Resumo
Monolithic stationary phases represent a new generation of chromatographic separation media. These phases consist of a continuous separation bed prepared by in situ polymerization or consolidation inside the column tubing. In recent years, their simple preparation procedure, unique properties and excellent performance have attracted quite remarkable attention in liquid chromatography and capillary electrochromatography. This review summarizes the preparation, characterization and applications of monolithic stationary phases. The analytical potential of these columns is demonstrated with separations involving various families of compounds in different separation modes.
monolithic stationary phases; high performance liquid chromatography; capillary electrochromatography
monolithic stationary phases; high performance liquid chromatography; capillary electrochromatography
REVISÃO
Fases estacionárias monolíticas para separações cromatográficas
Monolithic stationary phases for chromatographic separations
Anízio M. Faria; Carla B. G. Bottoli; Isabel C. S. F. Jardim; Carol H. Collins* * e-mail: chc@iqm.unicamp.br
Instituto de Química, Universidade Estadual de Campinas, CP 6154, 13084-971 Campinas - SP
ABSTRACT
Monolithic stationary phases represent a new generation of chromatographic separation media. These phases consist of a continuous separation bed prepared by in situ polymerization or consolidation inside the column tubing. In recent years, their simple preparation procedure, unique properties and excellent performance have attracted quite remarkable attention in liquid chromatography and capillary electrochromatography. This review summarizes the preparation, characterization and applications of monolithic stationary phases. The analytical potential of these columns is demonstrated with separations involving various families of compounds in different separation modes.
Keywords: monolithic stationary phases; high performance liquid chromatography; capillary electrochromatography.
INTRODUÇÃO
Apesar de mais de trinta anos de intensa pesquisa e do alto padrão alcançado atualmente pela tecnologia de colunas para cromatografia líquida, alguns problemas persistem e novos desenvolvimentos impulsionam pesquisas nesta área. O bom desempenho cromatográfico das colunas atuais é resultado do desenvolvimento da tecnologia de colunas, que permitiu a produção de partículas bem definidas e cada vez menores, concomitante à diminuição do comprimento e do diâmetro da coluna. Porém, as colunas recheadas com materiais particulados parecem ter alcançado seu limite de desempenho nos equipamentos convencionais. Embora seja tecnicamente possível uma maior redução do tamanho das partículas (< 3,0 µm), na prática esta possibilidade se torna inviável. A diminuição da permeabilidade das colunas recheadas com partículas muito pequenas provoca um aumento acentuado da resistência ao fluxo de fase móvel e, conseqüentemente, uma considerável queda de pressão ao longo da coluna, resultando em separações de baixa eficiência1-3. Portanto, novas estratégias vêm sendo delineadas para obtenção de colunas mais eficientes.
MacNair et al.4 propuseram um sistema de cromatografia líquida de ultra-alta pressão (ultra-CLAE) ("ultrahigh-pressure liquid chromatography"), que possibilita separações a pressões de até uma ordem de grandeza superior ao sistema convencional de cromatografia líquida. A utilização de eletrocromatografia capilar (ECC) ("capillary electrochromatography") também foi sugerida em substituição aos equipamentos convencionais, pois a vazão da fase móvel é controlada por um fluxo eletroosmótico (FEO) ("electroosmotic flow") fluxo de uma solução aquosa em um tubo com paredes e partículas eletrocarregadas sob a ação de campo elétrico alto o qual independe de pressão5. Eficiências de colunas superiores a duzentos mil pratos m-1 são obtidas por esta técnica2. Porém, a utilização de instrumentação comum de cromatografia líquida de alta eficiência (CLAE) ("high performance liquid chromatography"), obtendo eficiências de separação nestas proporções, também tornou-se possível com o desenvolvimento das colunas monolíticas6.
Uma fase monolítica é um meio contínuo de separação (fase ou suporte contendo uma "partícula única"), comumente em formato cilíndrico, que possui uma estrutura sólida e altamente porosa, de pequenos domínios e canais relativamente grandes, que fornecem altas permeabilidade e eficiência de coluna (Figura 1). Os domínios podem ser micro ou mesoporosos, ter estrutura dupla de poros (micro e mesoporos), ou podem ser não porosos7. [Segundo a IUPAC, os microporos são poros com diâmetros que não excedem a 2 nm (20 Å), os macroporos são definidos como poros com diâmetros superiores a 50 nm (500 Å), e os mesoporos são poros de tamanhos intermediários a estes, 2 a 50 nm]8. A fase móvel passa pela coluna principalmente através dos canais do monolito, nominalmente de 2 µm de diâmetro. A estrutura e geometria destes canais oferecem uma menor resistência à vazão da fase móvel, comparada aos materiais particulados. Portanto, é possível realizar análises em vazões elevadas (> 5 mL min-1) mas sem perdas significativas da eficiência de separação. Esta é uma das principais vantagens oferecidas pelas fases monolíticas, além da redução do tempo de análise, sem comprometer a resolução cromatográfica9,10.
Apesar de ser uma linha de pesquisa bastante atual, a primeira aplicação das fases monolíticas data do início da década de 70, quando Ross e Jefferson12 empregaram esponja de poliuretana como fase estacionária para cromatografia gasosa (CG) ("gas chromatography") e CLAE. Devido à excessiva dilatação e ao amolecimento em alguns solventes orgânicos, o interesse pelas fases estacionárias (FE) monolíticas tornou-se ínfimo a partir de então. O renascimento dos meios contínuos de separação aconteceu somente no fim da década de 80 quando Hjertén et al.13 usaram, de forma bem sucedida, géis de baixo grau de entrecruzamento, comprimidos, como uma FE contínua em separações de proteínas por CLAE. Mas, foi em 1992 que a tecnologia de colunas monolíticas começou a se desenvolver. Svec e Fréchet6 introduziram uma classe completamente nova de meio contínuo de separação baseada em monolitos poliméricos macroporosos, rígidos, produzidos por um processo de "moldagem" muito simples. As propriedades únicas do monolito polimérico resultante possibilitaram seu uso em uma ampla variedade de aplicações. Em 1996, Minakuchi et al.14, empregando o processo sol-gel, prepararam monolitos de sílica derivatizada com grupos octadecil como fase estacionária para CLAE.
A popularização das fases monolíticas aconteceu a partir de 2000, quando monolitos de sílica se tornaram disponíveis comercialmente. Ao longo destes últimos cinco anos os materiais monolíticos tiveram ampla difusão, com algumas centenas de trabalhos publicados. Apesar do emprego destas fases em várias técnicas, tais como microextração em fase sólida15, cromatografia por bioafinidade16, cromatografia por injeção seqüencial17, dentre outras, aproximadamente 95% das aplicações de fases monolíticas concentram-se em sistemas de CLAE e ECC.
A eletrocromatografia capilar é uma técnica de separação eletrocinética bastante atual, que possui características que permitem considerá-la um híbrido de eletroforese capilar (EC) ("capillary electrophoresis") e CLAE. Em ECC, a fase móvel é conduzida por um fluxo eletroosmótico (FEO) através do capilar, usando fases móveis e estacionárias típicas de CLAE, que fornecem as interações essenciais para as separações. Devido ao perfil planar do fluxo eletroosmótico, a ECC oferece eficiências de separação muito maiores que as obtidas em CLAE, a qual apresenta fluxo laminar. Diferente da eletroforese capilar, a ECC não se restringe somente a analitos ionizados, tanto espécies neutras como carregadas podem interagir com a fase estacionária, mostrando ser uma técnica de separação muito mais ampla18. As principais colunas empregadas em ECC são as recheadas com partículas e as tubulares abertas19,20, sendo as primeiras responsáveis pela quase totalidade. Porém, algumas dificuldades, por ex., o enchimento de capilares com materiais particulados e a fabricação de filtros para retenção de partículas dentro do capilar, limitaram maiores desenvolvimentos da ECC. Em particular, a fabricação dos filtros acarreta uma série de desvantagens, a citar: falta de reprodutibilidade, influência imprevisível no FEO e, principalmente, a formação de bolhas3. Devido à facilidade de preparação e à ausência de filtros retentores, uma vez que a fase estacionária se encontra aderida às paredes do capilar, as fases monolíticas vêm sendo cada vez mais usadas em ECC. Além disso, o processo de preparação simples destas fases estacionárias permite a incorporação de espécies iônicas, necessárias para garantir o FEO21, além da possibilidade de controle de suas propriedades, tais como porosidade, área superficial e funcionalidade.
As fases monolíticas vêm se desenvolvendo no campo de preparação de fases estacionárias, principalmente devido à facilidade de incorporação de diferentes grupos (moléculas hidrofóbicas, moléculas carregadas, moléculas para interações específicas, etc.) durante seu preparo. Uma outra vantagem destas fases é a possibilidade da preparação e funcionalização em uma única etapa22,23.
Dependendo da natureza do material monolítico duas categorias gerais podem ser identificadas: i) colunas monolíticas baseadas em polímeros orgânicos6,24,25 e, ii) colunas monolíticas baseadas em sílica7,14,26.
PREPARAÇÃO DE FASES ESTACIONÁRIAS MONOLÍTICAS
As fases estacionárias monolíticas podem ser preparadas tanto em colunas de dimensões similares às das convencionais utilizadas em CLAE, como também em colunas capilares de sílica fundida, empregadas em microcromatografia líquida de alta eficiência (µCLAE) e eletrocromatografia capilar. As fases estacionárias monolíticas preparadas para ECC são muito similares às originalmente desenvolvidas para CLAE. Porém, algumas peculiaridades, principalmente no que se refere à ECC, serão descritas distintamente, pois, como os mecanismos de separação diferem entre si, alguns tratamentos adicionais normalmente se fazem necessários.
Preparação de monolitos poliméricos orgânicos
A preparação de cilindros monolíticos poliméricos orgânicos é relativamente simples e realizada dentro da própria coluna cromatográfica, convencional ou capilar, através da polimerização in situ. Os materiais poliméricos são de maior interesse quando utilizados em colunas capilares, pois a dilatação e o encolhimento frente a solventes orgânicos são menos pronunciados que em colunas convencionais. Além disso, a condução da fase móvel por FEO, sem pressão, permite o uso de polímeros com menor grau de entrecruzamento, quando empregados em ECC, pois, não há o problema de resistência ao fluxo da fase móvel como ocorre em CLAE. Outra vantagem do uso de monolitos poliméricos em ECC é a boa estabilidade química, entre pH 2 e 12, visto que, freqüentemente, se realizam separações em condições de pH fora da faixa de estabilidade da sílica (pH de 2 a 8), para enriquecimento do fluxo eletroosmótico1,3,27.
Até o momento três tipos diferentes de polímeros orgânicos foram preparados na forma de suportes cromatográficos monolíticos em CLAE e ECC: poliacrilamida21,28, poliestireno-divinilbenzeno11,24,29-31 e polimetacrilato25,32-36. Os polimetacrilatos são os mais empregados, devido à ampla variedade de monômeros funcionais e a fácil derivatização após a polimerização. De modo geral, a preparação das fases monolíticas poliméricas orgânicas é fundamentalmente a mesma.
Normalmente em CLAE, uma mistura consistindo de monômeros, um agente de entrecruzamento, um iniciador e agentes porogênicos é colocada em um molde, coluna convencional ou capilar, com uma das extremidades vedadas, fechando-se a outra após o enchimento. A polimerização é iniciada por aquecimento, em temperaturas que variam de 50 a 80 °C25,31,32-36, ou por luz UV37-39. A principal vantagem da iniciação por luz UV é a polimerização local do monolito. O material utilizado para fechar as extremidades das colunas é substituído por conexões e ligado a uma bomba cromatográfica. Um solvente é bombeado através da coluna para remoção dos resíduos e agentes porogênicos, após a polimerização ter sido concluída. Em seguida, a coluna é seca com a passagem de gás nitrogênio, sendo subseqüentemente derivatizada para o modo cromatográfico de interesse24,37,40.
A derivatização ou funcionalização da fase estacionária monolítica é obtida pela modificação de grupos reativos da superfície do monolito com reagentes específicos, ou pela inserção de monômeros funcionais na superfície dos poros. No primeiro caso25,31,34, os reagentes são introduzidos na coluna (convencional ou capilar) logo após a etapa de preparação do monolito, deixando-a em repouso em local adequadamente aquecido. Os resíduos são removidos pelo bombeamento de um solvente apropriado. No segundo caso21,28,40, a funcionalização é realizada simultaneamente à preparação da matriz monolítica com a adição de um monômero funcional adequado à mistura de polimerização, seguindo o procedimento previamente descrito.
Petro et al.24 prepararam fases estacionárias monolíticas poliméricas em tubos de aço inoxidável de 50 mm x 8 mm de d.i. para CLAE. Os monolitos foram preparados pela polimerização de estireno e divinilbenzeno a 70 °C, dentro do tubo, empregando dodecanol como agente porogênico. As fases monolíticas apresentaram diâmetros de poros na faixa de 700 a 7000 nm, que permitiram vazões de até 20 mL min-1, sem queda excessiva de pressão ao longo da coluna, sendo só de 2,6 MPa. As fases monolíticas de poli(estireno-co-divinilbenzeno) foram empregadas na separação de padrões de poliestireno e os resultados foram similares aos obtidos com colunas comerciais de materiais particulados.
A mistura de polimerização pode ser preparada em uma solução aquosa ou na presença de solventes orgânicos. O grande inconveniente do preparo da mistura de polimerização em solução aquosa é a baixa solubilidade de um grande número de monômeros em água. Porém, esta limitação pode ser contornada pela adição de um agente compatibilizante na solução aquosa, por ex., surfactantes ou poli(oxietileno) (PEO). Em contraste às propriedades solubilizantes "fixas" da água, os solventes orgânicos possuem ampla faixa de polaridade, variando de altamente apolares até extremamente polares. Desta forma, a preparação de uma matriz monolítica em agentes porogênicos permite a utilização de monômeros de características mais diversificadas, conforme os objetivos propostos. Além disso, os agentes porogênicos são responsáveis pelo controle fino das propriedades porosas dos monolitos resultantes. Na mistura de polimerização podem ser adicionados um, dois ou até três desses agentes. Freqüentemente dois solventes são empregados, um responsável pela formação de mesoporos e outro, de estrutura molecular maior (decanol, dodecanol, etc.), pela moldagem dos macroporos.
Os maiores inconvenientes do uso de monolitos poliméricos orgânicos para CLAE convencional, em tubos de aço inoxidável de 4,6 mm de diâmetro interno são a dilatação acentuada em solventes orgânicos e a excessiva compressão quando são empregadas vazões altas.
Em ECC, a preparação de fases monolíticas poliméricas orgânicas pode ser dividida em três etapas:
a) Preparação das paredes internas do capilar
A coluna capilar é primeiramente lavada com uma base forte para que os grupos siloxanos da superfície interna dos capilares de sílica fundida sejam hidrolisados, aumentando a densidade de grupos silanóis que, por sua vez, servirão como âncoras para reações subseqüentes. Em seguida, a coluna capilar é preenchida com um agente silanizante bifuncional, tipicamente [3-(metacriloiloxi)propil]trimetoxissilano, e vedada. A coluna é deixada por um determinado tempo, com ou sem aquecimento, para que a reação ocorra. Os resíduos são removidos do interior do capilar pela passagem de um solvente apropriado. Devido ao tratamento, ligações do tipo ºSi-O-Si-Cº são formadas entre as paredes do capilar e, os grupos metacriloíla reativos (Figura 2) ficam disponíveis para subseqüente fixação da matriz monolítica à parede do capilar, durante a reação de polimerização11,32,38.
b) Preparação do monolito polimérico poroso dentro do capilar
A preparação do monolito polimérico para ECC é muito similar à descrita previamente para CLAE, utilizando uma mistura de monômeros, agente de entrecruzamento, um iniciador e agentes porogênicos. Porém, existem algumas diferenças quanto ao preenchimento das colunas cromatográficas com o monolito. Em CLAE, a mistura de polimerização ocupa todos os espaços dentro da coluna, ou seja, a coluna é completamente preenchida com o monolito polimérico. Em ECC, a detecção é feita "on column" necessitando de uma seção transparente à luz UV (cerca de 8,5 cm da extremidade do capilar) correspondente à janela de detecção. Portanto, um pequeno espaço deve estar livre de material monolítico para que a detecção aconteça em solução livre, pois, em geral, os monolitos são opacos. Algumas alternativas foram sugeridas para a criação da janela, tais como preencher o capilar com a mistura de polimerização até determinada extensão, parando o fluxo da mistura40,41 e deixando parte do capilar vazio; utilizar gel de poliacrilamida com baixo grau de entrecruzamento devido a sua transparência à luz UV42; fazer uma abertura no capilar (nominalmente 1 a 3 mm) pela combustão da camada externa de poliimida e subseqüente queima do monolito presente no local21,35; cortar uma pequena seção com uma lâmina34,43, ou até mesmo utilizar um fio de aço aquecido44. Antes, durante e após o preparo da janela deve-se bombear água pura ao longo da coluna para a remoção de resíduos ou cinzas do monolito.
A forma mais comum de preparação das fases monolíticas poliméricas é a polimerização via radical livre iniciada termicamente. Esta técnica é bem apropriada para preparação de estruturas monolíticas maiores, que preenchem completamente o volume interno de um molde fechado hermeticamente, tais como colunas de aço inoxidável ou PEEK. Além da simplicidade e da utilização de baixas temperaturas, este procedimento é perfeitamente adequado para capilares com comprimentos facilmente ajustáveis. Uma outra importante vantagem das fases monolíticas capilares com polimerização iniciada termicamente é a possibilidade da utilização de capilares de sílica fundida revestidos com poliimida, fornecendo, desta forma, uma maior resistência e flexibilidade à coluna monolítica. Porém, a dificuldade de se preparar o meio de separação dentro de um espaço restrito é o principal inconveniente da polimerização iniciada termicamente. Obviamente, é mais difícil ajustar o comprimento e a posição do monolito depois de completada sua polimerização, necessitando de métodos destrutivos para a confecção da janela de detecção.
Um aspecto importante na polimerização iniciada por luz UV é a utilização de materiais de recobrimento de capilares transparentes à mesma, comumente poli(tetrafluoretileno) PTFE37,39, ao invés de poliimida. Porém, estes materiais são menos resistentes e flexíveis que os capilares recobertos com poliimida. Por outro lado, a polimerização iniciada por luz UV é bastante apropriada para formação de monolitos dentro de um espaço específico na coluna ou em micro-dispositivos ("micro-chips")45. Usando uma máscara em um processo de foto-modelagem, a polimerização é restrita às áreas irradiadas, enquanto que os monômeros não se polimerizam nas áreas mascaradas - sem irradiação. Além disso, a polimerização foto-iniciada é rápida e realizada à temperatura ambiente37.
c) Funcionalização do monolito
A funcionalização da fase estacionária monolítica para ECC também é semelhante à realizada para CLAE. Assim como em CLAE, grupos funcionais, tais como os hidrofóbicos (contendo C8 ou C18, etc.) devem ser incorporados à superfície do monolito. Além disso, grupos carregados também devem ser inseridos no intuito de aumentar o fluxo eletroosmótico. Da mesma forma, esta etapa pode ser realizada simultaneamente à preparação do suporte monolítico. Sendo assim, devem ser adicionados dois monômeros funcionais à mistura de polimerização, um responsável pela interação e o outro (um monômero contendo moléculas carregadas), pela alimentação do fluxo eletroosmótico. Moléculas contendo cargas negativas21,32 ou positivas11,25,38 são introduzidas na matriz monolítica polimérica para fornecer fluxo eletroosmótico catódico ou anódico, respectivamente.
Bedair e El Rassi41 prepararam fases monolíticas de acrilato de estearila contendo grupos carregados positivamente para ECC no modo de fase reversa. A mistura de polimerização consistiu de um monômero com grupo de interação hidrofóbica (monoestearato diacrilato de pentaeritritol - PEDAS), um outro monômero com um grupo carregado positivamente (metilsulfato de [2-(acriloilóxi)etil]trimetilamônio - ETAS) e agentes porogênicos (cicloexanol, etilenoglicol e água). A mistura foi introduzida em um tubo capilar, com paredes internas previamente ativadas, e polimerizada a 60 °C por 18 h, resultando em colunas monolíticas poli(PEDAS-co-ETAS) de 250 ou 85 mm x 100 µm de d.i. As fases foram empregadas na separação de diferentes classes de compostos, tais como anilinas, proteínas hidrossolúveis, aminoácidos e pesticidas, por eletrocromatografia capilar utilizando fase móvel em pH 3,0. As fases apresentaram eluições rápidas com eficiências acima de 50.000 pratos m-1. A Figura 3 mostra a eluição de 14 pesticidas, em menos de 2 min empregando o monolito poli(PEDAS-co-ETAS).
Devido à facilidade de incorporação de grupos funcionais na matriz monolítica, diversas funcionalidades cromatográficas já foram avaliadas em aplicações de ECC e CLAE, tais como interação hidrofóbica (C4, C6, C8, C18, etc.)11,25,31,37,38 no modo de fase reversa, seletividade quiral34,46-48, troca iônica35,39, impressão molecular ("MIP molecularly imprinted polymer")49,50, etc. Em ECC, as fases monolíticas preparadas com grupos carregados, para garantir o fluxo eletroosmótico, são consideradas de modo misto, uma vez que, freqüentemente, se realizam separações no modo de fase normal (ou reversa) e como trocadoras de íons35,39.
Preparação de monolitos de sílica
A sílica monolítica como suporte cromatográfico é preparada na forma de um cilindro, pelo processo sol-gel. Diferentemente de monolitos poliméricos, os monolitos de sílica para uso em CLAE não são preparados dentro das colunas cromatográficas convencionais (aço inoxidável). Após a etapa de secagem no processo sol-gel, o cilindro monolítico sofre um encolhimento, perdendo o contato com a parede interna da coluna cromatográfica. Por ex., a preparação de um monolito em um molde de 9 mm de diâmetro interno gera um cilindro monolítico de 7 mm de diâmetro7,14. Portanto, materiais especiais devem ser utilizados para contornar este problema, como a utilização do plástico de poli(éter-éter-cetona) ["PEEK poly(ether-ether-ketone")], que se ajusta, com pressão e aquecimento, mais facilmente ao diâmetro do monolito de sílica46. Os monolitos de sílica preparados dentro de colunas capilares não apresentam este problema, já que a matriz monolítica é aderida à superfície interna pré-tratada da sílica18,51,52, similar aos monolitos poliméricos.
Em um procedimento típico de preparação de monolitos de sílica pelo processo sol-gel para CLAE, TMOS (tetrametoxissilano) ou TEOS (tetraetoxissilano) é adicionado a uma solução de poli(oxietileno) - agente porogênico - em água contendo um catalisador apropriado (ácido ou base). A mistura é agitada a 0 °C por um determinado tempo. A solução homogênea resultante é colocada dentro de um molde de policarbonato, deixando-a a uma determinada temperatura para que ocorram as reações de hidrólise e policondensação. A primeira etapa é a formação de um sol, suspensão coloidal de espécies sólidas em um líquido. Em seguida, é convertido em um gel, através da policondensação do sol levando a uma estrutura úmida. A amostra na forma de gel é então envelhecida por alguns dias, normalmente, na mesma temperatura da reação. O cilindro de sílica úmido formado é posteriormente lavado com água e colocado em uma solução aquosa de um solvente apropriado, para formar as estruturas mesoporosas. Sucessivas evaporações, secagem e tratamento térmico são realizados para decompor os constituintes orgânicos e estabilizar a superfície do gel hidrofílico de sílica. Após as etapas de envelhecimento e secagem, o monolito de sílica sofre um encolhimento dentro do molde. O cilindro monolítico é retirado do molde e encaixado em um tubo que, por sua vez, através de aquecimento e pressão externa, engloba e comprime o monolito em seu interior, assegurando a inexistência de espaços entre o monolito e o tubo. O monolito deve posteriormente ser funcionalizado para o modo de fase reversa através de reação "on column" com um reagente silano apropriado7,14,53. Alternativamente, a funcionalização pode acontecer antes do encapsulamento54.
Minakuchi et al.14 foram os primeiros a aplicarem os monolitos de sílica em separações cromatográficas. Em 1996, prepararam fases monolíticas de sílica pelo processo sol-gel, empregando poli(oxietileno) e NH4OH como agentes macro e microporogênicos. A mistura sol-gel foi adicionada a um molde de policarbonato obtendo-se um monolito de sílica de formato cilíndrico que, posteriormente, foi encapsulado em um tubo de PTFE 83 mm x 7 mm d.i. O monolito de sílica foi derivatizado para C18 e capeado com hexametildissilazano. A fase monolítica de sílica-C18 apresentou alta porosidade, aproximadamente 85%, com canais de 1,7 µm. O desempenho cromatográfico foi similar às colunas convencionais com partículas de 3 µm, porém com uma queda de pressão ao longo da coluna similar às colunas com partículas de 15 µm. Além disso, as fases monolíticas de sílica-C18 proporcionaram separações de uma série de alquilbenzenos até seis vezes mais rápidas quando comparadas a uma coluna comercial de partículas de 5 µm de sílica-C18.
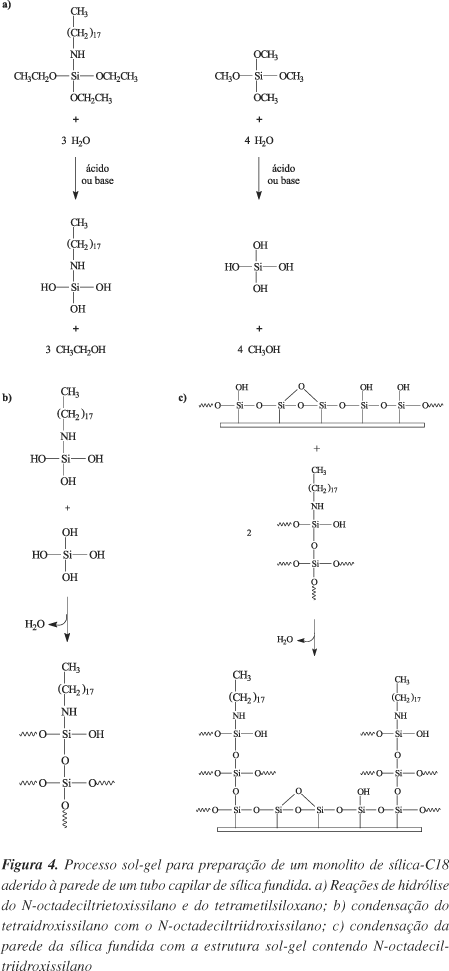
A preparação de monolitos de sílica para ECC é muito similar ao procedimento descrito para preparo de monolitos poliméricos orgânicos, com a diferença que o processo sol-gel é o responsável pela polimerização da sílica dentro do capilar18,52. As mesmas etapas estão envolvidas na preparação; preparação das paredes internas do capilar, preparação da matriz monolítica dentro do capilar e funcionalização do monolito. A Figura 4 apresenta um esquema referente à formação de um monolito de sílica pelo processo sol-gel dentro de um capilar de sílica fundida. A etapa de funcionalização foi realizada concomitantemente ao preparo da matriz de sílica pela adição de um silano funcional (N-octadeciltrietoxissilano) à reação sol-gel. A fixação de grupos carregados em monolitos de sílica não é sempre necessária, pois a própria superfície da sílica, através da ionização dos grupos silanóis, fornece cargas negativas para garantir um fluxo eletroosmótico catódico.
Ishizuka et al.52 empregaram colunas monolíticas capilares de sílica-C18, preparadas pelo processo sol-gel, no modo de µCLAE e ECC. A mistura sol-gel foi introduzida dentro de tubos capilares de 250 mm x 100 µm d.i., com paredes internas previamente ativadas. Os autores utilizaram poli(oxietileno) como agente porogênico. Após a polimerização da sílica, o bastão monolítico foi derivatizado com grupos C18. As fases monolíticas apresentaram canais de até ~8 µm, resultando em uma porosidade total de 86%, isto é, apenas 14% de material sólido dentro do capilar. As colunas monolíticas capilares foram empregadas na separação de uma série homóloga de seis alquilbenzenos em µCLAE e em ECC. O desempenho cromatográfico das fases foi bastante promissor, uma vez que eficiências de 50.000 e 130.000 pratos m-1 para o hexilbenzeno foram obtidas no modo de µCLAE e ECC, respectivamente.
Outras formas de preparação de colunas monolíticas de sílica para uso em ECC foram sugeridas, tais como a sinterização de partículas de sílica-C18 dentro do capilar55, formando uma estrutura única, ou a adesão de partículas de sílica-C18 dentro do capilar por uma solução sol-gel56. Devido à série de desvantagens originadas pelos monolitos produzidos, estes métodos não se estabeleceram.
Atualmente, os monolitos de sílica são mais empregados em aplicações de CLAE, pois, além de apresentarem maior resistência mecânica, não dilatam na presença de solventes orgânicos, como os materiais poliméricos. Em ECC, devido à maior facilidade e rapidez na preparação e da possibilidade de serem empregados em condições de pH fora da faixa de estabilidade da sílica, os monolitos poliméricos orgânicos encontram maiores aplicações que os monolitos de sílica.
CARACTERÍSTICAS DAS FASES MONOLÍTICAS
Estrutura, porosidade e propriedades das colunas com fases monolíticas
As fases estacionárias monolíticas são meios contínuos de separação cromatográfica, constituídos de pequenos domínios (esqueletos ou glóbulos) e longos canais, da ordem de micrômetros18,43. Os esqueletos de sílica e os longos canais dentro da estrutura monolítica apresentam-se em uma seqüência ordenada e ininterrupta, enquanto que, nos monolitos poliméricos, os domínios são normalmente constituídos de microglóbulos (Figura 5). Os longos canais, comumente maiores que 1 µm de diâmetro, são os principais responsáveis pela baixa resistência ao fluxo da fase móvel e, conseqüentemente, alta permeabilidade oferecida pelas fases monolíticas57. As baixas quedas de pressão nas colunas monolíticas permitem que as separações sejam realizadas em vazões de fase móvel tão altas quanto 9 mL min-1, reduzindo drasticamente o tempo de análise58,59. Os pequenos domínios de sólido dentro dos monolitos (1,0 1,5 µm) podem ou não conter poros. Estes poros contribuem para a área superficial específica do monolito e, também, na separação cromatográfica3. Os domínios não porosos são exclusivos de alguns monolitos poliméricos32. Porém, normalmente os glóbulos (domínios) das fases poliméricas possuem micro ou mesoporos, nominalmente 15% da porosidade total. Devido à presença de pequenas quantidades de micro e mesoporos, fases monolíticas poliméricas orgânicas apresentam pequenas áreas superficiais, entre 5 e 50 m2 g-1, dependendo da quantidade de microporos nos glóbulos30,35,39. Conseqüentemente, a capacidade de amostra destas fases é baixa. Nas fases monolíticas poliméricas orgânicas as porosidades intersticial e total são 60 e 70%, respectivamente2,33,40,60.
Os esqueletos de sílica nas fases monolíticas são constituídos de mesoporos (~13 nm) e são os principais responsáveis pela separação cromatográfica. Colunas monolíticas convencionais possuem uma porosidade intersticial que varia de 65 a 70% e porosidade total de 80%. Colunas monolíticas capilares de sílica possuem porosidade externa de 80% e total de 90%, enquanto que em colunas recheadas com partículas de sílica-C18 a porosidade externa é de aproximadamente 40%2,57. Devido à menor quantidade de sílica dentro das colunas, as fases estacionárias monolíticas apresentam menor área superficial específica quando comparadas às colunas de materiais particulados60. Normalmente, a área superficial de uma coluna monolítica de sílica varia de 50 a 300 m2 g-1.
Os valores de porosidade dos monolitos, principalmente dos poliméricos, podem não ser exatamente aqueles do momento de operacionalização, pois os monolitos encontram-se no estado úmido, enquanto que as medidas são freqüentemente conduzidas a seco60.
A porosidade de uma coluna monolítica é controlada pelos agentes porogênicos; quanto maior a percentagem de agentes porogênicos na mistura de polimerização, maior a porosidade da fase. Além disso, o tipo de agente também influencia nas propriedades porosas do monolito resultante. Freqüentemente, empregam-se dois agentes porogênicos na mistura de polimerização, um macroporogênico (dodecanol, decanol, etc.) e um microporogênico (hexano, butanol, propanol, etc.)29. Outro fator que afeta a porosidade de uma fase monolítica é o grau de entrecruzamento do polímero. Quanto maior a percentagem de agente de entrecruzamento na mistura de polimerização, menor será o tamanho dos poros na fase monolítica39. Desta forma, as propriedades porosas dos materiais monolíticos podem ser rigorosamente controladas durante seu preparo.
Além do controle das propriedades físicas, por ex., o grau de entrecruzamento, tamanho dos poros, diâmetro da coluna, etc., as propriedades químicas da superfície dos monolitos também podem ser rigorosamente controladas. Em princípio quaisquer moléculas, ionizadas ou não, hidrofóbicas ou hidrofílicas, de interação específica, etc., podem ser incorporadas às estruturas do monolito. Estas moléculas podem ser ligadas à superfície, após a polimerização ou reação sol-gel, ou serem incorporadas à matriz monolítica durante o preparo dos monolitos poliméricos orgânicos, ou à base de sílica com um monômero funcional, ou um silano funcional, respectivamente. Além disso, em eletrocromatografia capilar há a possibilidade de se realizar separações empregando fluxo eletroosmótico catódico ou anódico (fluxo invertido ou reverso) ou ambos, dependendo da molécula e do tipo de carga (negativa ou positiva) que foram incorporadas à matriz do monolito.
Características cromatográficas
A principal vantagem das colunas monolíticas frente às colunas recheadas com partículas é a possibilidade de realizar separações rápidas em vazões elevadas de fase móvel, sem comprometer a resolução cromatográfica. Estas vazões elevadas somente são possíveis devido à presença dos longos canais, de diâmetro maior que 1 µm, oferecendo uma baixa resistência ao fluxo de fase móvel. A separação cromatográfica ocorre principalmente nos mesoporos dos pequenos domínios, que são os principais responsáveis pelas altas eficiências obtidas com as fases monolíticas52. As eficiências das fases monolíticas são similares às obtidas com colunas recheadas com partículas de 3 µm, no entanto a queda de pressão ao longo de uma coluna monolítica é cerca de 65% menor2. As vantagens das fases monolíticas são mais claramente observadas quando se compara o número de pratos por unidade de pressão com as fases particuladas. As fases monolíticas apresentam cerca de 1 900 pratos bar-1, enquanto que as fases estacionárias com partículas de 3 µm geram apenas 310 pratos bar-1 57. Isto implica que colunas monolíticas apresentam eficiências similares às colunas recheadas, porém com uma pressão no sistema cromatográfico aproximadamente seis vezes menor.
As fases estacionárias monolíticas apresentam altura de prato tipicamente na faixa de 5 a 20 µm, tanto para CLAE como para ECC. O melhor desempenho das colunas monolíticas é obtido quando se utilizam vazões elevadas de fase móvel2,30,39,57,60. Desta forma, a contribuição do termo de transferência de massa da fase móvel é maior, enquanto que da fase estacionária é menor quando comparado às colunas de materiais particulados60. Um outro fator que contribui para as altas eficiências é a transferência de massa que ocorre por convecção, além de difusão (caminhos múltiplos), nas fases monolíticas. Este tipo de fluxo permite uma maior rapidez na transferência de massa e só ocorre em poros bastante largos3.
As propriedades cromatográficas das fases monolíticas assemelham-se às de materiais particulados em algumas ocasiões. Por ex., a atividade silanofílica, devido à existência de grupos silanóis residuais em fases monolíticas baseadas em sílica, é muito similar e muitas vezes até maior que em colunas recheadas59,61. Visando contornar tais inconvenientes, novas alternativas vêm sendo propostas no campo de preparo das fases monolíticas, tais como preparação de materiais híbridos orgânicos-inorgânicos62, preparação de monolitos de sílica recobertos com óxidos quimicamente mais estáveis63, etc.
Como cada fase monolítica é única, devido à preparação direta dentro da coluna cromatográfica, a reprodutibilidade de colunas é um fator de extrema importância e que vêm sendo sistematicamente avaliada. A reprodutibilidade da preparação das colunas foi verificada tanto para fases monolíticas fabricadas em laboratório como para as comerciais57, sendo o principal parâmetro avaliado o tempo de retenção ou a migração de vários compostos, não retidos e retidos. Dentre os diversos estudos, as estimativas dos desvios padrão relativo ("RSD relative standard deviations") para as retenções não excederam a 5% para todos os compostos avaliados28,33,57,58. Estes valores são aceitáveis, levando em consideração o modo de preparo das colunas monolíticas.
APLICAÇÕES
Cromatografia líquida de alta eficiência
A fácil preparação das fases monolíticas, em colunas com diâmetros internos que variam desde 0,02 até 8 mm, possibilitou a utilização destas fases para as mais diversas finalidades. As separações de compostos orgânicos sob condições de fase reversa representam quase a totalidade das aplicações das fases monolíticas em CLAE. Vários compostos foram analisados por CLAE (ou µCLAE) empregando colunas monolíticas poliméricas orgânicas ou à base de sílica, tais como hidrocarbonetos aromáticos43,52, compostos básicos61 e ácidos30, oligonucleotídeos30, polímeros24, etc. Porém, a maioria das aplicações de fases monolíticas em CLAE de fase reversa se concentrou em separações e análises de fármacos, peptídeos e proteínas.
A grande vantagem de se utilizar fases monolíticas na análise de fármacos é a possibilidade de separação muito rápida, devido às vazões elevadas permitidas com este tipo de coluna. Para tal aplicação, fases monolíticas à base de sílica são as mais empregadas. Por ex., Pavli e Kmetec64 utilizaram uma coluna monolítica comercial à base de sílica (Chromolith RP-18e, 100 x 4,6 mm d.i.) para separação dos princípios ativos e produtos de degradação oxidativa de um antibiótico. Comparando com uma coluna convencional de material particulado, o tempo de análise foi reduzido drasticamente (aproximadamente 8 vezes) a vazões de até 5 mL min-1. Análises de quatro antidepressivos, mianserin e metabólitos, foram realizadas por CLAE empregando uma coluna monolítica comercial de sílica-C18 (Chromolith RP-18e)10. Os autores validaram uma metodologia para determinação, após separações rápidas, destes antidepressivos em plasma humano. Foram obtidas separações em menos de 5 min com boas seletividades e detectabilidades para os quatro compostos. Assim como estes, vários outros trabalhos empregando colunas monolíticas para separações e determinações de fármacos foram descritos9,10,65-67.
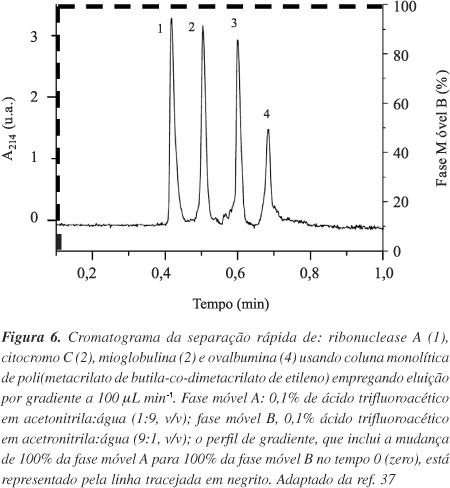
A fácil preparação de colunas miniaturizadas (20, 100, 200 µm de d.i.) possibilitou a utilização de fases monolíticas nas separações e determinações rápidas de diversos peptídeos e proteínas11,31. Lee et al.37 prepararam colunas capilares monolíticas (200 µm de d.i.) de poli(metacrilato de butila-co-dimetacrilato de etileno) para separações, em pouco mais de 40 s, de quatro proteínas (ribonuclease A, citocromo C, mioglobulina e ovalbumina), obtendo altas reprodutibilidades nas corridas cromatográficas (Figura 6). Além disso, colunas com estas dimensões permitem um melhor acoplamento do cromatógrafo a líquido com o espectrômetro de massas (CL-EM). Por ex., Premstaller et al.29 prepararam colunas monolíticas de poli(estireno-co-divinilbenzeno) de 20 µm de diâmetro interno e empregaram-nas em separações rápidas de proteínas do fotossistema II de espinafre e peptídeos trípticos de catalase bovina, com detecção por espectrometria de massas. Os autores obtiveram eficiências superiores às fases particuladas convencionais. Walcher et al.68 prepararam colunas monolíticas (200 µm de d.i.) de poli(estireno-co-divinilbenzeno) aplicando-as em separações de peptídeos, proteínas e ácidos nucléicos em um sistema CLAE-IES-EM ("IES ionização electrospray"). Os autores obtiveram boas separações, com 50 a 140 picos eluindo em 10 min. Estudos proteômicos e genômicos empregando CLAE vêm se difundindo com o advento das colunas monolíticas capilares29,68,69.
Além de aplicações no modo de fase reversa, fases monolíticas foram preparadas, pela incorporação de grupos carregados, para separações por troca iônica. Sýkora et al.25 prepararam colunas monolíticas de poli(metacrilato de glicidila-co-dimetacrilato de etileno) derivatizadas com dietilamina, fornecendo funcionalidades de troca aniônica para separação de uma série homóloga de oligonucleotídeos. Vazões de 4 mL min-1 foram empregadas para obter rápidas separações dos ácidos no modo cromatográfico de troca aniônica.
Separações de compostos quirais também já foram relatadas empregando colunas monolíticas. Chankvetadze et al.46 ligaram covalentemente 3,5-dimetilfenilcarbamato em moléculas de celulose e, por reação "on-column", no suporte de sílica de uma coluna monolítica comercial (Chromolith Performance Si, 100 x 4,6 mm) para separações enantiosseletivas. Chen et al.47 prepararam colunas monolíticas capilares à base de sílica pelo processo sol-gel e as derivatizaram com seletores quirais, tais como L-fenilalaninamida, L-alaninamida e L-prolinamida para separação enantiosseletiva de aminoácidos dansilados e hidroxiácidos por µCLAE. Todas as colunas monolíticas preparadas mostraram alta enantiosseletividade.
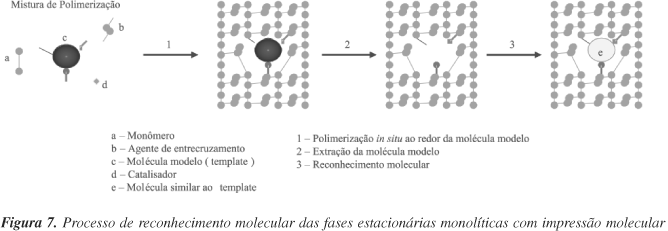
Fases estacionárias monolíticas com impressão molecular também foram aplicadas em CLAE para enantiosseparações. O modo de preparo das fases monolíticas com impressão molecular é bastante similar ao já descrito, exceto que uma molécula modelo é adicionada à mistura de polimerização e, após o término da reação, esta molécula é extraída por um solvente apropriado, deixando sua impressão molecular na matriz monolítica (Figura 7). Huang et al.50 prepararam seis fases estacionárias monolíticas com diferentes impressões moleculares, em colunas de 150 x 4 mm d.i., para separação de enantiômeros e diastereoisômeros de derivados de aminoácidos empregando CLAE. Enantiosseparações muito rápidas foram obtidas quando se utilizou eluição por gradiente a vazões elevadas. Huang et al.70 prepararam fases monolíticas com impressão molecular com cinchonina, N-(carbobenzilóxi)-L-triptofano (Cbz-L-Trp) e Fmoc-L-triptofano (Fmoc-L-Trp) em colunas curtas para separações rápidas de enantiômeros e diastereoisômeros de alcalóides cinchonina. Todas as separações foram obtidas com boas estereosseletividades em menos de 3 min empregando vazões de até 9 mL min-1.
Eletrocromatografia capilar
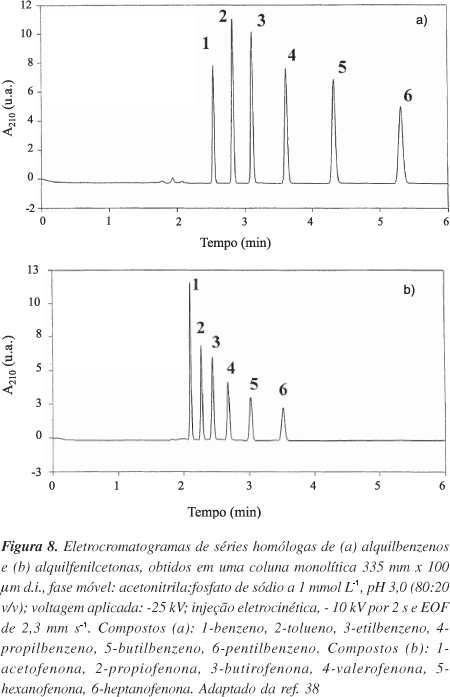
Assim como em CLAE, a maioria das aplicações das fases monolíticas em eletrocromatografia capilar ocorre no modo de fase reversa, porém, com eficiências de separação muito maiores que, normalmente, às obtidas em µCLAE empregando as mesmas colunas capilares. Por ex., Buszewski e Szumski32 compararam o desempenho de diversas colunas monolíticas capilares à base de polímeros metacrílicos usando ECC e µCLAE. Em todas as aplicações, as eficiências em ECC foram superiores às obtidas em µCLAE. Diversas moléculas já foram analisadas no modo de fase reversa da eletrocromatografia capilar, a citar: hidrocarbonetos aromáticos policíclicos18,35,36,55,56, compostos básicos33,35,36,63 e ácidos33,36,55,62, fármacos28, pesticidas41, entre outros. Estes compostos são freqüentemente separados com rapidez, devido à presença de moléculas na FM que fornecem altas velocidades de fluxo eletroosmótico e às altas eficiências cromatográficas, provenientes do perfil planar do FEO. Por ex., Bedair e El Rassi38 prepararam uma fase estacionária monolítica catiônica, baseada em poliacrilatos com grupos amino quaternários embutidos, para fornecer FEO anódico em altas velocidades. A coluna monolítica foi aplicada na separação de séries homólogas de seis alquilbenzenos e seis alquilfenilcetonas com eficiências superiores a 100.000 pratos m-1 em menos de 6 min. Os eletrocromatogramas estão apresentados na Figura 8.
Em contraste à boa separação de moléculas pequenas no modo de fase reversa de ECC, a separação de analitos macromoleculares ionizados (peptídeos, proteínas e aminoácidos) é difícil. Isto se deve, basicamente, a dois motivos: a forte migração eletroforética destas moléculas no campo elétrico em direção ao cátodo ou ânodo, dependendo do pH da fase móvel e, principalmente, a forte interação eletrostática destas moléculas com os grupos carregados das fases estacionárias, essenciais para o fluxo eletroosmótico, levando a longos tempos de eluição ou adsorção irreversível. Para contornar esta limitação e analisar polipeptídeos e proteínas por ECC, Gusev et al.11 prepararam fases estacionárias monolíticas de poli(estireno-co-divinilbenzeno) funcionalizadas com grupos dimetiloctilamina. A fase monolítica foi empregada na separação de insulina e três peptídeos angiotensina sintéticos em pH 3, garantindo que todas as macromoléculas e os grupos amino da superfície do monolito apresentassem cargas positivas e, portanto, fluxo eletroosmótico invertido. Desta forma, evitaram-se as interações eletrostáticas indesejáveis entre os polipeptídeos e os grupos amino, obtendo boas separações e picos essencialmente simétricos. De maneira similar, Fu et al.36 analisaram bi- e tripeptídeos em dois valores de pH (2 e 8) empregando uma coluna monolítica à base de polimetacrilatos com grupos carregados positiva ou negativamente, dependendo do pH da fase móvel. Em pH 2,0 a eluição dos seis peptídeos analisados foi aproximadamente três vezes mais rápida que em pH 8,0, além de apresentar melhores simetrias para os picos das macromoléculas, devido à inexistência de interações eletrostática entre as macromoléculas e os grupos iônicos da superfície do monolito. Outros trabalhos11,21,36,41, diante da facilidade de preparação, empregaram colunas monolíticas para separação de peptídeos, proteínas ou aminoácidos por ECC, obtendo resultados bastante satisfatórios e confirmando a potencialidade da eletrocromatografia capilar em análises de macromoléculas.
Além do modo de fase reversa, outras modalidades de ECC também foram avaliadas empregando fases estacionárias monolíticas. Lämmerhofer et al.39 empregaram fases monolíticas altamente hidrofílicas, à base de dimetilaminoetil metacrilato e hidroxietil metacrilato, no modo de fase normal em ECC. Estes autores promoveram a separação de compostos fenólicos, xantinas e aminas aromáticas, obtendo ótimas separações e eficiências acima de 80.000 pratos m-1. Allen e El Rassi71 prepararam fases monolíticas hidrofílicas à base de sílica contendo grupos ciano ligados à superfície e as empregaram na separação de compostos ácidos, básicos e neutros no modo de fase normal em ECC. Separações rápidas, em menos de 5 min, devido às altas velocidades do fluxo eletroosmótico, e eficiências de até 160.000 pratos m-1 foram obtidas para fenóis, mono e oligossacarídeos, nucleosídeos e clorofenóis.
Os grupos carregados (positiva ou negativamente), que são incorporados na fase monolítica para assegurar o fluxo eletroosmótico, podem agir também como sítios de interação e, conseqüentemente, além de interações hidrofílicas ou hidrofóbicas, podem ocorrer mecanismos de separação por troca iônica21,35,39. As fases monolíticas empregadas em ECC são comumente denominadas de ECC em modo misto. Vários trabalhos empregaram separações por troca iônicas. Por ex., Fu et al.35 prepararam colunas monolíticas a partir de metilsulfato de [2-(metacrilóxi)etil]trimetilamônio e dimetacrilato de etileno, denominada de poli(MEANS-co-EDMA), e as utilizaram nos modos de fase reversa e troca aniônica, devido à presença de grupos amino carregados. As colunas monolíticas de poli(MEANS-co-EDMA) promoveram separações de ácidos aromáticos com ordens de eluição de acordo com o pH da fase móvel. Separações rápidas, em menos de 3,5 min, ocorreram quando o mecanismo de interação prevalecente foi o de troca aniônica.
O controle adequado da porosidade das fases monolíticas em ECC permite a obtenção de colunas de eletrocromatografia capilar apropriadas para separações no modo de exclusão. A cromatografia por exclusão (CE "size exclusion chromatography") é um método isocrático de separação com base nas diferenças de volumes hidrodinâmicos dos analitos. Poucos trabalhos foram realizados em ECC empregando este modo de separação apesar de, em alguns casos, este mecanismo ocorrer de forma casual. Uma aplicação de fases monolíticas operando com mecanismos de exclusão em ECC foi mostrada por Peters et al.72. Fases monolíticas de poli(metacrilato de butila-co-dimetacrilato de etileno-co-ácido 2-acrilamido-2-metil-1-propanossulfônico) foram empregadas na separação de padrões de poliestireno de diferentes massas molares e, ao observar a ordem de eluição dos padrões, sugeriu-se que o principal mecanismo de separação cromatográfica foi a exclusão por tamanho.
A facilidade de preparação de monolitos com diferentes funcionalidades estimulou a preparação de fases estacionárias monolíticas quirais para enantiosseparações em ECC34. A simples adição de um monômero com função seletora quiral à mistura de polimerização produz excelentes colunas monolíticas quirais para ECC enantiosseletiva. Lämmerhofer et al.48 prepararam colunas monolíticas quirais à base de polimetacrilatos pela adição de um derivado da quinidina à mistura de polimerização. Esta coluna foi aplicada para separação de enantiômeros R,S de dinitrobenzoil-leucina (DNB-leucina), obtendo-se boa enantiosseparação e eficiências de até 75 000 pratos m-1.
Fases monolíticas com reconhecimento molecular seletivo também tiveram aplicações em ECC. O processo relativamente simples de preparo das fases monolíticas com impressão molecular tem proporcionado um número expressivo de aplicações, tanto em ECC como em CLAE. Liu et al.49 prepararam fases monolíticas pela adição da molécula modelo, ácido 4-hidroxibenzóico (4-HBA), à mistura de polimerização de ácido metacrílico e dimetacrilato de etilenoglicol. Após o término da polimerização as moléculas de 4-HBA foram removidas, deixando sua impressão molecular na matriz monolítica polimérica. O reconhecimento molecular destas fases monolíticas foi avaliado pela separação de tiouréia, 4-HBA e 2-HBA. O reconhecimento molecular, além de satisfatório, resultou em altas eficiências e tempos de análises mais curtos, comparados às colunas com impressão molecular convencionais.
CONCLUSÕES
As fases estacionárias monolíticas vêm sendo cada vez mais empregadas em separações cromatográficas, principalmente nos últimos cinco anos, após o advento da comercialização de monolitos de sílica. As separações rápidas e eficiências altas atraem os interesses de laboratórios de análises de rotina, devido à possibilidade de aumentar o número de análises/dia. Além disso, a facilidade de preparo e o fino controle das propriedades físicas e químicas das fases monolíticas, sem necessidade de equipamentos sofisticados e de custo elevado, também são fatores importantes que contribuem para este crescente interesse. Estas importantes vantagens, aliadas à preparação da matriz monolítica aderida às paredes de tubos capilares, eliminando a necessidade de filtros retentores, têm contribuído para uma maior aceitação da eletrocromatografia capilar, visto que a difusão desta técnica estava limitada pelos inconvenientes inerentes aos materiais particulados convencionais, como a dificuldade de enchimento de colunas capilares com partículas muito pequenas e a fabricação de filtros para reter as partículas dentro dos capilares.
Apesar das indiscutíveis vantagens oferecidas pelas fases monolíticas, os resultados cromatográficos não são tão surpreendentes quanto à sua estrutura. Grande parte dos problemas relacionados aos materiais particulados ainda permanece, como a atividade silanofílica, a estabilidade dos materiais à base de sílica, etc., e novos problemas surgiram, tais como a capacidade limitada de processamento de amostra devido a menor área superficial dos materiais monolíticos e a preparação individualizada das fases estacionárias, acarretando maiores variações coluna a coluna. Além disso, o excessivo encolhimento de monolitos de sílica durante o preparo e as desvantagens do uso de monolitos poliméricos fazem do seu uso em CLAE convencional uma opção pouco atrativa para pesquisas científicas, atraindo maiores interesses para análises de rotina. A fácil preparação de colunas miniaturizadas e o melhor acoplamento ao espectrômetro de massas são alguns dos atrativos para a intensificação de pesquisas na tecnologia de colunas monolíticas capilares visando seu melhoramento, principalmente para aplicações em µCLAE.
Diante das vantagens e limitações das fases monolíticas, estas devem ser consideradas como complementares às fases convencionais de materiais particulados e não como alternativos. Porém, os materiais monolíticos se encontram atualmente no "estágio inicial" de seu desenvolvimento e um futuro promissor é destinado a estes materiais, principalmente quando preparados em capilares para ECC ou µCLAE.
AGRADECIMENTOS
Aos Profs. F. Augusto e J. A. F. da Silva (IQ - Unicamp) pelas valiosas discussões e à Fapesp e ao CNPq pelo suporte financeiro.
Recebido em 28/2/05; aceito em 22/6/05; publicado na web em 8/12/05
- 1. Svec, F.; Peters, E. C.; Sýkora, D.; Fréchet, J. M. J.; J. Chromatogr., A 2000, 887, 3.
- 2. Tanaka, N.; Kobayashi, H.; Anal. Bioanal. Chem. 2003, 376, 298.
- 3. Zou, H.; Huang, X.; Ye, M.; Luo, Q.; J. Chromatogr., A 2002, 954, 5.
- 4. MacNair, J. E.; Patel, K. D.; Jorgenson, J. W.; Anal. Chem. 1999, 71, 700.
- 5. Dittmann, M. M.; Rozing, G. P.; J. Chromatogr., A 1996, 744, 63.
- 6. Svec, F.; Fréchet, J. M. J.; Anal. Chem. 1992, 64, 820.
- 7. Ishizuka, N.; Minakuchi, H.; Nakanishi, K.; Hirao, K.; Tanaka, N.; Colloids Surf., A 2001, 187, 273.
- 8. Rouquérol, J.; Avnir, D.; Fairbridge, C. W.; Everett, D. H.; Haynes, J. H.; Pericone, N.; Ramsay, J. D. F.; Sing, K. S. W.; Unger, K. K.; Pure Appl. Chem. 1994, 66, 1739.
- 9. Vallano, P. T.; Mazenko, R. S.; Woolf, E. J.; Matuszewski, B. K.; J. Chromatogr., B: Biomed. Sci. Appl. 2002, 779, 249.
- 10. Hefnawy, M. M.; Aboul-Enein, H. Y.; Anal. Chim. Acta 2004, 504, 291.
- 11. Gusev, I.; Huang, X.; Horváth, C.; J. Chromatogr., A 1999, 855, 273.
- 12. Ross, W. D.; Jefferson, R T.; J. Chromatogr. Sci. 1970, 8, 386.
- 13. Hjertén, S.; Liao, J. L.; Zhang, R.; J. Chromatogr. 1989, 473, 273.
- 14. Minakuchi, H.; Nakanishi, K.; Soga, N.; Ishizuka, N.; Tanaka, N.; Anal. Chem. 1996, 68, 3498.
- 15. Shintani, Y.; Zhou, X.; Furuno, M.; Minakuchi, H.; Nakanishi, K.; J. Chromatogr., A 2003, 958, 351.
- 16. Bedair, M.; El Rassi, Z.; J. Chromatogr., A 2004, 1044, 177.
- 17. Huclová, J.; Satínský, D.; Karlícek, R.; Anal. Chim. Acta 2003, 494, 133.
- 18. Hayes, J. D.; Malik, A.; Anal. Chem. 2000, 72, 4090.
- 19. Malik, A.; Electrophoresis 2002, 23, 3973.
- 20. Hilder, E. F.; Svec, F.; Fréchet, J. M. J.; Electrophoresis 2002, 23, 3934.
- 21. Hoegger, D.; Freitag, R.; J. Chromatogr., A 2003, 1004, 195.
- 22. Moravcova, D.; Jandera, P.; Urban, J.; Planeta, J.; J. Sep. Sci. 2004, 27, 789.
- 23. Moravcova, D.; Jandera, P.; Urban, J.; Planeta, J.; J. Sep. Sci. 2003, 26, 1005.
- 24. Petro, M.; Svec, F.; Fréchet, J. M. J.; J. Chromatogr., A 1996, 752, 59.
- 25. Sýkora, D.; Svec, F.; Fréchet, J. M. J.; J. Chromatogr., A 1999, 852, 297.
- 26. Miyabe, K.; Cavazzini, A.; Gritti, F.; Kele, M.; Guiochon, G.; Anal. Chem. 2003, 75, 6975.
- 27. Hilder, E. F.; Svec, F.; Fréchet, J. M. J.; J. Chromatogr., A 2004, 1044, 3.
- 28. Hoegger, D.; Freitag, R.; J. Chromatogr., A 2001, 914, 211.
- 29. Premstaller, A.; Oberacher, H.; Walcher, W.; Timperio, A. M.; Zolla, L.; Chervet, J. P.; Cavusoglu, N.; van Dorsselaer, A.; Huber, C. G.; Anal. Chem. 2001, 73, 2390.
- 30. Oberacher, H.; Premstaller, A.; Huber, C. G.; J. Chromatogr., A 2004, 1030, 201.
- 31. Quigley, C. L.; Marlin, N.; Smith, N. W.; J. Chromatogr., A 2004, 1030, 195.
- 32. Buszewski, B.; Szumski, M.; Chromatographia 2004, 60, s261.
- 33. Jiang, T.; Jiskra, J.; Claessens, H. A.; Cramers, C. A.; J Chromatogr., A 2001, 923, 215.
- 34. Preinerstorfer, B.; Bicker, W.; Lindner, W.; Lämmerhofer, M.; J. Chromatogr., A 2004, 1044, 187.
- 35. Fu, H.; Xie, C.; Xiao, H.; Dong, J.; Hu, J.; Zou, H.; J. Chromatogr., A 2004, 1044, 237.
- 36. Fu, H.; Xie, C.; Dong, J.; Huang, X.; Zou, H.; Anal. Chem. 2004, 76, 4866.
- 37. Lee, D.; Svec, F.; Fréchet, J. M. J.; J. Chromatogr., A 2004, 1051, 53.
- 38. Bedair, M.; El Rassi, Z.; J. Chromatogr., A 2003, 1013, 35.
- 39. Lämmerhofer, M.; Svec, F.; Fréchet, J. M. J.; Lindner, W.; J. Chromatogr., A 2001, 925, 265.
- 40. Grafnetter, J.; Coufal, P.; Tesarová, E.; Sucháncová, J.; Bosáková, Z.; Sevcík, J.; J. Chromatogr., A 2004, 1049, 43.
- 41. Bedair, M.; El Rassi, Z.; J. Chromatogr., A 2003, 1013, 47.
- 42. Fujimoto, C.; Fujise, Y.; Anal. Chem. 1996, 68, 2753.
- 43. Shu, X.; Chen, L.; Yang, B.; Guan, Y.; J. Chromatogr., A 2004, 1052, 205.
- 44. Ericson, C.; Liao, J.; Nakazato, K.; Hjertén, S.; J. Chromatogr., A 1997, 767, 33.
- 45. Rohr, T.; Hilder, E. F.; Donovan, J. J.; Svec, F.; Fréchet, J. M. J.; Macromolecules 2003, 36, 1677.
- 46. Chankvetadze, B.; Ikai, T.; Yamamoto, C.; Okamoto, Y.; J. Chromatogr., A 2004, 1042, 55.
- 47. Chen, Z.; Uchiyama, K.; Hobo, T.; J. Chromatogr., A 2002, 942, 83.
- 48. Lämmerhofer, M.; Peters, E. C.; Yu, C.; Svec, F.; Fréchet, J. M. J.; Anal. Chem. 2000, 72, 4614.
- 49. Liu, Z. S.; Xu, Y. L.; Yan, C.; Gao, R. Y.; Anal. Chim. Acta 2004, 523, 243.
- 50. Huang, X.; Zou, H.; Chen, X.; Luo, Q.; Kong, L.; J. Chromatogr., A 2003, 984, 273.
- 51. Motokawa, M.; Kobayashi, H.; Ishizuka, N.; Minakuchi, H.; Nakanishi, K.; Jinnai, H.; Hosoya, K.; Ikegami, T.; Tanaka, N.; J. Chromatogr., A 2002, 961, 53.
- 52. Ishizuka, N.; Minakuchi, H.; Nakanishi, K.; Soga, N.; Nagayama, H.; Hosoya, K.; Tanaka, N.; Anal. Chem. 2000, 72, 1275.
- 53. Siouffi, A. M.; J. Chromatogr., A 2003, 1000, 801.
- 54. Lubda, D.; Cabrera, K.; Nakanishi, K.; Minakuchi, H.; J. Sol-Gel Sci. Technol. 2002, 23, 185.
- 55. Aziaie, R.; Huang, X.; Farnan, D.; Horváth, C.; J. Chromatogr., A 1998, 806, 251.
- 56. Tang, Q.; Xin, B.; Lee, M.; J. Chromatogr., A 1999, 837, 35.
- 57. Engelhardt, H.; Götzinger, A.; Chromatographia 2004, 60, s207.
- 58. Svec, F.; LC-GC Europe 2003, 16, 24.
- 59. Martínez, J. M. H.; Méndez, A.; Bosch, E.; Rosés, M.; J. Chromatogr., A 2004, 1060, 135.
- 60. Ikegami, T.; Tanaka, N.; Curr. Opin. Chem. Biol. 2004, 8, 527.
- 61. McCalley, D.; J. Chromatogr., A 2002, 965, 51.
- 62. Yan, L.; Zhang, Q.; Zhang, J.; Zhang, L.; Li, T.; Feng, Y.; Zhang, L.; Zhang, W.; Zhang, Y.; J. Chromatogr., A 2004, 1046, 255.
- 63. Shi, Z. G.; Feng, Y. Q.; Xu, L.; Zhang, M.; Da, S. L.; Talanta 2004, 63, 593.
- 64. Pavli, V.; Kmetec, V.; J. Pharm. Biomed. Anal. 2004, 36, 257.
- 65. Schmidt, A. H.; J. Chromatogr., A 2005, 1073, 377.
- 66. Enein, H. Y. A.; Ali, I.; Talanta 2005, 65, 276.
- 67. Rocheleau, M. J.; Jean, C.; Bolduc, J.; Carazzato, D.; J. Pharm. Biomed. Anal. 2003, 31, 191.
- 68. Walcher, W.; Oberacher, H.; Troiani, S.; Hölzl, G.; Oefner, P.; Zolla, L.; Huber, C. G.; J. Chromatogr., B: Biomed. Sci. Appl. 2002, 782, 111.
- 69. Premstaller, A.; Oefner, P. J.; Oberacher, H.; Huber, C. G.; Anal. Chem. 2002, 74, 4688.
- 70. Huang, X.; Qin, F.; Chen, X.; Liu, Y.; Zou, H.; J. Chromatogr., B: Biomed. Sci. Appl. 2004, 804, 13.
- 71. Allen, D.; El Rassi, Z.; J. Chromatogr., A 2004, 1029, 239.
- 72. Peters, E. C.; Petro, M.; Svec, F.; Fréchet, J. M. J.; Anal. Chem. 1998, 70, 2296.
Datas de Publicação
-
Publicação nesta coleção
03 Abr 2006 -
Data do Fascículo
Abr 2006
Histórico
-
Recebido
28 Fev 2005 -
Aceito
22 Jun 2005