnética. Essa classe de polímeros, alguns dos quais estão mostrados na Figura 2, tem despertado o interesse dos estudiosos por suas propriedades elétricas e ópticas com potencial aplicação para fabricação de dispositivos emissores de luz (LEDs)22-33. Estes polímeros apresentam algumas propriedades interessantes para essa finalidade destacando-se, entre elas, grande estabilidade química, térmica e oxidativa18,34-38; alto rendimento quântico de fluorescência tanto em solução quanto no estado sólido37,39-57; possibilidade de formarem materiais líquido-cristalinos46,58-68 com altos parâmetros de ordem e temperaturas de transição termotrópica na faixa entre 100 a 170 ºC, ocorrendo entre as temperaturas de amolecimento e de decomposição térmica69-72. Dessas propriedades decorrem as potencialidades de aplicações dessa classe de polímeros. Por ex.: as altas estabilidades química e térmica dos poli(fluorenos) refletem-se na grande estabilidade espectral, que são descritas como sendo melhores que poli(p-fenilenovinileno) (PPV)48; a possibilidade de formação de fase líquido-cristalina que permite a fabricação de dispositivo eletroluminescente com emissão de luz altamente polarizada69, pois o ordenamento no cristal leva a uma forte anisotropia dos espectros de absorção e emissão61,70,73 e, conseqüentemente, esses materiais têm um grande potencial de aplicação em monitores de cristal líquido59,71; a cor da emissão37,74-76 depende da estrutura do polímero, bem como das ramificações na posição 9 (Figura 2) da estrutura central18,27-49,58-83, sendo que muitos tipos de estruturas podem ser sintetizadas18.
Entretanto, os polímeros eletroluminescentes apresentam um problema muito comum que é a baixa solubilidade, decorrente do tipo de estrutura química, o que cria dificuldades para preparação dos filmes finos poliméricos e de espessura uniforme, por quaisquer das técnicas usualmente empregadas (deposição por espalhamento centrífugo de soluções, deposição por jato de tinta, formação de filmes por Langmuir-Blodgett (LB) e por auto-montagem9,10,18,22,23,84). Para aumentar a solubilidade dos polímeros conjugados, a estratégia normalmente utilizada é a introdução de substituintes ligados à cadeia principal durante a síntese, sem prejudicar a conjugação da cadeia principal. No caso do poli(fluoreno), a solubilidade pode ser modificada pela funcionalização por diversos tipos de grupos na posição 9 do monômero. Essa funcionalização é feita, em muitos casos, pela adição de grupamentos flexíveis ou substituintes volumosos e, além de alterar a solubilidade, alteram-se também as propriedades físicas, mecânicas e de processamento dos materiais37,50-57,62,85,86. Portanto, as propriedades químicas, físicas e elétricas do poli(fluoreno) e de seus derivados dependem tanto da estrutura química rígida da cadeia principal, que é formada por unidades bifenílicas planares, quanto do tipo de substituição na posição 9 do anel (Figuras 1 e 2)57,85. A substituição na posição 9 reduz interações estéricas na estrutura conjugada principal do poli(fluoreno) sem distorção da cadeia principal, o que não ocorre no caso do PPV. Nesse último caso, as substituições podem levar a uma distorção da cadeia principal, reduzindo o comprimento médio de conjugação, devido ao fato de que no PPV os substituintes estão localizados nas posições orto em relação à cadeia arílica77,87. Portanto, os poli(fluorenos) podem ser considerados como uma nova classe de PPV na qual os pares de anéis de fenilenos estão fechados em um arranjo coplanar pela presença do carbono 9.
Na síntese de polímeros conjugados envolvendo união direta entre unidades aromáticas, como no caso dos poli(fluorenos), poli(p-fenilenos) ou poli(vinilcarbazóis), são usadas rotas clássicas da química orgânica, que utilizam metais como catalisadores, levando à formação de ligações C-C entre anéis aromáticos. As mais empregadas são as rotas de acoplamento de Yamamoto18,84,88 e Suzuki18,89-93 (Figura 3). A reação de Yamamoto envolve o acoplamento de arilas halogenadas por intermédio de reagente contendo níquel, como o bis(ciclooctadieno)níquel (Ni(COD)2). Se os monômeros forem iguais (Arila 1 = Arila 2) a reação de acoplamento fornecerá um homopolímero; se foram diferentes, será formado um copolímero estatístico. A reação de Suzuki promove o acoplamento entre arilas organoboradas e arilas halogenadas, por meio de catalisador contendo paládio. Se as arilas dibromadas e diboronadas forem iguais, o acoplamento fornecerá um homopolímero; se forem diferentes, será obtido um copolímero perfeitamente alternado, porque as arilas dibromadas ou diboronadas não homopolimerizam. É necessário ressaltar que a polimerização ocorre homogeneamente para todos os monômeros, respeitando a estequiometria do catalisador, que se disponibiliza novamente a cada ciclo. Assim como nos poli(fenilenos), as reações de Suzuki e Yamamoto18,84,88-93 têm sido aplicadas à síntese dos poli(fluorenos) gerando um grande número de estruturas, com variação na natureza das arilas. Uma variedade de cromóforos intercalados com o fluoreno tem sido utilizada, tais como fenileno, piridina, naftaleno, antraceno, estilbeno, cianovinileno e tiofeno, possibilitando emissões em todo o espectro visível18.
Propriedades dos dispositivos eletroluminescentes
Eletroluminescência é um processo de emissão gerado através de excitação eletrônica, pela passagem de uma corrente elétrica através do material. Esse processo pode ocorrer em materiais inorgânicos semicondutores, cristais orgânicos e polímeros orgânicos. Os fenômenos relacionados com a participação dos polímeros orgânicos em dispositivos eletroluminescentes são diferentes em vários aspectos quando comparados aos que envolvem a participação dos semicondutores inorgânicos94. Entre essas diferenças poderiam ser destacadas:
a) dificuldade de formação dos pares polarônicos95-98 porque a excitação eletrônica não leva a uma fácil separação de cargas. Essa separação requer uma energia de, aproximadamente 100 meV, muito maior do que comparada os poucos meV necessários para produzir a separação de cargas em um semicondutor inorgânico;
b) o transporte de carga ocorre por "saltos" entre estados localizados produzidos pelos elétrons p, e não através do transporte dentro da banda de condução, como no caso dos semicondutores inorgânicos. Por isso a mobilidade das cargas é mais baixa do que nos semicondutores inorgânicos;
c) a faixa espectral de absorção é relativamente estreita, comparada com o espectro visível da emissão solar e com as bandas produzidas por transições em semicondutores inorgânicos, em geral muito largas;
d) os coeficientes de absortividades molares dos polímeros conjugados são altos, de forma que altas densidades ópticas podem ser conseguidas, nos picos de absorção, em filmes com espessura menor que 100 nm;
e) muitos polímeros conjugados são susceptíveis de degradação na presença de oxigênio e/ou água, o que reduz a vida útil do material e prejudica o desempenho do dispositivo;
f) como semicondutores unidimensionais, a condução dá-se ao longo das ligações conjugadas e, por isso, suas propriedades eletrônicas e ópticas podem ser altamente anisotrópicas. Isto é potencialmente útil para alguns tipos de dispositivos, mas exige que as transferências de energia intercadeias sejam minimizadas, pois essas desativam os estados excitados delocalizados na cadeia principal.
As dificuldades de formação dos pares polarônicos e de transporte de cargas ocorrem pelo fato de que as forças intercadeias são tipicamente de van der Waals, sendo mais fracas que aquelas que ocorrem em cristais inorgânicos e, muito fracas quando comparadas com as ligações químicas covalentes. Além disso, os polímeros conjugados formam, em geral, sólidos amorfos com baixo grau de cristalinidade, o que dificulta a recombinação de cargas. Como conseqüência, os estados eletrônicos são, predominantemente, intracadeia, não se formando uma estrutura típica de bandas similar ao do sólido inorgânico. Como o material é em geral desordenado, ocorre uma baixa mobilidade dos portadores de carga. Do ponto de vista de modelos para descrever tanto os processos de transporte de cargas, quanto para cálculo de eficiência, diversas considerações e adaptações dos modelos desenvolvidos para semicondutores inorgânicos foram necessárias e estão disponíveis na literatura99-101.
Arquitetura dos dispositivos
Existem muitos tipos de montagens que são usadas em dispositivos eletroluminescentes poliméricos, que podem ser classificados como montagem em mono ou multicamada4. Nesse trabalho está detalhada apenas a arquitetura do dispositivo do tipo monocamada (Figura 4) composta por um suporte transparente (vidro ou um polímero inerte) sobre o qual é depositada uma fina camada metálica que constitui o cátodo. O cátodo é, geralmente, um metal com baixo potencial de ionização (alumínio, cálcio ou magnésio) e é responsável pela injeção de elétrons na matriz polimérica. Sobre o cátodo se deposita o filme polimérico emissor, filme esse que tem espessura entre 30 e 300 nm. É de suma importância que essa camada polimérica tenha espessura uniforme e baixa rugosidade superficial. Trabalhos já publicados indicam que os filmes poliméricos de maior eficiência possuem espessura em torno de 30 nm102. À outra face da camada polimérica deve se conectar o ânodo, que se constitui de uma liga metálica de alto potencial de ionização, sendo o responsável pela injeção de cargas positivas ("buracos"). A camada anódica deve ser muito fina e transparente, em geral é utilizado o óxido de estanho e índio (ITO), que por sua vez será depositada sobre um substrato também transparente (vidro ou filme polimérico inerte), por onde a luz será transmitida para o exterior. A escolha do cátodo e do ânodo deve ser compatível com a diferença dos níveis de energia do diagrama de bandas (função trabalho) do polímero eletroluminescente, para um bom desempenho do dispositivo.
Além disso, para serem obtidos dispositivos com bons desempenhos é absolutamente essencial que sejam controladas a morfologia e a espessura de todas as camadas depositadas. A deposição das camadas poliméricas por diferentes técnicas (deposição centrífuga, jato de tinta, automontagem, etc) permite que sejam controlados todos os parâmetros (espessura, uniformidade, rugosidade superficial, defeitos produzidos por tensões, etc) e, com isso, se otimizam as propriedades ópticas e elétricas dos dispositivos. Para cada material polimérico utilizado há necessidade de se adequarem às características das camadas e os tipos de materiais a serem utilizados como cátodo e ânodo.
As cargas positivas são injetadas pelo ITO e as cargas negativas são injetadas pelo eletrodo cátodo. Essas cargas fluem através do filme polimérico emissor em direções opostas formando pares polarônicos (regiões com excesso de cargas negativas e com deficiência das mesmas buracos ou lacunas) que, ao interagirem coulombicamente, se recombinam formando uma espécie eletronicamente excitada, denominada éxciton. O decaimento radiativo do éxciton é a eletroluminescência do dispositivo.
Devido à grande separação dos níveis eletrônicos de energia, os polímeros eletroluminescentes, no geral, apresentam potenciais de oxidação altos e afinidades eletrônicas baixas. Em conseqüência, as transições eletrônicas ocorrem com altas energias e, por isso, no caso dos poli(fluorenos) a emissão é de cor azul. Como no processo de passagem da corrente através do material pela injeção de carga entre ânodo e cátodo nos dispositivos eletroluminescentes poliméricos, os elétrons precisam ser excitados dos estados eletrônicos fundamentais para os excitados, e isso envolve valores altos de energia, dificultando o processo. Para minimizar esse problema e melhorar o desempenho desses dispositivos, acrescenta-se a esses uma camada transportadora de cargas adjacente ao ânodo e uma camada transportadora de elétrons adjacente ao cátodo, ficando entre elas a camada polimérica emissora. Dessa forma o dispositivo passa a ter pelo menos outras três camadas, além dos eletrodos. Deve-se destacar que, também, nesse caso ocorre a necessidade de se otimizar a espessura dessas camadas, realizando-se processos adequados de deposição, sem que a deposição de uma camada altere a morfologia das outras.
Propriedades fotofísicas dos polímeros eletroluminescentes
Já se demonstrou que os dispositivos eletroluminescentes poliméricos podem emitir várias cores, com grande eficiência, brilho e uniformidade4. A cor da emissão depende da diferença de energia entre os estados eletrônicos p-p* do polímero conjugado, e decorre de vários fatores, incluindo-se a estrutura química, o comprimento da conjugação, a morfologia do material no caso de sólidos ou, a concentração da solução.
A estrutura química do material tem forte influência sobre a região espectral da emissão, justamente porque a partir dela se definem os níveis de energia, o potencial de ionização e a afinidade eletrônica. Como já dito, a cor da emissão é controlada pela diferença de energia entre a banda de condução e a de valência, também chamada energia do "gap" (Eg), enquanto que o processo de injeção de carga é controlado pela diferença entre as funções trabalho dos respectivos eletrodos e a afinidade eletrônica (Ea) do cátodo e do potencial de ionização (Ia) do ânodo de injeção18,103. Essa função trabalho é a quantidade de energia necessária para retirar um elétron da banda de valência para o nível do vácuo. A emissão (foto ou eletroluminescente) é caracterizada por uma transição eletrônica entre o LUMO e um orbital molecular preenchido de maior energia (HOMO). Como os dois tipos de emissão se relacionam apenas com os estados eletrônicos do polímero e envolvem os mesmos estados eletrônicos, deve-se esperar que os espectros de foto e de eletroemissão sejam iguais e isso é o que, em geral, é observado. Para polímeros eletroluminescentes, esta diferença de energia está entre 1,4 e 3,3 eV, correspondendo a comprimentos de onda compreendidos entre 890 e 370 nm, ou seja, transições ocorrendo na região visível do espectro eletromagnético. A diferença principal entre os processos de fotoemissão e de eletroemissão está na forma com que o estado eletrônico excitado é gerado. Na fotoemissão, a excitação ocorre pela absorção de um fóton por uma espécie no estado eletrônico fundamental, enquanto que na eletroemissão a excitação ocorre por recombinação dos chamados pares polarônicos, produzindo um éxciton. Como a excitação não está localizada em um único grupo do polímero, se denomina o decaimento do grupo excitado, como o do éxciton (um fônon delocalizado sobre várias unidades monoméricas conjugadas intracadeias). Essa nomenclatura surge por analogia ao decaimento radiativo de materiais semicondutores inorgânicos.
Mostrou-se, portanto, que a emissão depende dos níveis eletrônicos envolvidos no processo, mas esse não é o único fator que interfere no espectro. O perfil da banda espectral de emissão, entretanto, depende de muitos outros fatores, a começar pelas regras de seleção espectroscópicas. Para polímeros conjugados orgânicos, se o decaimento radiativo ocorrer a partir de um estado eletrônico excitado que tenha a mesma multiplicidade de spins do estado eletrônico fundamental, será denominado um éxciton de estado singlete; de modo diferente, se envolve o decaimento entre estados com spins diferentes o éxciton será de estado triplete. Como para polímeros orgânicos emissores de luz o estado eletrônico fundamental é um estado singlete, o decaimento de um estado eletrônico excitado singlete para o estado eletrônico fundamental será uma transição permitida por multiplicidade de spins. Esse processo radiativo envolvendo estados de mesma multiplicidade de spins é denominado emissão de fluorescência. Da mesma forma que em compostos orgânicos, isto não garante que a transição eletrônica será permitida, pois existem outras regras de seleção, que envolvem a simetria dos orbitais participantes da transição eletrônica. Em geral, os polímeros eletroluminescentes apresentam um alto rendimento quântico de fotoemissão em solução diluída, o que sugere que as transições eletrônicas devam ser permitidas por simetria e por multiplicidade de spins eletrônicos1,2,5,7,27. Esse também é o caso dos poli(fluorenos).
As bandas de absorção e de emissão podem apresentar estrutura vibrônica, que reflete a forma com que os modos vibracionais se acoplam com os estados eletrônicos para participar da transição. O acoplamento vibrônico em polímeros orgânicos conjugados segue as mesmas regras de seleção que as de moléculas orgânicas luminescentes104-106. Isto quer dizer que serão acoplados os modos normais que apresentam simetria que leve a uma representação irredutível totalmente simétrica do integrando do operador no momento de dipolo de transição. Em muitos casos, os modos normais que se acoplam envolvem estiramentos das ligações conjugadas do cromóforo emissor. A relação observada de intensidades entre as bandas vibrônicas relaciona-se diretamente com os acoplamentos vibrônicos, mostrando que a probabilidade de transição entre os níveis ocorre de acordo com o Princípio de Franck-Condon107-109.
O parâmetro que quantifica a probabilidade de transição entre os níveis vibrônicos e relaciona a energia de relaxação entre estados vibrônicos a uma dada temperatura com a freqüência de acoplamento elétron-fônon é o de Huang-Rhys98-108. Assumindo um forte acoplamento com o modo vibracional de número de onda  , a relativa intensidade I(n) dos vários estados vibrônicos no espectro de fotoluminescência pode ser descrita pelo parâmetro de Huang-Rhys, S, dado pela Equação 1102,103,107,109-111
, a relativa intensidade I(n) dos vários estados vibrônicos no espectro de fotoluminescência pode ser descrita pelo parâmetro de Huang-Rhys, S, dado pela Equação 1102,103,107,109-111

onde I(n) é a intensidade do pico máximo de fluorescência e n é o acoplamento vibrônico.
Através da razão entre as intensidades relativas com a progressão vibrônica, o termo exponencial é cancelado, assim como o termo polinomial de S para n = 1 e n = 2, de modo que:

Considerando o espectro normalizado [I(1)/I(2)], obtém-se por substituições a Equação 3:

Utilizando-se os valores das intensidades das bandas e o número de onda obtido através do espectro vibracional, pode-se obter o parâmetro de Huang-Rhys (S) e a energia de relaxação de decaimento eletrônico (ERel), através da Equação 1102-104,109,110.
Quando polímeros conjugados são utilizados, além dos estados eletrônicos envolvidos, existem outras variáveis que controlam a região espectral e o perfil da banda. Entre essas varáveis estão o comprimento efetivo de conjugação e a distribuição desses tamanhos. Quanto maior o tamanho efetivo da conjugação, mais deslocados para valores menores de energias estarão os espectros eletrônicos de absorção e de emissão. O tamanho efetivo de conjugação é definido pelo valor médio de uma distribuição aleatória de segmentos com diferentes tamanhos de unidades adjacentes conjugadas108,111-113. Esta distribuição estatística segue o modelo gaussiano e é conseqüência de defeitos na cadeia principal, gerados pela presença de ligações cis, de sítios oxidados, de grupos carbonila, de ligações alifáticas, ou por torções conformacionais geradas por influência de solventes, por pressão ou mesmo pela temperatura114. O comprimento de conjugação ou tamanho de um segmento conjugado "n" é adequadamente definido como o número de repetições monoméricas de ligações simples e duplas alternadas e não interrompidas ao longo da molécula. O comprimento efetivo da conjugação está limitado pelo surgimento, aleatório, de torções entre as unidades monoméricas adjacentes. Esse ângulo de torção interfere na delocalização dos elétrons p, fazendo com que o comprimento da conjugação seja muito menor que o comprimento físico da macromolécula. Isso gera uma variação estatística do comprimento da cadeia e ocasiona um alargamento espectral da banda de absorção115,116. Uma distribuição mais larga de tamanhos de segmentos conjugados da cadeia polimérica resulta em uma distribuição mais larga de energias de excitação e de emissão, de modo que as bandas de absorção e emissão sofrerão um alargamento inomogêneo29. Esses efeitos são intrínsecos aos materiais e dependem da rota sintética escolhida. Disto resulta que diferentes cromóforos irão decair a partir de estados eletrônicos de Franck-Condon com energias diferentes, produzindo espectros alargados29,117,118. O controle do comprimento da conjugação permite, portanto, alguma modulação na posição da banda espectral, e o controle da distribuição de comprimentos de conjugação permite a modulação da largura da banda de emissão.
Outra fonte de alargamento espectral é a desordem conformacional das cadeias. Quando o espectro de emissão de polímeros emissores é comparado com o espectro de emissão de moléculas pequenas similares, esses são sempre mais alargados19-21,119-122. Esse problema aparece, também, em polímeros conjugados18-21. No caso dos poli(fluorenos), a desordem conformacional ocorre pelo fato de que quando substituintes na posição 9 modificam a cadeia do polímero (Figuras 1, 2) esses segmentos podem se orientar de várias maneiras em relação à cadeia principal, produzindo pequenas modificações espectrais. Em conseqüência, as moléculas decaem de estados de Franck-Condon levemente diferentes e, sendo o espectro observado uma soma de todos eles, as bandas vibrônicas se sobrepõem, levando a uma perda de resolução espectral. Por isso muitas vezes não se obtém uma boa resolução espectral, mesmo se trabalhado com instrumentos com alta resolução.
Diferentemente das emissões de fluorescência de moléculas pequenas, a emissão de fluorescência em polímeros conjugados não é necessariamente imagem especular do espectro eletrônico de absorção ou de excitação. As razões para isso são muitas, mas se devem, principalmente, ao fato de que o grupo que absorve é, em geral, um segmento de cadeia mais curto que, por migração, transfere a energia para um segmento mais longo e esse é, em geral, o que emite.
O rendimento quântico de fotoluminescência resulta da competição entre as eficiências dos processos radiativos e não radiativos, enquanto que no caso da eletroluminescência ele resulta da recombinação dos pares polarônicos intracadeia. Portanto, apesar dos espectros poderem ser muito semelhantes, os rendimentos quânticos não o são. É ainda importante enfatizar que, enquanto o rendimento quântico de fotoluminescência pode ser determinado inequivocamente em soluções diluídas, tanto o rendimento quântico de fluorescência quanto o de eletroluminescência são difíceis de serem obtidos no estado sólido, quer devido à ausência de padrões, quer devido à complexidade dos processos de transferência de energia no estado sólido. Em geral, os artigos comentam que a eficiência da fotoluminescência dos polímeros eletroluminescentes é muito menor no estado sólido do que em solução.
Quando os sistemas estão em soluções concentradas ou no estado sólido, deslocamentos espectrais são observados, decorrentes dos processos de agregação. Em geral, os espectros de absorção e de emissão deslocam-se para energias menores em relação às bandas de absorção e de emissão dos cromóforos isolados. No estado sólido os agregados são dímeros ou espécies maiores, envolvendo interações intermoleculares ou intercadeias entre espécies no estado eletrônico fundamental. Em conseqüência, tanto o espectro eletrônico de absorção quanto o de emissão são alterados. Em solução existe a possibilidade de aparecerem espécies que interagem durante o tempo de vida do estado eletrônico excitado. Essas espécies são denominadas excímeros, se as duas espécies que estão interagindo são idênticas. Como a interação se dá envolvendo uma molécula no estado eletrônico excitado, a presença de excímeros pode ser detectada somente pela presença de uma banda larga e sem estrutura vibrônica no espectro de emissão. Os estados eletrônicos dos excímeros são dissociativos no estado eletrônico fundamental e esta é a razão pela qual não podem ser observadas experimentalmente suas bandas por espectroscopia eletrônica de absorção. Um excímero é, portanto, definido como um complexo formado por duas moléculas idênticas que possuem interação repulsiva no estado eletrônico fundamental, mas atrativa quando uma delas está no estado eletrônico excitado. A emissão leva, portanto, à dissociação do complexo. Quando as moléculas são diferentes, o termo normalmente usado é exciplexo123-128.
Tem havido uma grande controvérsia na literatura sobre a possibilidade de agregação de polímeros conjugados, principalmente aqueles com substituintes nas posições 9 no caso dos poli(fluorenos). Entretanto, diversos trabalhos recentes demonstraram, inequivocamente, que a agregação existe e que é responsável pelas diferenças espectrais e fotofísicas19-21,129-131. Umas das formas de se acompanhar o processo de agregação é através da obtenção dos espectros (absorção, excitação e emissão) em soluções em diferentes concentrações.
A concentração do polímero em solução, portanto, é um importante fator de alteração do perfil espectral104,132. Soluções concentradas podem levar à agregação do polímero e, ao formar um dímero, o espectro deve sofre um desdobramento excitônico, que, de acordo com o mecanismo proposto por Kasha, produzirá efeitos espectrais diferentes dependendo da geometria dos mesmos. Espera-se, portanto, que em sistemas em que ocorra a formação de dímeros levem a deslocamentos dos espectros de absorção e de emissão para energias menores se envolverem uma sobreposição dos centros emissores com uma orientação relativa dos momentos dipolares em direções opostas. Sistemas deste tipo são, portanto cofaciais, sendo mantidos por interações envolvendo as nuvens p. A agregação de cadeias tende a reduzir o desempenho do dispositivo eletroluminescente, porque o rendimento quântico da foto e da eletroluminescência se reduz drasticamente105,106,116.
Foi observado que poli(fluorenos) com massa molar baixa apresentam maior probabilidade de formação de excímeros e agregados, sendo este efeito atribuído em parte ao aumento da mobilidade das cadeias esperada para cadeias menores18,133. A redução na intensidade de emissão é atribuída à possibilidade de ocorrência de vários tipos de processos, entre eles, migração da excitação, à desativação através dos processos de transferência de energia intermoleculares, etc134.
Os tempos de vida de emissão de polímeros conjugados luminescentes são, em geral, funções temporais complexas, principalmente no estado sólido. Por se tratarem de transições permitidas por regras de seleção, e por envolverem complexos processos de transferência e migração de energia, os tempos de decaimentos são, em geral, rápidos (poucos nanosegundos ou mais rápidos). A presença de grupos substituintes afeta tanto o perfil do decaimento, que passam a ser funções temporais complexas (biexponenciais e triexponenciais), como também os valores de tempo de vida. As modificações nos tempos de vida geralmente são devidas a processos de transferência de energia e de relaxações não radiativas do estado eletrônico excitado. Entre esses processos, estão os de transferência de energia via mecanismo de Förster, transferência de energia radiativa, migração de energia, aniquilação singlete-singlete, entre outros106,123-128,135,136. Em geral, os poli(fluorenos) apresentam decaimentos de fluorescência monoexponencias em soluções diluídas, com tempos de vida que variam na faixa de 80 ps até 5 ns (Tabela 1).
Os processos de agregação também podem ser acompanhados por medidas de tempos de decaimento da emissão. Uma das técnicas que podem ser empregadas é a de espectroscopia de emissão resolvida no tempo ("time-resolved spectroscopy, TRES"), na qual se excita um conjunto de cromóforos com um comprimento de onda fixo de excitação, e se medem os tempos de decaimento da emissão em um comprimento de onda de emissão fixo, com diferentes atrasos em relação ao pulso inicial de excitação. Isto permite se verificar a evolução na formação das diferentes espécies excitadas, a partir da excitação de uma delas. Variando-se os comprimentos de ondas de excitação e de emissão pode-se acompanhar a cinética de formação dessas espécies excitadas, bem como os processos de transferência de energia entre as mesmas. Isto é particularmente útil no caso de sistemas na fase sólida.
Processos de transferência de energia
Vários mecanismos de transferência de energia podem ocorrer quando duas moléculas interagem e pelo menos uma delas está em um estado eletrônico excitado. Esses mecanismos de transferência podem ser radiativos ou não radiativos. Dentre os não radiativos pode-se citar o mecanismo de Förster (interação dipolar de longa distância) e o mecanismo de Dexter (interação a distâncias curtas, envolvendo a troca de elétrons). Entre os mecanismos radiativos pode-se citar o de transferência de energia radiativa (trivial). A eficiência de cada um desses processos é função da distância de separação entre as espécies que estão interagindo, variando-se a forma de dependência em cada caso.
Transferência de energia via mecanismo de Förster
O processo de desativação de uma molécula no estado eletrônico excitado (doador D*) para uma outra no estado eletrônico fundamental (receptor A) ocorre de modo não radiativo através do mecanismo de Förster, de acordo com o esquema.

Como o elétron no estado excitado de D* sofre uma transição para o orbital molecular de energia mais baixa de D, há uma mudança no momento de dipolo elétrico, o qual produz uma alteração no campo elétrico local. A interação dipolar entre D e A é proporcional ao momento de transição dipolo elétrico  e inversamente proporcional ao cubo da distância de separação entre as duas espécies. Portanto, a probabilidade de transferência de energia de acordo com esse mecanismo é um processo no qual a velocidade de transferência depende da distância de separação entre o doador e o receptor. A eficiência desse processo depende também da sobreposição entre o espectro eletrônico de absorção do receptor e o de emissão do doador. A formulação desenvolvida por Förster baseia-se em um modelo de interação fraca, onde a transferência de energia é mais lenta que a relaxação vibracional do doador e do receptor, e considera somente o termo dipolo-dipolo de interação coulômbica entre eles106,124-128,137,138. Define-se o raio de Förster (R0) como a distância entre o doador e o receptor na qual a probabilidade de transferência é igual à probabilidade de decaimento radiativo. Para distâncias maiores que R0 o processo de transferência pelo mecanismo de Förster é muito pouco eficiente. O raio de Förster é, portanto, uma propriedade característica de um par de moléculas doador-receptor.
e inversamente proporcional ao cubo da distância de separação entre as duas espécies. Portanto, a probabilidade de transferência de energia de acordo com esse mecanismo é um processo no qual a velocidade de transferência depende da distância de separação entre o doador e o receptor. A eficiência desse processo depende também da sobreposição entre o espectro eletrônico de absorção do receptor e o de emissão do doador. A formulação desenvolvida por Förster baseia-se em um modelo de interação fraca, onde a transferência de energia é mais lenta que a relaxação vibracional do doador e do receptor, e considera somente o termo dipolo-dipolo de interação coulômbica entre eles106,124-128,137,138. Define-se o raio de Förster (R0) como a distância entre o doador e o receptor na qual a probabilidade de transferência é igual à probabilidade de decaimento radiativo. Para distâncias maiores que R0 o processo de transferência pelo mecanismo de Förster é muito pouco eficiente. O raio de Förster é, portanto, uma propriedade característica de um par de moléculas doador-receptor.
Por ser um processo não radiativo, a existência desse tipo de transferência de energia acarreta uma diminuição na intensidade total do espectro de emissão do doador. Uma das formas de se calcular a eficiência desse mecanismo de transferência de energia é através da espectroscopia de emissão fotoestacionária, medindo-se a intensidade de emissão do doador na presença e na ausência do receptor. Quanto maior a eficiência do processo, maior a redução na intensidade da emissão. Uma outra evidência para ocorrência do mecanismo de transferência não radiativa do tipo Förster é a redução no tempo de vida do decaimento de fluorescência do doador em presença do receptor. Nesse caso, ocorre uma redução do tempo de vida no estado eletrônico excitado do doador. Em conseqüência, é possível se determinar a eficiência desse processo de transferência não radiativa através da determinação experimental dos tempos de decaimento da fluorescência do doador na presença e na ausência do receptor.
Transferência de energia radiativa
O processo de transferência radiativa de energia ocorre entre o doador e o receptor após a excitação do primeiro, de acordo com o mecanismo128,139:

A diferença entre o processo trivial e o processo não radiativo que ocorre pelo mecanismo de Förster reside no fato da mudança no tempo de vida do doador de energia em função da concentração do receptor ser muito pequena e que a eficiência do processo independe da viscosidade do meio. O mecanismo trivial não requer uma interação energética entre o doador e o receptor e tem uma dependência com o inverso da distância receptor-doador ao quadrado. Além do mais, a eficiência do processo trivial também depende da sobreposição espectral entre os espectros de emissão do doador e absorção do receptor106,124,125,128:

onde FD representa a intensidade de emissão do doador, integrada sobre toda a banda de emissão e eA a absortividade molar do receptor, integrada sobre toda sua banda de absorção, sendo ambas em número de onda.
O processo trivial pode ocorrer entre moléculas idênticas, o que pode acarretar os efeitos observados de auto-absorção e re-emissão, bem como o efeito de filtro interno em soluções concentradas, em líquidos e cristais, bem como em sistemas que possuem grandes volumes. Freqüentemente é o processo dominante em processos de transferência de energia em soluções diluídas. Tanto em um mecanismo quanto no outro, os dados a serem analisados são os mesmos, mas as conclusões a serem obtidas são diferentes106,123,128-141. No caso da transferência radiativa pode ocorrer auto-absorção e re-emissão por parte do receptor e distorções no espectro de emissão do doador, sendo este último um parâmetro importante para distinguir o processo trivial do processo via Förster. Existem meios de se verificar se está ocorrendo um ou outro processo, tais como variação da concentração do doador na ausência do receptor, medidas de fluorescência por excitação frontal, etc142-145.
Outros tipos de mecanismos de transferência de energia no estado sólido
Além do mecanismo trivial e do proposto por Förster, têm sido propostos outros mecanismos para explicar como ocorre a interação entre moléculas excitadas com outras no estado fundamental no estado sólido. Entre esses mecanismos estão o de difusão clássico e o de delocalização quântica, que fornecem subsídios para entender a interação à longa distância146. Em geral, o mecanismo de difusão não tem grande importância para regimes ultra-rápidos, sendo somente relevante para processos que ocorrem a tempos longos, onde as moléculas podem se difundir durante o processo de interação. Em longos tempos, esse processo deve ser levado em consideração, pois afeta as distâncias entre as moléculas envolvidas para maiores ou menores valores de distâncias e R0. O mecanismo de delocalização quântica fornece informações de como espécies excitadas interagem na mesma cadeia, favorecendo a interação entre duas cadeias poliméricas. Este mecanismo estabelece que a excitação se propaga a grandes distâncias (aproximadamente 1000 unidades monoméricas considerando o MEH-PPV), o que favorece o processo de desativação bimolecular entre as espécies envolvidas no processo de transferência de energia114,147-149. Definir o tipo de mecanismo de desativação e tentar minimizá-lo é um dos objetivos a serem perseguidos nos estudos para otimizar o desempenho de dispositivos, pois, independentemente do mecanismo, esses processos de transferência levam à perda de eficiência do mesmo.
CONCLUSÕES
O progresso na área de pesquisa acadêmica ou tecnológica em materiais eletroluminescentes nos últimos anos tem sido expressivo e crescente, tendo como etapa inicial a síntese e a caracterização de novos materiais, como propriedades planejadas para produzir emissão em uma certa região espectral. Neste contexto, o poli(fluoreno) e seus derivados têm sido extensamente estudados e despontam como materiais promissores para essa aplicação, em função da sua versatilidade estrutural e estabilidades química, térmica e fotoquímica. As extensas faixas de comprimentos de onda de absorção e de emissão e a facilidade de se modificar sua estrutura para permitir novas características físico-químicas torna esses materiais muito atraentes. Os altos valores de rendimento quântico e os tempos de decaimentos situados em uma ampla faixa de tempos de vida torna-os promissores para aplicações nos vários tipos de sistemas, entre eles a tela plana de televisores e visores de relógios, telefones celulares e computadores. Se os dispositivos forem montados sobre substratos adequados, é possível a fabricação de dispositivos flexíveis e coloridos.
Como em toda área em franco desenvolvimento científico e tecnológico, várias questões desafiantes ainda persistem quando se trata de OPLEDs, sendo os pontos mais críticos a estabilidade da eletroemissão por períodos longos de tempo, a susceptibilidade desses materiais à oxidação e conseqüente degradação foto e termoquímica, a estabilidade morfológica em ciclos sucessivos de operação, a otimização dos contatos elétricos entre componentes metálicos e não metálicos dos dispositivos, etc.
AGRADECIMENTOS
À FAPESP, CNPq, MCT/PADCT/IMMP e ao FAEPEX/Unicamp pelo apoio financeiro e pelas bolsas recebidas. Agradecimentos também a A. M. Machado pelos comentários e sugestões.
REFERÊNCIAS
1. Shirakawa, H.; Louis, E. J.; MacDiarmid, A. G.; Chiang, C. K.; Heeger, A. J.; J. Chem. Soc. Chem. Commun. 1977, 16, 578.
2. Chiang, C. K.; Ficher, C. R.; Park, Y. W.; Heeger, A. J.; Shirakawa, H.; Louis, E. J.; Gau, S. C.; MacDiarmid, A. G.; Phys. Rev. Lett. 1977, 39, 1098.
3. Burroughes, J. H.; Bradley, D. D. C.; Brown. A. R.; Marks, R. N.; Mackay, K.; Friend, R. H.; Burns, P. L.; Holmes, A. B.; Nature 1990, 347, 539.
4. Miyata, S.; Nalma, H. S.; Organic Electroluminescent Materials and Devices, Gordon and Breach Science Publishers: Tokyo, 1997.
5. Friend, R. H.; Gymer, R. W.; Homes, A. B.; Burroughes, J. H.; Marks, R. N.; Taliani, C.; Bradley, D. D. C.; dos Santos, D. A.; Brédas, J. L.; Logdlund, M.; Salaneck, W. R.; Nature 1999, 397, 121.
6. Yu, G.; Gao, J.; Hummelen, J. C.; Wudl, F.; Heeger, A. J.; Science 1995, 270, 1789.
7. Halls, J. J. M.; Walsh, C. A.; Greenham, N. C.; Marseglia, E. A.; Friend, R. H.; Moratti, S. C.; Holmes, A. B.; Nature 1995, 376, 498.
8. Brabec, C. J.; Sariciftci, N. S.; Hummelen, J. C.; Adv. Funct. Mater. 2001, 11, 15.
9. Garnier, F.; Hajlaoui, R.; Yassar, A.; Srivastava, P.; Science 1994, 265, 1684.
10. Yang, Y.; Heeger, A. J.; Nature 1994, 372, 344.
11. Sirringhaus, H.; Tessler, N.; Friend, R. H.; Science 1998, 280, 1741.
12. Tessler, N.; Denton, G. J.; Friend, R. H.; Nature 1996, 382, 695.
13. Hide, F.; Schwartz, B. J.; DiazGarcia, M. A.; Heeger, A. J.; Chem. Phys. Lett. 1996, 256, 424.
14. Berggren, M.; Dodabalapur, A.; Slusher, R. E.; Bao, Z.; Nature 1997, 389, 466.
15. Gupta, R.; Stevenson, M.; McGehee, M. D.; Dogariu, A.; Srdanov, V.; Park, J. Y.; Heeger, A. J.; Synth. Met. 1999, 102, 875.
16. Tessler, N.; Ho, P. K. H.; Cleave, V.; Pinner, D. J.; Friend, R. H.; Yahioglu, G.; Le Barny, P.; Gray, J.; de Souza, M.; Rumbles, G.; Thin Solid Films 2000, 363, 64.
17. Crone, B.; Dodabalapur, A.; Lin, Y. Y.; Filas, R. W.; Bao, Z.; LaDuca, A.; Sarpeshkar, R.; Katz, H. E.; Li, W.; Nature 2000, 403, 521.
18. Akcelrud, L.; Prog. Polym. Sci. 2003, 28, 875.
19. Machado, A. M.; Motta Neti, J. D.; Cossiello, R. F.; Atvars, T. D. Z.; Karasz, F. E.; Akcelrud, L.; Polymer 2005, 46, 2452.
20. Deus, J. F.; Andrade, M. L.; Atvars, T. D. Z.; Akcelrud, L.; Chem. Phys. 2004, 297, 117.
21. Deus, J. F.; Corradini, W. A.; Atvars, T. D. Z.; Akcelrud, L.; Macromolecules 2004, 37, 6938.
22. Pei, J.; Liu, X.; Chen, Z.; Zhang, X.; Lai, Y.; Huang, W.; Macromolecules 2003, 36, 323.
23. Burrows, H. D.; Melo, J. S.; Serpa, C.; Arnaut, L. G.; Miguel, M. G.; Monkman, A. P.; Hamblett, I.; Navaratnam, S.; Chem. Phys. 2002, 285, 3.
24. Rothe, C.; Palsson, L. O.; Monkman, A. P.; Chem. Phys. 2002, 285, 95.
25. Tirapattur, S.; Belletête, M.; Drolet, N.; Leclerc, M.; Durocher, G.; Macromolecules 2002, 35, 8889.
26. Zeng, G.; Yu, W.; Chua, S.; Huang, W.; Macromolecules 2002, 35, 6907.
27. Ding, J.; Day, M.; Robertson, G.; Roovers, J.; Macromolecules 2002, 35, 3474.
28. Palsson, L.; Wang, C.; Russell, D. L.; Monkman, A. P.; Bryce, M. R.; Rumbles, G.; Samuel, I. D. W.; Chem. Phys. 2002, 279, 229.
29. Zhan, X.; Liu, Y.; Wu, X.; Wang, S.; Zhu, D.; Macromolecules 2002, 35, 2529.
30. Lupton, J. M.; Klein, J.; Chem. Phys. Lett. 2002, 363, 204.
31. Rothe, C.; Hintschich, S. I.; Palsson, L.; Monkman, A. P.; Guentner, R.; Scherf, U.; Chem. Phys. Lett. 2002, 360, 111.
32. Cornil, J.; Gueli, I.; Dkhissi, A.; Garcia, J. C. S.; Hennebicq, E.; Calbert, J. P.; Lemaur, V.; Beljonne, D.; Brédas, J. L.; J. Chem. Phys. 2003, 118, 6615.
33. Pasini, M.; Destri, S.; Porzio, W.; Botta, C.; Giovanella, U.; J. Mater. Chem. 2003, 13, 807.
34. Herz, L. M.; Silva, C.; Phillips, R. T.; Setayesh, S.; Müllen, K.; Chem. Phys. Lett. 2001, 347, 318.
35. Meskers, S. C. J.; Hübner, J.; Oestreich, M.; Bässler, H.; Chem. Phys. Lett. 2001, 339, 223.
36. Buckley, A. R.; Rahn, M. D.; Hill, J.; Cabanillas-Gonzalez, J.; Foz, A. M.; Bradley, D. D. C.; Chem. Phys. Lett. 2001, 339, 331.
37. Geng, Y.; Trajkovska, A.; Katsis, D.; Ou, J. J.; Culligan, S. W.; Chen, S. H.; J. Am. Chem. Soc. 2002, 124, 8337.
38. Meskers, S. C. J.; Hübner, J.; Oestreich, M.; Bässler, H.; J. Phys. Chem. B 2001, 105, 9139.
39. Lemmer, U.; Hein, S.; Mahrt, R. F.; Scherf, U.; Hopmeir, M.; Wiegner, U.; Gobel, R. O.; Mullen, K.; Bassler, H.; Chem. Phys. Lett. 1995, 240, 371.
40. Charas, A.; Morgado, J.; Martinho, J. M. G.; Alcácer, L.; Lim, S. F.; Friend, R. H.; Cacialli, F.; Polymer 2003, 44, 1843.
41. Yu, W. L.; Pei, J.; Huang, W.; Heeger, A. J.; Adv. Mater. 2000, 12, 828.
42. Virgili, T.; Lidzey, D. G.; Bradley, D. D. C.; Adv. Mater. 2000, 12, 58.
43. Whitehead, K. S.; Grell, M.; Bradley, D. D. C.; Jandke, M.; Strohriegl, P.; Appl. Phys. Lett. 2000, 76, 2946.
44. Grell, M.; Knoll, W.; Lupo, D.; Meisel, A.; Miteva, T.; Neher, D.; Nothofer, H. G.; Scherf, U.; Yasuda, A.; Adv. Mater. 1999, 11, 671.
45. Yang, Y.; Pei, Q.; Appl. Phys. Lett. 1997, 81, 3294.
46. Lee, J.; Klaerner, G.; Miller, R. D.; Chem. Mater. 1999, 11, 1083.
47. Klaerner, G.; Lee, J. K.; Davey, M. H.; Miller, R. D.; Adv. Mater. 1999, 11, 115.
48. Kreyenschimdt, M.; Klaerner, G.; Fuhrer, T.; Ashenhurst, J.; Karg, S.; Chen, W.; Lee, V. Y.; Scott, J. C.; Miller, R. D.; Macromolecules 1998, 31, 1099.
49. Bliznyuk, V. N.; Carter, S. A.; Scott, J. C.; Klaerner, G.; Miller, R. D.; Macromolecules 1999, 32, 361.
50. Yang, Y.; Pei, Q.; J. Am. Chem. Soc. 1996, 118, 7416.
51. He, Y.; Gong, S.; Hattori, R.; Kanicki, J.; Appl. Phys. Lett. 1999, 74, 2265.
52. Yu, W.; Cao, Y.; Pei, J.; Huang, W.; Heeger, A. J.; Appl. Phys. Lett. 1999, 75, 3270.
53. Yu, W.; Pei, J.; Cao, Y.; Huang, W.; Heeger, A. J.; Chem. Commun. 1999, 18, 1837.
54. Liu, B.; Yu, W.; Lai, Y.; Huang, W.; Chem. Commun. 2000, 7, 551.
55. Sainova, D.; Miteva, T.; Nothofer, H.; Scherf, U.; Glowacki, I.; Ulanski, J.; Fujikawa, H.; Neher, D.; Appl. Phys. Lett. 2000, 76, 1810.
56. Weinfurtner, K.; Fujikawa, H.; Tokito, S.; Taga, Y.; Appl. Phys. Lett. 2000, 76, 2502.
57. Jenekhe, S. A.; Osaheni, J. A.; Science 1994, 265, 765.
58. Liu, B.; Yu, W.; Lai, Y.; Huang, W.; Macromolecules 2000, 33, 8945.
59. Grell, M.; Bradley, D. D. C.; Ungar, G.; Hill, J.; Whitehead, K. S.; Macromolecules 1999, 32, 5810.
60. Cadby, A. J.; Lane, P. A.; Wohlgenannt, M.; An, C.; Vardeny, Z. V.; Bradley, D. D. C.; Synth. Met. 2000, 111-112, 515.
61. Bauer, C.; Urbasch, G.; Giessen, H.; Meisel, A.; Nothofer, H.; Neher, D.; Scherf, U.; Mahrt, R. F.; Chem. Phys. Chem. 2000, 3, 142.
62. Zhan, X.; Lui, Y.; Yu, G.; Wu, X.; Zhu, D.; Sun, R.; Wang, D.; Epstein, A. J.; J. Mater. Chem. 2001, 11, 1606.
63. Leclerc, M.; J. Polym. Sci., Part A: Polym. Chem. 2001, 39, 2867.
64. Kameshima, H.; Nemoto, N.; Endo, T.; J. Polym. Sci., Part A: Polym. Chem. 2001, 39, 3143.
65. Suh, Y.; Ko, S. W.; Jung, B.; Shim, H.; Opt. Mater. 2002, 21, 109.
66. Bliznyuk, V. N.; Carter, S. A.; Scott, J. C.; Klärner, G.; Miller, R. D.; Miller, D. C.; Macromolecules 1999, 32, 361.
67. Cadby, A. J.; Lane, P. A.; Mellor, H.; Martin, S. J.; Grell, M.; Giebeler, C.; Bradley, D. D. C.; Wohlgenannt, M.; An, C.; Vardeny, Z. V.; Phys. Rev. B: Condens. Matter Matter. Phys. 2000, 62, 15604.
68. Lee, J. I., Lee, V. Y., Miller, R. D.; ETRI J. 2002, 24, 409.
69. Grell, M.; Redecker, M.; Whitehead, K. S.; Bradley, D. D. C.; Inbasekaran, M.; Woo, E. P.; Wu, W.; Liq. Cryst., no prelo.
70. Grell, M.; Bradley, D. D. C.; Inbasekaran, M.; Woo, E. P.; Adv. Mater. 1997, 9, 798.
71. Grell, M.; Bradley, D. D. C.; Adv. Mater., no prelo.
72. Schartel, B.; Wachtendorf, V.; Grell, M.; Bradley, D. D. C.; Hennecke, M.; Phys. Rev. B: Condens. Matter Mater. Phys. 1999, 60, 277.
73. Long, X.; Grell, M.; Malinowski, A.; Bradley, D. D. C.; Opt. Mater. 1998, 9, 70.
74. Weinfurtner, K. H.; Weissortel, F.; Harmgarth, G.; Salbeck, J.; Proc. SPIE. 1998, 3476, 40.
75. Geng, Y. H.; Katsis, D.; Culligan, S. W.; Ou, J. J.; Chen, S. H.; Rothberg, L. J.; Chem. Mater. 2002, 14, 463.
76. Katsis, D.; Geng, Y. H.; Culligan, S. W.; Trajkovska, A.; Chen, S. H.; Rothberg, L.; Chem. Mater. 2002, 14, 1332.
77. Andersson, M. R.; Yu, G.; Heeger, A. J.; Synth. Met. 1997, 85, 1275.
78. Xu, B.; Holdcroft, S.; Macromolecules 1993, 26, 4457.
79. Swanson, L. S.; Shinar, J.; Ding, Y. W.; Barton, T. J.; Synth. Met. 1993, 55, 1.
80. Greenham, N. C.; Brown, A. R.; Bradley, D. D. C.; Adv. Mater. 1994, 6, 491.
81. Greenham, N. C.; Brown, A. R.; Bradley, D. D. C.; Friend, R. H.; Synth. Met. 1993, 57, 4134.
82. Braun, D.; Gustafsson, G.; McBranch, D.; Heeger, A. J.; J. Appl. Phys. 1992, 72, 564; Mao, H.; Xu, B.; Holdcroft, S.; Macromolecules 1993, 26, 1163.
83. Uchida, M.; Ohmori, Y.; Morishima, C.; Yoshino, K.; Synth. Met. 1993, 57, 4168.
84. Tada, K; Sonoda, T.; Yokota, Y.; Kobashi, K.; Yoshino, K.; J. Appl. Phys. 1998, 84, 5635.
85. Klaerner, G.; Miller, R. D.; Macromolecules 1998, 31, 2007.
86. Jiang, X.; Liu, S.; Ma, H.; Jen, A. K. Y.; Appl. Phys. Lett. 2000, 76, 1813.
87. Rehahn, M.; Schluter, A. D.; Wegner, G.; Feast, W. J.; Polymer 1989, 30, 1054.
88. Yamamoto, T.; Morita, A.; Miyazaki, Y.; Maruyama, T.; Wakayama, H.; Zhou, Z. H.; Nakamura, Y.; Kanbara, T.; Macromolecules 1992, 25, 1214.
89. Kannan R.; He, G. S.; Yuan, L.; Faming, X.; Prasad, P. N.; Dombroskie, A. G.; Reinhardt, B. A; Baur, J. W.; Vaia, R. A.; Tan, L. S.; Chem. Mater. 2001, 13, 1896.
90. Ding, J.; Day, M.; Robertson, G.; Roovers, J.; Macromolecules 2002, 35, 3474.
91. Ranger, M.; Rondeau, D.; Leclerc, M.; Macromolecules 1997, 30, 7686.
92. Miyaura, M.; Suzuki, A.; Chem. Rev. 1995, 95, 2457.
93. Setayesh, S.; Marsitzky, D.; Müllen, K.; Macromolecules 2000, 33, 2016.
94. Nelson, J.; Current Opinion in Solid State and Materials Science 2002, 6, 87.
95. Silva, G. B.; Santos, L. F.; Faria, R. M.; Grae, C. F. O.; Physica B 2001, 308310, 1078.
96. Mahrt, R. F.; Pauck, T.; Lemmer, U.; Siegner, U.; Hopmeier, M.; Hennig, R.; Bassler, H.; Gobel, E. O.; Bolivar, P. H.; Wegmann, G.; Kurz, H.; Scherf, U.; Mullen, K.; Phys. Rev. B: Condens. Matter Mater. Phys. 1996, 54, 1759.
97. Yan, M.; Rothberg, L. J.; Kwock, E. W.; Miller, T. M.; Phys. Rev. Lett. 1995, 75, 1992.
98. Beljonne, D.; Cornil, J.; Sirringhaus, H.; Brown, P. J.; Shkunov, M.; Friend, R. H.; Brédas, J. L.; Adv. Funct. Mater. 2001, 11, 229.
99. Halls, J. J. M.; Baigent, D. R.; Cacialli, F.; Greenham, N C.; Friend, R. H.; Moratti, S C.; Holmes, A. B.; Thin. Solid Films 1996, 276, 13.
100. Romijn, I. G.; Pasveer, W. F.; Martens, H. C. F.; Brom, H. B.; Michels, M. A. J.; Synth. Met. 2001, 119, 439
101. Parker, I. D.; J. Appl. Phys. 1994, 75, 1657.
102. Petritsch, K.; Friend, R. H.; Synth. Met. 1999, 102, 976.
103. Barashkov, N. N.; Gunder, O. A.; Fluorescent Polymers, Ellis Horwood: New York, 1994.
104. Schaller, R. D.; Lee, L. F.; Johnson, J. C.; Haber, L. H.; Saykally, R. J.; Vieceli; J.; Benjamin, I.; Nguyen, T.; Schwartz, B. J.; J. Phys. Chem. B 2002, 74, 10.
105. Kasha, M.; Rawls, H. R.; El-Bayoumi, M. A.; Pure Appl. Chem. 1965, 11, 371.
106. Birks, J. B.; Photophysics of Aromatic Molecules, John Wiley & Sons Ltd.: New York, 1970.
107. Blatchford, J. W.; Jessen, S. W.; Lin, L. B.; Gustafson, T. L.; Fu, D. K.; Wang, H. L.; Swager, T. M.; Macdiarmid, A. G.; Epstein, A. J.; Phys. Rev. B: Condens. Matter Matter. Phys. 1996, 54, 9180.
108. Lim, S. H.; Bjorklund, T. G.; Bardeen, C. J.; Chem. Phys. Lett. 2001, 342, 555.
109. Hagler, T. W.; Pakbaz, K.; Heeger, A. J.; Phys. Rev. B: Condens. Matter Matter. Phys. 1994, 49, 10968.
110. Guha, S.; Rice, J. D.; Yau, Y. T.; Martin, C. M.; Chandrasekhar, M.; Chandrasekhar, H. R.; Guentner, R.; Freitas, P. S.; Ucherf, U.; Phys. Rev. B: Condens. Matter Matter. Phys. 2002, 67, 125204.
111. Nguyen, T. Q.; Schwartz, B. J.; Schaller, R. D.; Johnson, J. C.; Lee, L. F.; Haber, L. H.; Saykally, R. J.; J. Phys. Chem. B 2001, 105, 5153.
112. Gettinger, C. L.; Heeger, A. J.; J. Chem. Phys. 1994, 101, 74.
113. Tikhoplav, R. K.; Hess, B. C.; Synth. Met. 1999, 101, 236.
114. Köhler, A.; dos Santos, D. A.; Beljonne, D.; Shuai, Z.; Brédas, J. L.; Holmes, A. B.; Kraus, A.; Müllen, K.; Friend, R. H.; Nature 1998, 392, 903.
115. Oberski, J. M.; Greiner, A.; Bassler, H.; Chem. Phys. Lett. 1991, 184, 391.
116. Chang, R.; Hsu, J. H.; Fann, W. S.; Liang, K. K.; Chang, C. H.; Hayashi, M.; Yu, Y.; Lin, S. H.; Chang, E. C.; Chuang, K. R.; Chen, S. A.; Chem. Phys. Lett. 2000, 317, 142.
117. Pichler, K.; Halliday, D. A.; Bradley, D. D. C.; Brurn, P. L.; Friend, R. H.; Holmes, A. B.; J. Phys.: Condens. Matter 1993, 5, 7155.
118. Yu, J.; Hsu, J. H.; Chuang, K. R.; Chão, C. I.; Chen, S. A.; Kao, F. J.; Fann, W. S.; Lin, S. H.; Synth. Met. 1996, 82, 159.
119. Andrade, M. L.; Atvars, T. D. Z.; J. Phys. Chem. B 2004, 108, 3975.
120. Andrade, M. L.; Atvars, T. D. Z.; Macromolecules 2004, 37, 6938.
121. Xu, J.; Luo, C.; Atvars, T. D. Z.; Weiss, R. G.; Res. Chem. Intermed. 2004, 30, 509.
122. Brown, G.; Guardala, N. A.; Atvars, T. D. Z.; Weiss, R. G.; J. Polym. Sci., Part B: Polym. Phys. 2004, 42, 2957.
123. Kalyanasundaram, K.; Photochemistry in Microheterogeneous Systems, Academic Press: Orlando, 1992.
124. Scaiano, J. C.; Handbook of Organic Photochemistry, CRC Press: Boca Raton, 1989, vols. I e II.
125. Gilbert, A.: Baggott, J.: Essentials of Molecular Photochemistry, Blackwell Scientific Publications: Oxford, 1991.
126. Lakowicz; J. R.; Principles of Fluorescence Spectroscopy, 2nd ed., Academic Press: New York, 1999.
127. Vekshin, N. L.; Energy Transfer in Macromolecules, SPIE Optical Engineering Press: Washington, 1997.
128. Rohatgi-Mukherjee, K. K.; Fundamentals of Photochemistry, revised edition, New Age International Limited: New Delhi, 1978.
129. Cossiello, R. F.; Kowalski, E.; Rodrigues, P. C.; Akcelrud, L.; Bloise, A. C.; de Azevedo, E. R.; Bonagamba, T. J.; Atvars, T. D. Z.; Macromolecules 2005, 38, 925.
130. Cossiello, R. F.; Akcelrud, L.; Atvars, T. D. Z.; J. Braz. Chem. Soc. 2005, 16, 74.
131. Aguiar, M.; Akcelrud, L.; Pinto, M. R.; Atvars, T. D. Z.; Karsz, F. E.; Saltiel, J.; J. Photoscience 2003, 10, 149.
132. Sariciftci, N. S.; Primary Photoexcitations in Conjugated Polymers: Molecular exciton versus semiconductor band model, World Scientific Publishing Co.: Singapure, 1997.
133. Borsenberger, P. M.; Weiss, D. S.; Organic Photoreceptors for Imaging Systems, Marcel Dekker: New York, 1998.
134. Yu, J.; Fann, W. S.; Kao, F. J.; Yang, D. Y.; Lin, S. H.; Synth. Met. 1994, 66, 143.
135. Förster, T., Discuss. Faraday Soc. 1959, 27, 7.
136. Berkovic, G., J. Chem. Educ. 1984, 61, 832.
137. Suppan, P.; Chemistry and Light, Royal Society of Chemistry: Cambridge, 1994.
138. Lakowicz; J. R.; Topics in Fluorescence Spectroscopy, Plenum Press: New York, 1991, vol. 2.
139. Adam, W.; Cilento, G.; Chemical and Biological Generation of Excited States; Academic Press: New York, 1982.
140. Turro, N. J.; Modern Molecular Photochemistry, 2nd ed., Menlo Park: Califórnia, 1978.
141. Kopecky J.; Organic Photochemistry: A Visual Approach, VCH Publishers: New York, 1992.
142. Marinari, A.; Saltiel, J.; Mol. Photochem. 1976, 7, 225.
143. Conte, J. C.; Martinho, J. M. G.; J. Lumin. 1981, 22, 273.
144. Martinho, J. M. G.; Conte, J. C.; J. Lumin. 1981, 22, 285.
145. Martinho, J. M. G.; Pereira, V. R.; Silva, G.; Conte, J. C.; J. Photochem. 1985, 30, 383.
146. Dogariu, A.; Vacar, D.; Heeger, A. J.; Phys. Rev. B: Condens. Matter Matter. Phys. 1998, 58, 10218.
147. Powell, R. C.; Soos, Z. G.; J. Lumin. 1975, 11, 1.
148. Eisenthal, K. B.; Siegel, S.; J. Chem. Phys. 1964, 41, 652.
149. Yang, C. Y.; Hide, F.; Diaz-Garcia, M. A.; Heeger, A. J.; Cao, Y.; Polymer 1997, 39, 2299.
150. Belletête, M.; Morin, J.; Beaupré, S.; Ranger, M.; Leclerc, M.; Durocher, G.; Macromolecules 2001, 34, 2288.
151. Xia, C.; Advincula, R. C.; Macromolecules 2001, 34, 5854.
152. Jin, S.; Kang, S.; Kim, M.; Chan, Y. U.; Kim, J. Y.; Lee, K.; Gal, Y.; Macromolecules 2003, 36, 3841.
153. Park, J. H.; Ko, H. C.; Kim, J. H.; Lee, H.; Synth. Met. 2004, 144, 193.
154. Belletête, M.; Morin, J.; Beaupré, S.; Leclerc, M.; Durocher, G.; Synth. Met. 2002, 126, 43.
155. Kulkarni, A. P.; Kong, X.; Jenekhe, S. A.; J. Phys. Chem. B. 2004, 108, 8689.
Recebido em 29/9/04; aceito em 21/6/05; publicado na web em 1/12/05
- 1. Shirakawa, H.; Louis, E. J.; MacDiarmid, A. G.; Chiang, C. K.; Heeger, A. J.; J. Chem. Soc. Chem. Commun. 1977, 16, 578.
- 2. Chiang, C. K.; Ficher, C. R.; Park, Y. W.; Heeger, A. J.; Shirakawa, H.; Louis, E. J.; Gau, S. C.; MacDiarmid, A. G.; Phys. Rev. Lett 1977, 39, 1098.
- 3. Burroughes, J. H.; Bradley, D. D. C.; Brown. A. R.; Marks, R. N.; Mackay, K.; Friend, R. H.; Burns, P. L.; Holmes, A. B.; Nature 1990, 347, 539.
- 4. Miyata, S.; Nalma, H. S.; Organic Electroluminescent Materials and Devices, Gordon and Breach Science Publishers: Tokyo, 1997.
- 5. Friend, R. H.; Gymer, R. W.; Homes, A. B.; Burroughes, J. H.; Marks, R. N.; Taliani, C.; Bradley, D. D. C.; dos Santos, D. A.; Brédas, J. L.; Logdlund, M.; Salaneck, W. R.; Nature 1999, 397, 121.
- 6. Yu, G.; Gao, J.; Hummelen, J. C.; Wudl, F.; Heeger, A. J.; Science 1995, 270, 1789.
- 7. Halls, J. J. M.; Walsh, C. A.; Greenham, N. C.; Marseglia, E. A.; Friend, R. H.; Moratti, S. C.; Holmes, A. B.; Nature 1995, 376, 498.
- 8. Brabec, C. J.; Sariciftci, N. S.; Hummelen, J. C.; Adv. Funct. Mater 2001, 11, 15.
- 9. Garnier, F.; Hajlaoui, R.; Yassar, A.; Srivastava, P.; Science 1994, 265, 1684.
- 10. Yang, Y.; Heeger, A. J.; Nature 1994, 372, 344.
- 11. Sirringhaus, H.; Tessler, N.; Friend, R. H.; Science 1998, 280, 1741.
- 12. Tessler, N.; Denton, G. J.; Friend, R. H.; Nature 1996, 382, 695.
- 13. Hide, F.; Schwartz, B. J.; DiazGarcia, M. A.; Heeger, A. J.; Chem. Phys. Lett 1996, 256, 424.
- 14. Berggren, M.; Dodabalapur, A.; Slusher, R. E.; Bao, Z.; Nature 1997, 389, 466.
- 15. Gupta, R.; Stevenson, M.; McGehee, M. D.; Dogariu, A.; Srdanov, V.; Park, J. Y.; Heeger, A. J.; Synth. Met 1999, 102, 875.
- 16. Tessler, N.; Ho, P. K. H.; Cleave, V.; Pinner, D. J.; Friend, R. H.; Yahioglu, G.; Le Barny, P.; Gray, J.; de Souza, M.; Rumbles, G.; Thin Solid Films 2000, 363, 64.
- 17. Crone, B.; Dodabalapur, A.; Lin, Y. Y.; Filas, R. W.; Bao, Z.; LaDuca, A.; Sarpeshkar, R.; Katz, H. E.; Li, W.; Nature 2000, 403, 521.
- 18. Akcelrud, L.; Prog. Polym. Sci 2003, 28, 875.
- 19. Machado, A. M.; Motta Neti, J. D.; Cossiello, R. F.; Atvars, T. D. Z.; Karasz, F. E.; Akcelrud, L.; Polymer 2005, 46, 2452.
- 20. Deus, J. F.; Andrade, M. L.; Atvars, T. D. Z.; Akcelrud, L.; Chem. Phys 2004, 297, 117.
- 21. Deus, J. F.; Corradini, W. A.; Atvars, T. D. Z.; Akcelrud, L.; Macromolecules 2004, 37, 6938.
- 22. Pei, J.; Liu, X.; Chen, Z.; Zhang, X.; Lai, Y.; Huang, W.; Macromolecules 2003, 36, 323.
- 23. Burrows, H. D.; Melo, J. S.; Serpa, C.; Arnaut, L. G.; Miguel, M. G.; Monkman, A. P.; Hamblett, I.; Navaratnam, S.; Chem. Phys. 2002, 285, 3.
- 24. Rothe, C.; Palsson, L. O.; Monkman, A. P.; Chem. Phys. 2002, 285, 95.
- 25. Tirapattur, S.; Belletête, M.; Drolet, N.; Leclerc, M.; Durocher, G.; Macromolecules 2002, 35, 8889.
- 26. Zeng, G.; Yu, W.; Chua, S.; Huang, W.; Macromolecules 2002, 35, 6907.
- 27. Ding, J.; Day, M.; Robertson, G.; Roovers, J.; Macromolecules 2002, 35, 3474.
- 28. Palsson, L.; Wang, C.; Russell, D. L.; Monkman, A. P.; Bryce, M. R.; Rumbles, G.; Samuel, I. D. W.; Chem. Phys. 2002, 279, 229.
- 29. Zhan, X.; Liu, Y.; Wu, X.; Wang, S.; Zhu, D.; Macromolecules 2002, 35, 2529.
- 30. Lupton, J. M.; Klein, J.; Chem. Phys. Lett. 2002, 363, 204.
- 31. Rothe, C.; Hintschich, S. I.; Palsson, L.; Monkman, A. P.; Guentner, R.; Scherf, U.; Chem. Phys. Lett. 2002, 360, 111.
- 32. Cornil, J.; Gueli, I.; Dkhissi, A.; Garcia, J. C. S.; Hennebicq, E.; Calbert, J. P.; Lemaur, V.; Beljonne, D.; Brédas, J. L.; J. Chem. Phys 2003, 118, 6615.
- 33. Pasini, M.; Destri, S.; Porzio, W.; Botta, C.; Giovanella, U.; J. Mater. Chem. 2003, 13, 807.
- 34. Herz, L. M.; Silva, C.; Phillips, R. T.; Setayesh, S.; Müllen, K.; Chem. Phys. Lett. 2001, 347, 318.
- 35. Meskers, S. C. J.; Hübner, J.; Oestreich, M.; Bässler, H.; Chem. Phys. Lett. 2001, 339, 223.
- 36. Buckley, A. R.; Rahn, M. D.; Hill, J.; Cabanillas-Gonzalez, J.; Foz, A. M.; Bradley, D. D. C.; Chem. Phys. Lett. 2001, 339, 331.
- 37. Geng, Y.; Trajkovska, A.; Katsis, D.; Ou, J. J.; Culligan, S. W.; Chen, S. H.; J. Am. Chem. Soc. 2002, 124, 8337.
- 38. Meskers, S. C. J.; Hübner, J.; Oestreich, M.; Bässler, H.; J. Phys. Chem. B 2001, 105, 9139.
- 39. Lemmer, U.; Hein, S.; Mahrt, R. F.; Scherf, U.; Hopmeir, M.; Wiegner, U.; Gobel, R. O.; Mullen, K.; Bassler, H.; Chem. Phys. Lett 1995, 240, 371.
- 40. Charas, A.; Morgado, J.; Martinho, J. M. G.; Alcácer, L.; Lim, S. F.; Friend, R. H.; Cacialli, F.; Polymer 2003, 44, 1843.
- 41. Yu, W. L.; Pei, J.; Huang, W.; Heeger, A. J.; Adv. Mater 2000, 12, 828.
- 42. Virgili, T.; Lidzey, D. G.; Bradley, D. D. C.; Adv. Mater 2000, 12, 58.
- 43. Whitehead, K. S.; Grell, M.; Bradley, D. D. C.; Jandke, M.; Strohriegl, P.; Appl. Phys. Lett. 2000, 76, 2946.
- 44. Grell, M.; Knoll, W.; Lupo, D.; Meisel, A.; Miteva, T.; Neher, D.; Nothofer, H. G.; Scherf, U.; Yasuda, A.; Adv. Mater 1999, 11, 671.
- 45. Yang, Y.; Pei, Q.; Appl. Phys. Lett. 1997, 81, 3294.
- 46. Lee, J.; Klaerner, G.; Miller, R. D.; Chem. Mater. 1999, 11, 1083.
- 47. Klaerner, G.; Lee, J. K.; Davey, M. H.; Miller, R. D.; Adv. Mater 1999, 11, 115.
- 48. Kreyenschimdt, M.; Klaerner, G.; Fuhrer, T.; Ashenhurst, J.; Karg, S.; Chen, W.; Lee, V. Y.; Scott, J. C.; Miller, R. D.; Macromolecules 1998, 31, 1099.
- 49. Bliznyuk, V. N.; Carter, S. A.; Scott, J. C.; Klaerner, G.; Miller, R. D.; Macromolecules 1999, 32, 361.
- 50. Yang, Y.; Pei, Q.; J. Am. Chem. Soc 1996, 118, 7416.
- 51. He, Y.; Gong, S.; Hattori, R.; Kanicki, J.; Appl. Phys. Lett 1999, 74, 2265.
- 52. Yu, W.; Cao, Y.; Pei, J.; Huang, W.; Heeger, A. J.; Appl. Phys. Lett 1999, 75, 3270.
- 53. Yu, W.; Pei, J.; Cao, Y.; Huang, W.; Heeger, A. J.; Chem. Commun 1999, 18, 1837.
- 54. Liu, B.; Yu, W.; Lai, Y.; Huang, W.; Chem. Commun 2000, 7, 551.
- 55. Sainova, D.; Miteva, T.; Nothofer, H.; Scherf, U.; Glowacki, I.; Ulanski, J.; Fujikawa, H.; Neher, D.; Appl. Phys. Lett 2000, 76, 1810.
- 56. Weinfurtner, K.; Fujikawa, H.; Tokito, S.; Taga, Y.; Appl. Phys. Lett 2000, 76, 2502.
- 57. Jenekhe, S. A.; Osaheni, J. A.; Science 1994, 265, 765.
- 58. Liu, B.; Yu, W.; Lai, Y.; Huang, W.; Macromolecules 2000, 33, 8945.
- 59. Grell, M.; Bradley, D. D. C.; Ungar, G.; Hill, J.; Whitehead, K. S.; Macromolecules 1999, 32, 5810.
- 60. Cadby, A. J.; Lane, P. A.; Wohlgenannt, M.; An, C.; Vardeny, Z. V.; Bradley, D. D. C.; Synth. Met. 2000, 111-112, 515.
- 61. Bauer, C.; Urbasch, G.; Giessen, H.; Meisel, A.; Nothofer, H.; Neher, D.; Scherf, U.; Mahrt, R. F.; Chem. Phys. Chem 2000, 3, 142.
- 62. Zhan, X.; Lui, Y.; Yu, G.; Wu, X.; Zhu, D.; Sun, R.; Wang, D.; Epstein, A. J.; J. Mater. Chem. 2001, 11, 1606.
- 63. Leclerc, M.; J. Polym. Sci., Part A: Polym. Chem. 2001, 39, 2867.
- 64. Kameshima, H.; Nemoto, N.; Endo, T.; J. Polym. Sci., Part A: Polym. Chem. 2001, 39, 3143.
- 65. Suh, Y.; Ko, S. W.; Jung, B.; Shim, H.; Opt. Mater. 2002, 21, 109.
- 66. Bliznyuk, V. N.; Carter, S. A.; Scott, J. C.; Klärner, G.; Miller, R. D.; Miller, D. C.; Macromolecules 1999, 32, 361.
- 67. Cadby, A. J.; Lane, P. A.; Mellor, H.; Martin, S. J.; Grell, M.; Giebeler, C.; Bradley, D. D. C.; Wohlgenannt, M.; An, C.; Vardeny, Z. V.; Phys. Rev. B: Condens. Matter Matter. Phys. 2000, 62, 15604.
- 68. Lee, J. I., Lee, V. Y., Miller, R. D.; ETRI J. 2002, 24, 409.
- 70. Grell, M.; Bradley, D. D. C.; Inbasekaran, M.; Woo, E. P.; Adv. Mater. 1997, 9, 798.
- 72. Schartel, B.; Wachtendorf, V.; Grell, M.; Bradley, D. D. C.; Hennecke, M.; Phys. Rev. B: Condens. Matter Mater. Phys. 1999, 60, 277.
- 73. Long, X.; Grell, M.; Malinowski, A.; Bradley, D. D. C.; Opt. Mater 1998, 9, 70.
- 74. Weinfurtner, K. H.; Weissortel, F.; Harmgarth, G.; Salbeck, J.; Proc. SPIE. 1998, 3476, 40.
- 75. Geng, Y. H.; Katsis, D.; Culligan, S. W.; Ou, J. J.; Chen, S. H.; Rothberg, L. J.; Chem. Mater 2002, 14, 463.
- 76. Katsis, D.; Geng, Y. H.; Culligan, S. W.; Trajkovska, A.; Chen, S. H.; Rothberg, L.; Chem. Mater. 2002, 14, 1332.
- 77. Andersson, M. R.; Yu, G.; Heeger, A. J.; Synth. Met. 1997, 85, 1275.
- 78. Xu, B.; Holdcroft, S.; Macromolecules 1993, 26, 4457.
- 79. Swanson, L. S.; Shinar, J.; Ding, Y. W.; Barton, T. J.; Synth. Met. 1993, 55, 1.
- 80. Greenham, N. C.; Brown, A. R.; Bradley, D. D. C.; Adv. Mater. 1994, 6, 491.
- 81. Greenham, N. C.; Brown, A. R.; Bradley, D. D. C.; Friend, R. H.; Synth. Met. 1993, 57, 4134.
- 82. Braun, D.; Gustafsson, G.; McBranch, D.; Heeger, A. J.; J. Appl. Phys. 1992, 72, 564;
- Mao, H.; Xu, B.; Holdcroft, S.; Macromolecules 1993, 26, 1163.
- 83. Uchida, M.; Ohmori, Y.; Morishima, C.; Yoshino, K.; Synth. Met. 1993, 57, 4168.
- 84. Tada, K; Sonoda, T.; Yokota, Y.; Kobashi, K.; Yoshino, K.; J. Appl. Phys 1998, 84, 5635.
- 85. Klaerner, G.; Miller, R. D.; Macromolecules 1998, 31, 2007.
- 86. Jiang, X.; Liu, S.; Ma, H.; Jen, A. K. Y.; Appl. Phys. Lett 2000, 76, 1813.
- 87. Rehahn, M.; Schluter, A. D.; Wegner, G.; Feast, W. J.; Polymer 1989, 30, 1054.
- 88. Yamamoto, T.; Morita, A.; Miyazaki, Y.; Maruyama, T.; Wakayama, H.; Zhou, Z. H.; Nakamura, Y.; Kanbara, T.; Macromolecules 1992, 25, 1214.
- 89. Kannan R.; He, G. S.; Yuan, L.; Faming, X.; Prasad, P. N.; Dombroskie, A. G.; Reinhardt, B. A; Baur, J. W.; Vaia, R. A.; Tan, L. S.; Chem. Mater 2001, 13, 1896.
- 90. Ding, J.; Day, M.; Robertson, G.; Roovers, J.; Macromolecules 2002, 35, 3474.
- 91. Ranger, M.; Rondeau, D.; Leclerc, M.; Macromolecules 1997, 30, 7686.
- 92. Miyaura, M.; Suzuki, A.; Chem. Rev 1995, 95, 2457.
- 93. Setayesh, S.; Marsitzky, D.; Müllen, K.; Macromolecules 2000, 33, 2016.
- 94. Nelson, J.; Current Opinion in Solid State and Materials Science 2002, 6, 87.
- 95. Silva, G. B.; Santos, L. F.; Faria, R. M.; Grae, C. F. O.; Physica B 2001, 308310, 1078.
- 96. Mahrt, R. F.; Pauck, T.; Lemmer, U.; Siegner, U.; Hopmeier, M.; Hennig, R.; Bassler, H.; Gobel, E. O.; Bolivar, P. H.; Wegmann, G.; Kurz, H.; Scherf, U.; Mullen, K.; Phys. Rev. B: Condens. Matter Mater. Phys. 1996, 54, 1759.
- 97. Yan, M.; Rothberg, L. J.; Kwock, E. W.; Miller, T. M.; Phys. Rev. Lett 1995, 75, 1992.
- 98. Beljonne, D.; Cornil, J.; Sirringhaus, H.; Brown, P. J.; Shkunov, M.; Friend, R. H.; Brédas, J. L.; Adv. Funct. Mater 2001, 11, 229.
- 99. Halls, J. J. M.; Baigent, D. R.; Cacialli, F.; Greenham, N C.; Friend, R. H.; Moratti, S C.; Holmes, A. B.; Thin. Solid Films 1996, 276, 13.
- 100. Romijn, I. G.; Pasveer, W. F.; Martens, H. C. F.; Brom, H. B.; Michels, M. A. J.; Synth. Met 2001, 119, 439
- 101. Parker, I. D.; J. Appl. Phys 1994, 75, 1657.
- 102. Petritsch, K.; Friend, R. H.; Synth. Met 1999, 102, 976.
- 103. Barashkov, N. N.; Gunder, O. A.; Fluorescent Polymers, Ellis Horwood: New York, 1994.
- 104. Schaller, R. D.; Lee, L. F.; Johnson, J. C.; Haber, L. H.; Saykally, R. J.; Vieceli; J.; Benjamin, I.; Nguyen, T.; Schwartz, B. J.; J. Phys. Chem. B 2002, 74, 10.
- 105. Kasha, M.; Rawls, H. R.; El-Bayoumi, M. A.; Pure Appl. Chem 1965, 11, 371.
- 106. Birks, J. B.; Photophysics of Aromatic Molecules, John Wiley & Sons Ltd.: New York, 1970.
- 107. Blatchford, J. W.; Jessen, S. W.; Lin, L. B.; Gustafson, T. L.; Fu, D. K.; Wang, H. L.; Swager, T. M.; Macdiarmid, A. G.; Epstein, A. J.; Phys. Rev. B: Condens. Matter Matter. Phys. 1996, 54, 9180.
- 108. Lim, S. H.; Bjorklund, T. G.; Bardeen, C. J.; Chem. Phys. Lett 2001, 342, 555.
- 109. Hagler, T. W.; Pakbaz, K.; Heeger, A. J.; Phys. Rev. B: Condens. Matter Matter. Phys 1994, 49, 10968.
- 110. Guha, S.; Rice, J. D.; Yau, Y. T.; Martin, C. M.; Chandrasekhar, M.; Chandrasekhar, H. R.; Guentner, R.; Freitas, P. S.; Ucherf, U.; Phys. Rev. B: Condens. Matter Matter. Phys 2002, 67, 125204.
- 111. Nguyen, T. Q.; Schwartz, B. J.; Schaller, R. D.; Johnson, J. C.; Lee, L. F.; Haber, L. H.; Saykally, R. J.; J. Phys. Chem. B 2001, 105, 5153.
- 112. Gettinger, C. L.; Heeger, A. J.; J. Chem. Phys 1994, 101, 74.
- 113. Tikhoplav, R. K.; Hess, B. C.; Synth. Met 1999, 101, 236.
- 114. Köhler, A.; dos Santos, D. A.; Beljonne, D.; Shuai, Z.; Brédas, J. L.; Holmes, A. B.; Kraus, A.; Müllen, K.; Friend, R. H.; Nature 1998, 392, 903.
- 115. Oberski, J. M.; Greiner, A.; Bassler, H.; Chem. Phys. Lett 1991, 184, 391.
- 116. Chang, R.; Hsu, J. H.; Fann, W. S.; Liang, K. K.; Chang, C. H.; Hayashi, M.; Yu, Y.; Lin, S. H.; Chang, E. C.; Chuang, K. R.; Chen, S. A.; Chem. Phys. Lett 2000, 317, 142.
- 117. Pichler, K.; Halliday, D. A.; Bradley, D. D. C.; Brurn, P. L.; Friend, R. H.; Holmes, A. B.; J. Phys.: Condens. Matter 1993, 5, 7155.
- 118. Yu, J.; Hsu, J. H.; Chuang, K. R.; Chão, C. I.; Chen, S. A.; Kao, F. J.; Fann, W. S.; Lin, S. H.; Synth. Met 1996, 82, 159.
- 119. Andrade, M. L.; Atvars, T. D. Z.; J. Phys. Chem. B 2004, 108, 3975.
- 120. Andrade, M. L.; Atvars, T. D. Z.; Macromolecules 2004, 37, 6938.
- 121. Xu, J.; Luo, C.; Atvars, T. D. Z.; Weiss, R. G.; Res. Chem. Intermed. 2004, 30, 509.
- 122. Brown, G.; Guardala, N. A.; Atvars, T. D. Z.; Weiss, R. G.; J. Polym. Sci., Part B: Polym. Phys 2004, 42, 2957.
- 123. Kalyanasundaram, K.; Photochemistry in Microheterogeneous Systems, Academic Press: Orlando, 1992.
- 124. Scaiano, J. C.; Handbook of Organic Photochemistry, CRC Press: Boca Raton, 1989, vols. I e II.
- 125. Gilbert, A.: Baggott, J.: Essentials of Molecular Photochemistry, Blackwell Scientific Publications: Oxford, 1991.
- 126. Lakowicz; J. R.; Principles of Fluorescence Spectroscopy, 2nd ed., Academic Press: New York, 1999.
- 127. Vekshin, N. L.; Energy Transfer in Macromolecules, SPIE Optical Engineering Press: Washington, 1997.
- 128. Rohatgi-Mukherjee, K. K.; Fundamentals of Photochemistry, revised edition, New Age International Limited: New Delhi, 1978.
- 129. Cossiello, R. F.; Kowalski, E.; Rodrigues, P. C.; Akcelrud, L.; Bloise, A. C.; de Azevedo, E. R.; Bonagamba, T. J.; Atvars, T. D. Z.; Macromolecules 2005, 38, 925.
- 130. Cossiello, R. F.; Akcelrud, L.; Atvars, T. D. Z.; J. Braz. Chem. Soc 2005, 16, 74.
- 131. Aguiar, M.; Akcelrud, L.; Pinto, M. R.; Atvars, T. D. Z.; Karsz, F. E.; Saltiel, J.; J. Photoscience 2003, 10, 149.
- 132. Sariciftci, N. S.; Primary Photoexcitations in Conjugated Polymers: Molecular exciton versus semiconductor band model, World Scientific Publishing Co.: Singapure, 1997.
- 133. Borsenberger, P. M.; Weiss, D. S.; Organic Photoreceptors for Imaging Systems, Marcel Dekker: New York, 1998.
- 134. Yu, J.; Fann, W. S.; Kao, F. J.; Yang, D. Y.; Lin, S. H.; Synth. Met 1994, 66, 143.
- 135. Förster, T., Discuss. Faraday Soc. 1959, 27, 7.
- 136. Berkovic, G., J. Chem. Educ. 1984, 61, 832.
- 137. Suppan, P.; Chemistry and Light, Royal Society of Chemistry: Cambridge, 1994.
- 138. Lakowicz; J. R.; Topics in Fluorescence Spectroscopy, Plenum Press: New York, 1991, vol. 2.
- 139. Adam, W.; Cilento, G.; Chemical and Biological Generation of Excited States; Academic Press: New York, 1982.
- 140. Turro, N. J.; Modern Molecular Photochemistry, 2nd ed., Menlo Park: Califórnia, 1978.
- 141. Kopecky J.; Organic Photochemistry: A Visual Approach, VCH Publishers: New York, 1992.
- 142. Marinari, A.; Saltiel, J.; Mol. Photochem 1976, 7, 225.
- 143. Conte, J. C.; Martinho, J. M. G.; J. Lumin 1981, 22, 273.
- 144. Martinho, J. M. G.; Conte, J. C.; J. Lumin 1981, 22, 285.
- 145. Martinho, J. M. G.; Pereira, V. R.; Silva, G.; Conte, J. C.; J. Photochem 1985, 30, 383.
- 146. Dogariu, A.; Vacar, D.; Heeger, A. J.; Phys. Rev. B: Condens. Matter Matter. Phys 1998, 58, 10218.
- 147. Powell, R. C.; Soos, Z. G.; J. Lumin 1975, 11, 1.
- 148. Eisenthal, K. B.; Siegel, S.; J. Chem. Phys 1964, 41, 652.
- 149. Yang, C. Y.; Hide, F.; Diaz-Garcia, M. A.; Heeger, A. J.; Cao, Y.; Polymer 1997, 39, 2299.
- 150. Belletête, M.; Morin, J.; Beaupré, S.; Ranger, M.; Leclerc, M.; Durocher, G.; Macromolecules 2001, 34, 2288.
- 151. Xia, C.; Advincula, R. C.; Macromolecules 2001, 34, 5854.
- 152. Jin, S.; Kang, S.; Kim, M.; Chan, Y. U.; Kim, J. Y.; Lee, K.; Gal, Y.; Macromolecules 2003, 36, 3841.
- 153. Park, J. H.; Ko, H. C.; Kim, J. H.; Lee, H.; Synth. Met. 2004, 144, 193.
- 154. Belletête, M.; Morin, J.; Beaupré, S.; Leclerc, M.; Durocher, G.; Synth. Met. 2002, 126, 43.
- 155. Kulkarni, A. P.; Kong, X.; Jenekhe, S. A.; J. Phys. Chem. B. 2004, 108, 8689.
Datas de Publicação
-
Publicação nesta coleção
03 Abr 2006 -
Data do Fascículo
Abr 2006
Histórico
-
Aceito
21 Jun 2005 -
Recebido
29 Set 2004