Resumo
Novel and quantitative mass spectrometry methods for rapid and accurate enantiomeric excess determination are presented. These methodologies use electrospray ionization (ESI) and mass spectrometry (MS) to detect and analyze, via collision-induced dissociation (CID), mass-selected transition metal complexes that promote enantio especific interactions. The data from CID are conveniently treated by the kinetic method, a sensitive linear free energy method of treating mass spectrometric results. Four different variations of this methodology are described: single ratio method (S R), quotient ratio method (Q R), fixed ligand method (S Rfixed), and quotient ratio method with fixed ligand (Q Rfixed). These individual methods are compared and their main features discussed in detail.
enantiomeric excess determinattion; electrospray ionization mass spectrometry; kinetic method
enantiomeric excess determinattion; electrospray ionization mass spectrometry; kinetic method
DIVULGAÇÃO
Análise quiral por espectrometria de massas através da utilização do método cinético
Chiral analysis by mass spectrometry using the kinetic method
Daniella Vasconcellos AugustiI; Rodinei AugustiII, * * e-mail: augusti@ufmg.br
IFundação Ezequiel Dias, Instituto Octávio Magalhães, Divisão de Vigilância Sanitária, 30510-010 Belo Horizonte - MG
IIDepartamento de Química, Instituto de Ciências Exatas, Universidade Federal de Minas Gerais, CP 702, 31270-901 Belo Horizonte - MG
ABSTRACT
Novel and quantitative mass spectrometry methods for rapid and accurate enantiomeric excess determination are presented. These methodologies use electrospray ionization (ESI) and mass spectrometry (MS) to detect and analyze, via collision-induced dissociation (CID), mass-selected transition metal complexes that promote enantio especific interactions. The data from CID are conveniently treated by the kinetic method, a sensitive linear free energy method of treating mass spectrometric results. Four different variations of this methodology are described: single ratio method (SR), quotient ratio method (QR), fixed ligand method (SRfixed), and quotient ratio method with fixed ligand (QRfixed). These individual methods are compared and their main features discussed in detail.
Keywords: enantiomeric excess determinattion; electrospray ionization mass spectrometry; kinetic method.
INTRODUÇÃO
A descoberta de moléculas assimétricas por Pasteur1, em 1848, foi o ponto inicial para o estudo de moléculas quirais. Poucos tópicos em Química, Biologia ou Ciências Farmacêuticas têm atraído maior interesse que a quiralidade. A presença exclusiva de L- aminoácidos e D-açúcares nos seres vivos, tema que tem intrigado os cientistas há mais de um século, mostra que a quiralidade teve grande influência no desenvolvimento da vida na Terra2.
A importância de drogas quirais puras é reconhecida desde o começo dos anos 60, a partir do famoso episódio conhecido como "A Tragédia da Talidomida", quando se verificou que o enantiômero levógero deste composto causava má formação nos fetos das gestantes que o consumiam3. Deste então, antes que uma droga quiral possa ser comercializada, são exigidos3 testes farmacológicos e toxicológicos com os enantiômeros puros.
A produção de drogas quirais tem imposto uma demanda crescente no desenvolvimento de métodos rápidos de análise enantiomérica em áreas que envolvem estudos farmacológicos e clínicos4. A determinação de excessos enantioméricos em drogas quirais é comumente realizada por cromatografia líquida de alta eficiência (HPLC), onde se usa uma coluna quiral contendo ciclodextrina como fase estacionária5-7. Sistemas similares têm sido utilizados em eletroforese capilar (CE)8, onde as pequenas dimensões da coluna e a rapidez da separação fazem desta técnica um método atrativo para quantificação quiral9-12. A ressonância magnética nuclear (NMR) utiliza reagentes quirais, mas requer uma quantidade grande de amostra e não tolera impurezas.
RESOLUÇÃO QUIRAL POR ESPECTROMETRIA DE MASSAS
Dentre as pesquisas por métodos rápidos de determinação de excesso enantiomérico está a espectrometria de massas (MS), uma técnica muito sensível e seletiva13. Nos últimos anos, progressos importantes têm sido obtidos, permitindo que esta técnica se torne uma das principais ferramentas na análise quiral. O reconhecimento quiral e a determinação de excesso enantiomérico (ee) através da espectrometria de massas podem ser classificados em quatro tipos principais de experimentos: (a) ionização química (CI-MS)14-16 e espectrometria de massas com bombardeamento por átomos rápidos (FAB-MS)17-21 adutos diasteroisoméricos são gerados usando-se uma referência quiral e, em seguida, analisados em um espectrômetro de massas de estágio simples. Um enantiômero do analito é isotopicamente marcado e, assim, a mistura correspondente do aduto diasteroisomérico pode ser resolvida. Esses adutos são do tipo hospedeiro-convidado e suas interações são forças não-covalentes, como ligação de hidrogênio ou forças de van der Waals. Recentemente, essa técnica vem sendo ampliada através da utilização da ionização "electrospray" (ESI)22-25; (b) reconhecimento quiral baseado em reações íon-molécula na fase gasosa, freqüentemente envolvendo reações de troca. Adutos diasteroisoméricos do tipo hospedeiro-convidado são gerados a partir de um hospedeiro quiral, como a b-ciclodextrina26. Tais adutos são selecionados por massa e o analito quiral é substituído em uma reação com um gás neutro (quiral ou aquiral). A distinção quiral é possível porque a velocidade de troca varia com a quiralidade do analito incorporado dentro do hospedeiro quiral23,27-29; (c) a terceira metodologia é baseada na dissociação induzida por colisão (CID) de adutos diasteroisoméricos formados pelo analito e uma referência quiral em um experimento de espectrometria de massas sequencial (MS/MS)30-35 e, (d) o assunto explorado neste trabalho, usa também a espectrometria de massas sequencial, mas emprega o método cinético para quantificação dos efeitos quirais36,37.
O MÉTODO CINÉTICO
Em 1977 Cooks e colaboradores38, utilizando um equipamento do tipo MIKES "mass-analyzed ion kinetic energy spectroscopy" com fonte de ionização química, estudaram a dissociação metaestável de dímeros prótonados de aminas alifáticas. Como produtos, as respectivas aminas protonadas foram obtidas com abundâncias relativas altamente sensíveis à diferença nas afinidades protônicas das aminas. Este foi o primeiro trabalho usando o método cinético39,40. Desde então, tem sido empregado para estimar valores relativos de quantidades termoquímicas41-44, tais como afinidade protônica39,45, distinção isomérica36, basicidade em fase gasosa38, energia de ionização46, afinidade por cátions metálicos43,47 e, mais recentemente, para determinar excessos enantioméricos37,40.
Trata-se de um procedimento aproximado, geralmente aplicado em determinações termoquímicas relativas e tem como base as diferentes velocidades de dissociações unimoleculares competitivas (k1 e k2) de complexos selecionados por massa. A Equação 1 mostra, por ex., um complexo contendo ligações de hidrogênio (A---H+---B) que fornece, após dissociação, os dois monômeros individuais protonados38,39,48.

Tratamentos teóricos baseados na teoria de dissociação unimolecular leva à equação simplificada do método cinético (Equação 2)38,39,45,49-53

Nesta equação, [AH+] e [BH+] são as abundâncias das bases protonadas, D(DGB) é a diferença na basicidade em fase gasosa dos compostos A e B, e Teff é a temperatura efetiva. Essa relação é uma simples conseqüência de se escrever as expressões de velocidades individuais e cancelar para o reagente comum (A---H+---B)54, 55. A basicidade em fase gasosa (DGB) é definida como o oposto da variação da energia livre (-DG) para a reação (Equação 3):

Na equação simplificada do método cinético (Equação 2), as seguintes aproximações estão implícitas: as diferenças de entropia entre os canais de dissociação competitivos são negligenciadas; a energia de ativação para as reações reversas é nula ou desprezível; inexistência de outras formas isoméricas para o complexo ativado.
O método é sensível para pequenas diferenças em valores termoquímicos (< 1 KJ mol-1) e é também aplicável para compostos não voláteis e polares, incluindo peptídeos e outras biomoléculas difíceis de serem analisadas por outros métodos56. Além disso, o método não exige que os compostos de interesse estejam puros.
DETERMINAÇÃO DE EXCESSOS ENANTIOMÉRICOS UsANDO O MÉTODO CINÉTICO E ESPECTROMETRIA DE MASSAS SEQÜENCIAL COM FONTE DE IONIZAÇÃO "ELECTROSPRAY"
O método da razão simples (SR)
Um enantiômero, D ou L, de um analito quiral (AD ou AL) e um composto de referência quiral (ref*) são complexados com um íon bivalente de metal de transição (MII) formando os complexos triméricos [M(AD)(ref*)2 H]+ ou [M(AL)(ref*)2 H]+, os quais são detectados através de análise por ESI-MS ("electrospray ionization mass spectrometry")57,58. A seguir, tais complexos são selecionados por massa e dissociados por colisão com um gás inerte ("CID- collision-induced dissociation") produzindo os respectivos íons diméricos [M(ref*)2 H]+ e [M(ref*)(AD(L)) H]+, como mostrado na Equação 4:

onde kA e kref são as respectivas velocidades de dissociação dos complexos triméricos.
A razão entre as abundâncias destes fragmentos (RD e RL) é definida de acordo com as Equações 5 e 6, respectivamente:

Nestas condições, a razão de seletividade quiral (RC) é definida de acordo com a Equação 7:

Quanto maior a diferença de RC da unidade, maior o grau de seletividade quiral observado. Se RC = 1, não há discriminação quiral, indicando que a combinação específica envolvendo o cátion metálico e o composto de referência não é capaz de promover distinção quiral entre os dois enantiômeros, AD e AL.
Um diagrama de energia para a dissociação dos complexos [M(AD)(ref*)2 H]+ e [M(AL)(ref*)2 H]+ é mostrado na Figura 1. Neste diagrama, a diferença em energia D(DG) entre os íons diasteroisoméricos [M(ref*)(AD) H]+ e [M(ref*)(AL) H]+ é responsável pela diferença entre RD e RL, o que leva a um valor de RC¹ 1. Se tal diferença em energia não é observada obtém-se, conseqüentemente, um valor de RC = 1.
A relação entre a razão R (R = [M(A)(ref*) H]+ / [M(ref*)2 H]+) e o excesso enantiomérico (ee) do analito quiral (A) pode ser obtida a partir da equação do método cinético (Equação 2). O logaritmo natural da razão R é proporcional às diferenças de energia livre para as dissociações competitivas que fornecem os dois íons diméricos (Equação 4). Tal relação pode ser visualizada a seguir (Equação 8):

onde R é a constante dos gases ideais, Teff é a temperatura efetiva do complexo ativado (temperatura média dos dois complexos ativados para as duas reações competitivas), e D(DG) (Figura 1) é definido como a diferença nas energias livre entre as reações 9 e 10, para as quais as energias de ativação reversas são consideradas desprezíveis:

Quando o analito A é constituído por enantiômeros puros, D ou L, D(DG) torna-se D(DG)D ou D(DG)L. Para uma mistura enantiomérica com um excesso do enantiômero D, por ex., pode-se escrever (Equação 11):

Combinando-se as Equações 11 e 8 obtém-se a expressão que prevê uma relação linear entre o excesso enantiomérico (ee) e o ln (R) (Equação 12):

Finalmente, combinando-se as Equações 12 e 8 obtém-se a Equação 13, que coloca ln(R) como uma função de ln(RL) e ln(RD):

Deste modo, em um gáfico ln(R) versus ee, o intercepto representa a média entre os valores de ln(RL) e ln(RD), enquanto que a inclinação corresponde à metade da diferença entre ln(RD) e ln(RL).
O sucesso do método depende da escolha adequada da referência, bem como do metal de transição. O primeiro passo para quantificar misturas quirais é a construção da curva analítica entre ln(R) e o ee sendo possível determinar rapidamente a relação entre os componentes da mistura, utilizando um espectrômetro de massas seqüencial. Como uma relação linear entre ln(R) e o ee é esperada, em análises rápidas é possível construir a curva analítica com apenas duas amostras com conhecidos ee, como por ex., os enantiômeros D e L puros ou a mistura racêmica.
A Figura 2 mostra, por ex., um espectro ESI-MS59 de uma solução em água/metanol 1:1, contendo o aminoácido modificado N-Ac-L-Phe, utilizado como referência quiral, o cátion metálico Cu2+ e o açúcar D-ribose. Note a presença do complexo trimérico desejado [Cu(N-Ac-L-Phe)2(D-Ribose)-H]+ de m/z 626. Outras espécies também foram detectadas, as quais estão indicadas nesta mesma Figura 2. Um espectro similar (não mostrado) foi obtido a partir de uma solução contendo N-Ac-L-Phe, Cu2+ e L-ribose, onde se verificou, principalmente, a formação do complexo trimérico [Cu (N-Ac-L-Phe)2(L-Ribose)-H]+ também de m/z 626.
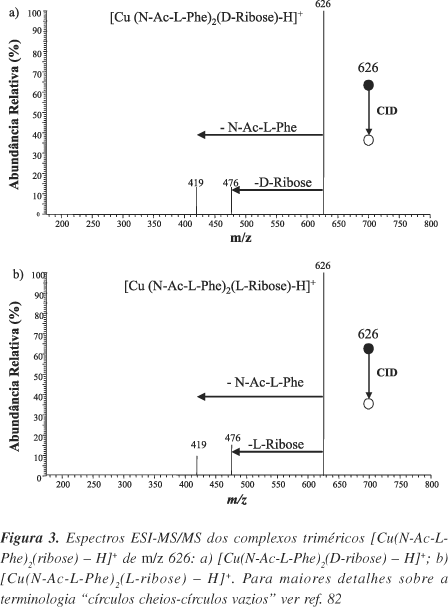
A seleção por massa dos complexos [Cu (N-Ac-L-Phe)2(D-Ribose)-H]+ e [Cu (N-Ac-L-Phe)2(L-Ribose)-H]+ e fragmentação por colisão com hélio (CID) gerou os espectros MS/MS mostrados na Figura 3.
Em ambos espectros observa-se a presença de dois fragmentos provenientes da perda de ribose, [Cu (N-Ac-L-Phe)2-H]+ de m/z 476, e de N-Ac-L-Phe, [Cu (N-Ac-L-Phe) (L-Ribose)-H]+ de m/z 419. As diferentes razões (R) entre as intensidades dos íons de m/z 419 e 476 para cada complexo trimérico são causadas pelas diferentes interações entre analito (D e L-ribose) e referência quiral (N-Ac-L-Phe). O fator de seletividade quiral (RC) pode ser calculado medindo-se as intensidades dos íons de m/z 419 e 476 em ambos espectros e aplicando-se a Equação 7. A razão R entre as intensidades dos íons de m/z 419 e 476 depende da proporção dos enantiômeros do analito quiral em solução. Deste modo, preparando-se várias soluções com excessos enantioméricos ee conhecidos e medindo-se as respectivas razões R, pode-se facilmente construir uma curva de calibração entre ln(R) versus ee. Finalmente, esta curva de calibração pode ser utilizada para determinação do excesso enantiomérico de soluções desconhecidas. A principal desvantagem do método da razão simples envolve a necessidade de se utilizar, para construção da curva analítica, duas amostras do analito com proporções enantioméricas exatamente conhecidas as quais, em muitas situações, não podem ser obtidas.
O método da razão simples60 tem sido aplicado com sucesso na determinação de excessos enantioméricos de soluções de aminoácidos61-64, a-hidroxi-ácidos65,66, peptídeos67-70, drogas quirais71-74, vitaminas75, açúcares76 e outras substâncias77.
VARIAÇÕES DO MÉTODO
Existem alguns casos em que o método da razão simples (SR) não pode ser aplicado. Visando superar tais limitações, trabalhos recentes foram publicados empregando métodos alternativos para determinação de excessos enatioméricos, os quais são descritos a seguir.
Método do quociente (QR)
O método do quociente (QR) foi introduzido por Cooks e Tao78, em 2002. Neste método, complexos triméricos [M(ref*D)(A)2 H]+ e [M(ref*L)(A)2 H]+ são gerados a partir de duas soluções contendo um íon metálico (MII), um analito quiral (A) e uma referência quiral (ref*D ou ref*L). A seleção por massa e dissociação (CID) de tais complexos, em dois experimentos MS/MS distintos realizados em condições idênticas, gera os íons diméricos [M(A)2 H]+ e [M(ref D(L))(A) H]+ pela perda da referência ou do analito, respectivamente. A diferença de energia requerida para gerar as formas diasteroisoméricas [M(ref*D)(A) H]+ e [M(ref *L)(A) H]+ resulta em diferentes abundâncias, as quais são medidas em relação à abundância do fragmento [M(A)2 H]+. O quociente das razões (RR), equivalente ao R no método da razão simples, depende da composição enantiomérica do analito A (Equação 14):

Quando o analito é enantiomericamente puro, RR torna-se RRD ou RRL. Deste modo, o fator de seletividade quiral RRC é calculado pelo quociente das razões RR (Equações 15 e 16):

ou seja,

Quanto mais distante o valor de RRC estiver da unidade, melhor será a seletividade quiral. Se RRC = 1, temos um sistema que não consegue distinguir as interações estereoquímicas nas condições empregadas.
A relação entre RR e o ee é derivada do método cinético. Por analogia com o método da razão simples, ln (RR) é proporcional à diferença de energia livre para as dissociações competitivas, que fornecem os produtos diméricos (Equação 17):

onde R é a constante dos gases ideais, Teff é a temperatura efetiva média dos dois complexos ativados para as duas reações competitivas e D(DG) é definido como a diferença na energia livre da Equação 18, para a qual a energia de ativação reversa é considerada desprezível:

Quando uma mistura dos dois enantiômeros de A (AD e AL) é examinada, D(DG) é a soma das trocas de energia livre D(DG)D ou D(DG)L das duas reações independentes, Equações 19 e 20, respectivamente.

Os complexos diméricos [M(ref*L)AD H]+ e [M(ref*D)AL H]+, bem como [M(ref*D)AD H]+ e [M(ref*L)AL H]+, são pares enantioméricos. Portanto, as reações acima possuem o mesmo valor absoluto de D(DG), porém com sinais opostos (Equação 21):

Para uma mistura enantiomérica, em que o excesso enantiomérico do enantiômero D é expresso por ee62, pode-se escrever (Equação 22):

e a relação entre RR e o ee pode ser expressa combinando as Equações 17 e 22, para se obter a Equação 23:

Quando a amostra é uma mistura racêmica (ee = 0%), de acordo com as Equações 21 e 23, ln(RR) será sempre zero (Equação 24):

A Equação 23 prevê a relação linear do ln(RR) e o ee. A inclinação da curva analítica depende da magnitude de D(DGD) ou D(DGL). Uma distinção quiral adequada (valores de D(DGD) ou D(DGL) elevados) fornece uma inclinação elevada. Análogo ao método da razão simples, uma curva analítica com dois pontos pode ser usada para análises de amostras desconhecidas, ressaltando-se que neste método toda mistura racêmica fornecerá ln(RR) = 0. Ou seja, na prática precisa-se apenas de uma amostra com pureza óptica conhecida para a construção da curva analítica, o que vem a ser a maior vantagem do método, uma vez que nem sempre os dois enantiômeros puros estão disponíveis comercialmente. O Esquema 1 mostra uma comparação sucinta entre os métodos da razão simples (a) e do quociente (b). No método do quociente, duas referências enantiomericamente puras são empregadas em dois experimentos consecutivos, enquanto que no método da razão simples uma única referência quiral é necessária.
Método do ligante fixo (SRfixo)
Uma nova versão do método da razão simples foi desenvolvida por Cooks e Wu79, em 2003. Como no método da razão simples, os autores prepararam uma solução em água/metanol 1:1 contendo um analito quiral (A), uma referência quiral (ref*), um metal bivalente (MII) e um ligante quelante ligado fortemente ao metal (ligante fixo, Lfixo).
Neste experimento, complexos do tipo [(M(Lfixo-H)(ref*)(A)]+ foram gerados por ESI, selecionados por massa e fragmentados por CID. Tal processo levou a dois produtos de dissociação competitivos, através da perda da referência (ref*) ou do analito (A), como mostrado na Equação 25:

As mesmas considerações e analogias empregadas no método da razão simples também são válidas para o método do ligante fixo, levando a uma relação análoga entre ln R e o ee (Equações 26 a 28)

Em comparação com o método da razão simples, o complexo trimérico [(M(Lfixo-H)(ref*)(A)]+ perde os mesmos fragmentos neutros, i.e. A e ref*, em um experimento CID. O diferencial deste método é que o ligante fixo permanece ligado ao cátion metálico central, mesmo após a fragmentação do complexo trimérico. A vantagem está na facilidade de otimização do sistema e, portanto, um reconhecimento quiral superior é prontamente obtido. Comparando-se os mesmos sistemas, o método do ligante fixo mostrou uma maior seletividade quiral em relação ao da razão simples79.
Método do quociente com ligante fixo (QRfixo)
Mais recentemente, Cooks e Wu80 publicaram uma nova metodologia para determinação de excesso enantiomérico, denominada método do quociente com ligante fixo (QRfixo). Nesta metodologia, aplica-se o método do quociente fazendo-se uso de um ligante fixo, como suscintamente demonstrado no Esquema 2.
O método QRfixo requer duas análises consecutivas em experimentos MS/MS. Assim como no método do quociente (QR), somente uma amostra com ee conhecido é necessária para construção da curva de calibração, já que toda mistura racêmica fornecerá ln(RRfixo) = 0.
Em comparação com o método do quociente, o método QRfixo apresenta as seguintes vantagens: melhor exatidão na quantificação devido à simplicidade da cinética de dissociação e melhor seletividade quiral, através da alteração de propriedades, tais como tamanho e funcionalidade do ligante fixo; possibilidades múltiplas para a construção de curvas de calibração com dois pontos que podem ser usadas para averiguar os dados, um ponto importante em aplicações farmacêuticas. Por ex., a utilização do dipeptídeo Ala-Ala como ligante fixo em suas quatro configurações (AlaL-AlaL; AlaL-AlaD; AlaD-AlaL; AlaD-AlaD) permite a geração de quatro curvas de calibração que podem ser analisadas e a melhor delas utilizada na determinação do excesso enantiomérico de um analito quiral (A), através da determinação da respectiva razão RRfixo (Equação 29):

e, pelas trocas de quiralidade dos ligantes fixos ou das referências, as interações quirais no complexo são refinadas, permitindo maximizar a diferenciação quiral.
CONCLUSÃO E PERSPECTIVAS
Os métodos para determinação de excesso enantiomérico descritos neste artigo envolvem uma aplicação original e genial do método cinético. Tais metodologias, de acordo com as características descritas a seguir, apresentam grande potencial de aplicação, embora estudos adicionais com amostras reais sejam necessários para que possam ser definitivamente validadas e aplicadas pelos laboratórios de análise: são métodos rápidos, onde cada análise tem duração de cerca de 3 min; são métodos simples pois não se utilizam análises por via úmida, não havendo necessidade de derivatizações e pré-concentrações; não há necessidade da utilização de colunas quirais; são de baixo custo, desde que um espectrômetro de massas comercial com uma fonte ESI e capacidade para realizar experimentos MS/MS possa ser utilizado; sensíveis, pois podem ser utilizados na determinação de amostras contendo uma pequena fração de um dos enantiômeros; podem ser empregados em condições totalmente adversas, como na superfície de outros planetas e meteoros, onde a detecção de um excesso enantiomérico de moléculas vitais, como açúcares e aminoácidos, pode ser uma prova definitiva da presença de vida. Trabalhos recentes81 já mostram a possibilidade de se ampliar tal metodologia na quantificação de misturas contendo três constituintes, como por ex. D, L e meso-ácido tartárico. Porém, trabalhos futuros serão necessários para averiguar a possibilidade de quantificação de outras misturas multi-componentes, contendo enantiômeros e diastereoisômeros.
AGRADECIMENTOS
Ao CNPq e FAPEMIG pelas bolsas e auxílio financeiro concedidos.
59. Augusti, D. V.; Augusti, R.; resultados não publicados.
Recebido em 17/12/04; aceito em 21/7/05; publicado na web em 20/1/06
- 1. Pasteur, L. C. R.; Hebd. Seance Acad. Sci. 1848, 26, 535.
- 2. Stryer, L.; Biochemistry, Spectrum: Heidelberg, 1996.
- 3. Barreiro, E. J.; Ferreira, V. F.; Costa, P. R. R.; Quim. Nova 1997, 20, 647.
- 4. Kyranos, J. N.; Hogan, J. C.; Anal. Chem. 1998, 70, 389a.
- 5. Armstrong, D. W.; Ward, T. J.; Armstrong, R. D.; Beesley, T. E.; Science 1986, 232, 1132.
- 6. Schurig, V.; Nowotny, H. P.; Angew. Chem., Int. Ed. 1990, 29, 939.
- 7. Glausch, A.; Hahn, J.; Schurig, V.; Chemosphere 1995, 30, 2079.
- 8. Jorgenson, J. W.; Lukacs, K. D.; J. Chromatogr. 1981, 218, 209.
- 9. Penn, S. G.; Bergstrom, E. T.; Goodall, D. M.; Loran, J. S.; Anal. Chem. 1994, 66, 2866.
- 10. Snopeck, J.; Jelineck, L.; Smolková, E.; J. Cromatogr., A 1992, 609, 1.
- 11. Kuhn, R.; Hoffstetterkuhn, S.; Chromatographia 1992, 34, 505.
- 12. Terabe, S.; Otsuka, K.; Nishi, H.; J. Chromatogr., A 1994, 666, 295.
- 13. Hoffmann, E.; Stroobant, V.; Mass Spectrometry: Principles and Applications, John Wiley & Sons: Chichester, 2002.
- 14. Fales, H. M.; Wright, G. J.; J. Am. Chem. Soc. 1977, 99, 2339.
- 15. Nikolaev, E. N.; Denisov, E. V.; Nikolaeva, M. I.; Futrell, J. H.; Rakov, V. S.; Winkler, F. J.; Adv. Mass Spectrom. 1998, 14, 279.
- 16. Kostyanovsky, R. G.; Nikolaev, E. N.; Kharybin, O. N.; Kadorkina, G. K.; Kostyanovsky, V. R.; Mendeleev Commun. 2003, 97.
- 17. Sawada, M.; Mass Spectrom. Rev. 1997, 16, 73.
- 18. Sawada, M.; Takai, Y.; Yamada, H.; Hirayama, S.; Kaneda, T.; Tanaka, T.; Kamada, K.; Mizooku, T.; Takeuchi, S.; Ueno, K.; Hirose, K.; Tobe, Y.; Naemura, K.; J. Am. Chem. Soc. 1995, 117, 7726.
- 19. Pocsfalvi, G.; Liptak, M.; Huszthy, P.; Bradshaw, J. S.; Izatt, R. M.; Vekey, K.; Anal. Chem. 1996, 68, 792.
- 20. Sawada, M.; Yamaoka, H.; Takai, Y.; Kawai, Y.; Yamada, H.; Azuma, T.; Fujioka, T.; Tanaka, T.; Int. J. Mass Spectrom. 1999, 193, 123.
- 21. Sawada, M.; Shizuma, M.; Takai, Y.; Yamada, H.; Kaneda, T.; Hanafusa, T.; J. Am. Chem. Soc. 1992, 114, 4405.
- 22. Sawada, M.; Shizuma, M.; Takai, Y.; Adachi, H.; Takeda, T.; Uchiyama, T.; Chem. Commun. 1998, 1453.
- 23. Ahn, S.; Ramirez, J.; Grigorean, G.; Lebrilla, C. B.; J. Am. Soc. Mass Spectrom. 2001, 12, 278.
- 24. Sawada, M.; Takai, Y.; Yamada, H.; Yoshikawa, M.; Arakawa, R.; Tabuchi, H.; Takada, M.; Tanaka, J.; Shizuma, M.; Yamaoka, H.; Hirose, K.; Fukuda, K.; Tobe, Y.; Eur. J. Mass Spectrom. 2004, 10, 27.
- 25. Sawada, M.; Takai, Y.; Yamada, H.; Nishida, J.; Kaneda, T.; Arakawa, R.; Okamoto, M.; Hirose, K.; Tanaka, T.; Naemura, K.; J. Chem. Soc., Perkin Trans. 2 1998, 701.
- 26. He, P.; Lu, H. J.; Guo, Y. L.; Anal. Lett. 2003, 36, 493.
- 27. Chu, I. H.; Dearden, D. V.; Bradshaw, J. S.; Huszthy, P.; Izatt, R. M.; J. Am. Chem. Soc. 1993, 115, 4318.
- 28. Dearden, D. V.; Dejsupa, C.; Liang, Y. J.; Bradshaw, J. S.; Izatt, R. M.; J. Am. Chem. Soc. 1997, 119, 353.
- 29. Ramirez, J.; He, F.; Lebrilla, C. B.; J. Am. Chem. Soc. 1998, 120, 7387.
- 30. Smith, G.; Leary, J. A.; J. Am. Chem. Soc. 1996, 118, 3293.
- 31. Ho, Y. H.; Squires, R. R.; J. Am. Chem. Soc. 1992, 114, 10961.
- 32. Tabet, J. C.; Tetrahedron 1987, 43, 3413.
- 33. Lu, H. J.; Guo, Y. L.; J. Am. Soc. Mass Spectrom. 2003, 14, 571.
- 34. Lu, H. J.; Guo, Y. L.; Anal. Chim. Acta 2003, 482, 1.
- 35. Gronert, S.; Fagin, A. E.; Okamoto, K.; J. Am. Soc. Mass Spectrom. 2004, 15, 1509.
- 36. Shen, W. Y.; Wong, P. S. H.; Cooks, R. G.; Rapid Commun. Mass Spectrom. 1997, 11, 71.
- 37. Vekey, K.; Czira, G.; Anal. Chem. 1997, 69, 1700.
- 38. Cooks, R. G.; Kruger, T. L.; J. Am. Chem. Soc. 1977, 99, 1279.
- 39. Cooks, R. G.; Patrick, J. S.; Kotiaho, T.; Mcluckey, S. A.; Mass Spectrom. Rev. 1994, 13, 287.
- 40. Cooks, R. G.; Wong, P. S. H.; Acc. Chem. Res. 1998, 31, 379.
- 41. Augusti, R.; Turowski, M.; Cooks, R. G.; J. Mass Spectrom. 2004, 39, 558.
- 42. Meurer, E. C.; Gozzo, F. C.; Augusti, R.; Eberlin, M. N.; Eur. J. Mass Spectrom. 2003, 9, 295.
- 43. Eberlin, M. N.; Kotiaho, T.; Shay, B. J.; Yang, S. S.; Cooks, R. G.; J. Am. Chem. Soc. 1994, 116 , 2457.
- 44. Denault, J. W.; Feng, W.; Cooks, R. G.; Gozzo, F. C.; Eberlin, M. N.; J. Phys. Chem. A 2000, 104, 11290.
- 45. Mcluckey, S. A.; Cameron, D.; Cooks, R. G.; J. Am. Chem. Soc. 1981, 103, 1313.
- 46. Wong, P. S. H.; Ma, S. G.; Cooks, R. G.; Anal. Chem. 1996, 68, 4254.
- 47. Yang, S. S.; Wong, P.; Ma, S. G.; Cooks, R. G.; J. Am. Soc. Mass Spectrom. 1996, 7, 198.
- 48. Armentrout, P. B.; J. Mass Spectrom. 1999, 34, 74.
- 49. Wright, L. G.; Mcluckey, S. A.; Cooks, R. G.; Wood, K. V.; Int. J. Mass Spectrom. 1982, 42, 115.
- 50. Boand, G.; Houriet, R.; Gaumann, T.; J. Am. Chem. Soc. 1983, 105, 2203.
- 51. Brodbeltlustig, J. S.; Cooks, R. G.; Talanta 1989, 36, 255.
- 52. Majumdar, T. K.; Clairet, F.; Tabet, J. C.; Cooks, R. G.; J. Am. Chem. Soc. 1992, 114, 2897.
- 53. Klots, C. E.; J. Phys. Chem. A 1997, 101, 5378.
- 54. Ervin, K. M.; Int. J. Mass Spectrom. 2000, 196, 271.
- 55. Cooks, R. G.; Koskinen, J. T.; Thomas, P. D.; J. Mass Spectrom. 1999, 34, 85.
- 56. Caprioli, R. M.; J. Mass Spectrom. 1999, 34, 73.
- 57. Cole, R. B.; Electrospray Ionization Mass Spectrometry, John Wiley & Sons Inc.: Chichester, 1997.
- 58. Moraes, M. C. B.; do Lago, C. L.; Quim. Nova 2003, 26, 556.
- 60. Tao, W. A.; Cooks, R. G.; Anal. Chem. 2003, 75, 25a.
- 61. Tao, W. A.; Cooks, R. G.; Nikolaev, E. N.; Eur. J. Mass Spectrom. 2002, 8, 107.
- 62. Tao, W. A.; Zhang, D. X.; Nikolaev, E. N.; Cooks, R. G.; J. Am. Chem. Soc. 2000, 122, 10598.
- 63. Tao, W. A.; Zhang, D. X.; Wang, F.; Thomas, P. D.; Cooks, R. G.; Anal. Chem. 1999, 71, 4427.
- 64. Wu, L. M.; Tao, W. A.; Cooks, R. G.; J. Mass Spectrom. 2003, 38, 386.
- 65. Tao, W. A.; Wu, L. M.; Cooks, R. G.; Chem. Commun. 2000, 2023.
- 66. Wu, L. M.; Tao, W. A.; Cooks, R. G.; Anal. Bioanal. Chem. 2002, 373, 618.
- 67. Tao, W. A.; Cooks, R. G.; Angew. Chem., Int. Ed. 2001, 40, 757.
- 68. Tao, W. A.; Wu, L. M.; Cooks, R. G.; J. Am. Soc. Mass Spectr. 2001, 12, 490.
- 69. Wu, L. M.; Lemr, K.; Aggerholm, T.; Cooks, R. G.; J. Am. Soc. Mass Spectrom. 2003, 14, 152.
- 70. Wu, L. M.; Meurer, E. C.; Young, B.; Yang, P. X.; Eberlin, M. N.; Cooks, R. G.; Int. J. Mass Spectrom. 2004, 231, 103.
- 71. Tao, W. A.; Gozzo, F. C.; Cooks, R. G.; Anal. Chem. 2001, 73, 1692.
- 72. Shizuma, M.; Takai, Y.; Kawamura, M.; Takeda, T.; Sawada, M.; J. Chem. Soc., Perkin Trans. 2 2001, 1306.
- 73. Augusti, D. V.; Augusti, R.; Carazza, F.; Cooks, R. G.; Chem. Commun. 2002, 2242.
- 74. Tao, W. A.; Wu, L. M.; Cooks, R. G.; Wang, F.; Begley, J. A.; Lampert, B.; J. Med. Chem. 2001, 44, 3541.
- 75. Augusti, D. V.; Lago, R. M.; Augusti, R.; J. Braz. Chem. Soc. 2004, 15, 786.
- 76. Augusti, D. V.; Carazza, F.; Augusti, R.; Tao, W. A.; Cooks, R. G.; Anal. Chem. 2002, 74, 3458.
- 77. Yu, C. T.; Guo, Y. L.; Chen, G. Q.; Zhong, Y. W.; J. Am. Soc. Mass Spectrom. 2004, 15, 795.
- 78. Tao, W. A.; Clark, R. L.; Cooks, R. G.; Anal. Chem. 2002, 74, 3783.
- 79. Wu, L. M.; Cooks, R. G.; Anal. Chem. 2003, 75, 678.
- 80. Wu, L. M.; Meurer, E. C.; Cooks, R. G.; Anal. Chem. 2004, 76, 663.
- 81. Wu, L. M.; Clark, R. L.; Cooks, R. G.; Chem. Commun. 2003, 136.
- 82. Eberlin, M. N.; Mass Spectrom. Rev. 1997, 16, 113.
Datas de Publicação
-
Publicação nesta coleção
03 Abr 2006 -
Data do Fascículo
Abr 2006
Histórico
-
Aceito
21 Jul 2005 -
Recebido
17 Dez 2004